

Abstract
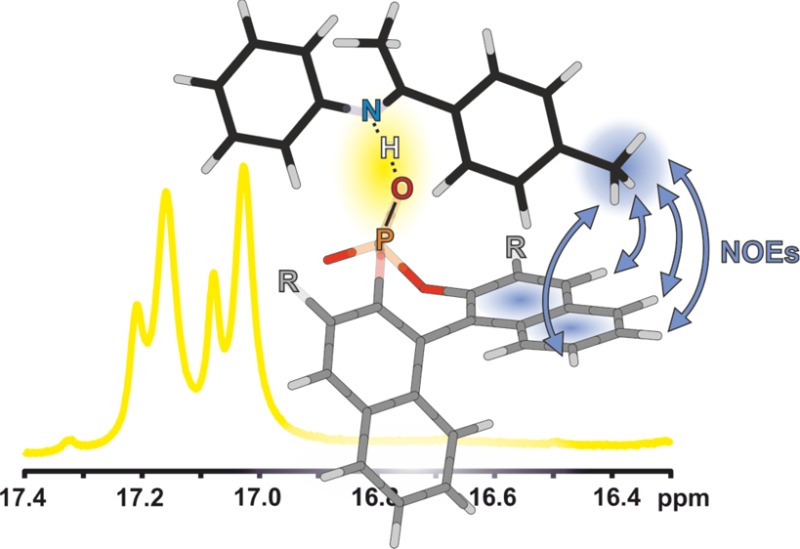
Despite the huge success of enantioselective Brønsted acid catalysis, experimental data about structures and activation modes of substrate/catalyst complexes in solution are very rare. Here, for the first time, detailed insights into the structures of imine/Brønsted acid catalyst complexes are presented on the basis of NMR data and underpinned by theoretical calculations. The chiral Brønsted acid catalyst R-TRIP (3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate) was investigated together with six aromatic imines. For each investigated system, an E-imine/R-TRIP complex and a Z-imine/R-TRIP complex were observed. Each of these complexes consists of two structures, which are in fast exchange on the NMR time scale; i.e., overall four structures were found. Both identified E-imine/R-TRIP structures feature a strong hydrogen bond but differ in the orientation of the imine relative to the catalyst. The exchange occurs by tilting the imine inside the complex and thereby switching the oxygen that constitutes the hydrogen bond. A similar situation is observed for all investigated Z-imine/R-TRIP complexes. Here, an additional exchange pathway is opened via rotation of the imine. For all investigated imine/R-TRIP complexes, the four core structures are highly preserved. Thus, these core structures are independent of electron density and substituent modulations of the aromatic imines. Overall, this study reveals that the absolute structural space of binary imine/TRIP complexes is large and the variations of the four core structures are small. The high mobility is supposed to promote reactivity, while the preservation of the core structures in conjunction with extensive π–π and CH−π interactions leads to high enantioselectivities and tolerance of different substrates.
Introduction
Since the development of the first BINOL-derived phosphoric acids in 2004 by Akiyama and Terada,1,2 Brønsted acid catalysts have emerged as a viable catalyst class for a variety of enantioselective reactions.3−7 Due to their excellent yields and ee values, these catalysts are able to provide a complementary technique to transition metal catalyzed reactions. For the Brønsted acid catalyzed enantioselective reduction of imines, Rueping et al. postulated a catalytic cycle that involves the formation of a binary complex as a precatalytic state consisting of catalyst 1b and the imine (see Figure 1a).4 Subsequent addition of a reduction agent, e.g., Hantzsch ester, leads to a ternary complex. However, the reasons for the high enantioselectivity induced by these catalysts have remained unclear, as structural investigations on the key reactive intermediates are very challenging.
Figure 1.

(a) Catalytic cycle for the transfer hydrogenation, as proposed by Rueping et al.4 (b) Brønsted acid catalysts 1a–c successfully applied for enantioselective reduction of imines or reductive amination.3−5
A DFT study by Himo on the transfer hydrogenation of imines proposed that the enantioselectivity is dictated by the kinetics of the hydride transfer step in the ternary complex.8 Using a truncated phosphoric acid as a catalyst model, the transition state involving the Z-imine was predicted to react faster than the E-imine. Goodman et al. could further confirm the Z-imine transition state as the reactive pathway by using catalysts 1a and 1b (see Figure 1b) in their calculations.9
So far, the only detailed experimental data about the binary complex was reported by MacMillan and co-workers, who were able to identify the structure of the E-imine/1a complex by a single crystal X-ray analysis.3 In 2013, W. Tang et al. elucidated the structure of a ternary 1c/imine/iridium complex with the help of two NOE signals between methyl groups.10 Additional experimental investigations of solvent and temperature effects on the interaction between catalyst and imine were performed by Rueping et al.11 With a combination of dielectric spectroscopy, quantum chemistry, and 1H NMR, they showed the important role of close-contact ion-pairs in a transfer hydrogenation reaction. With the help of vibrational circular dichroism, Merten et al. investigated the influence of different solvents on the transmission of stereochemical information from the chiral Brønsted catalyst anion to a MnIII–salen complex in an asymmetric epoxidation reaction.12 Our first NMR study about the hydrogen bond properties of achiral imine/diphenylphosphate complexes proposed the coexistence of ion paired and neutral hydrogen bond complexes.13 However, our recent theoretical and NMR investigations on the hydrogen bond situation in binary complexes containing chiral phosphoric acids revealed the ion paired complexes as the dominating species.14 A landscape of asymmetric single-well potential surfaces was revealed, composed of ion-paired complexes. However, to the best of our knowledge, no detailed experimental structure analysis of any Brønsted acid catalyst/substrate complex in solution has been presented so far.
In this paper, detailed structural information on chiral Brønsted acid/imine complexes is described on the basis of NMR data and theoretical calculations. A high mobility of the imine inside the substrate/catalyst complex is observed for both E- and Z-imine. For each isomer, two orientations of the imine in relation to the catalyst can be observed. These two basic structures are in fast exchange on the NMR time scale. Interchanging the orientations is possible by either rotating the imine by 180° or tilting the imine and thereby switching the oxygen that constitutes the hydrogen bond. For E-imine/catalyst complexes, only tilting is observed. For the Z-imine/catalyst complex, which is postulated to be the reactive species, the reduced steric hindrance of the imine enables both exchange pathways, canceling the asymmetry of the binary complex. These structural characteristics are conserved in all complexes despite varying substrates.
Results and Discussion
Model Systems and Methods
For our structural investigations, six imine/catalyst complexes were selected. Catalyst 1c was used, because it provided the best combination of solubility, 1H chemical shift dispersion, and performance, compared to 1a,b (see Figure 1b). Typical aromatic imines used in synthesis 2a–c and 3a–c were chosen, to provide different electronic properties (see Figure 2).4,52a–c derived from aniline were selected, because this allowed for a simple 15N labeling, which enables the unambiguous assignment of α-methyl groups and NH protons. In addition, 3a,c derived from anisidine were investigated to show the effect of an inverse relative electron density of the two aromatic substituents. The trifluoromethyl substituents of 2c and 3a enable 19F NMR investigations, and imine 3c allows for the direct comparison with the single crystal structure5 by means of theory. Toluene, dichloromethane (CD2Cl2), and freonic mixtures15 of CDCl2F and CDClF2 were tested as solvents. After extensive optimization of sample preparations, NMR parameters, and solvent screening, we were able to improve the line widths tremendously and altogether enable an investigation by means of NMR (for spectra, see the Supporting Information). Due to the better solubility of the imine/1c complexes in CD2Cl2, all relevant structural investigations were performed in this solvent. Low temperature measurements were used in order to reduce conformational averaging and exchange. The NMR measurements at 180 and 130 K were done in CD2Cl2 and freonic mixtures, respectively (see the Supporting Information).
Figure 2.

Brønsted acid catalyst 1c and imines with different functional groups. Imines 2a–c and 3a–b were investigated by means of NMR and theory. Imine 3c was used for theoretical calculations exclusively.
For the complexes 2a–c/1c and 3a–c/1c, theoretical calculations were performed to identify the energetic minima. All complexes were optimized in the continuum of CD2Cl2 (SMD) at the DFT level of theory using Grimme’s D3 empirical dispersion parameter (TPSS-D3/def2-SVP).16−20 As the experiment was performed at 180 K, the dielectric constant of the continuum medium was raised (from 8.93 at RT to 16.20 at 180 K) to adapt to the experimental condition. In all cases, only ion pair minima were obtained, which is in full agreement with the dominating species found in our recent NMR studies.14 Vibrational analysis were performed at the TPSS-D3/def2-SVP level of theory. In all cases, no imaginary vibrational mode is obtained. Subsequently, single point calculations (SCS-MP2/CBS//TPSS-D3/def2-SVP)21 and additional solvent corrections (SMD)17 on the optimized geometry were performed to determine the energy differences between conformational minima. Softwares used were Gaussian 09, version D.01, for geometry optimization and frequency analysis and ORCA 3.0.3 for single points.22,23
Computational Study
Four types of energetic minima with different arrangements of the imines were identified in our theoretical calculations. Two structures Type I E and Type II E containing E-isomers and two structures Type I Z and Type II Z containing Z-isomers of the imine (Figure 3a) are representatively illustrated by E,Z-2a/1c. Each of these four structures has at least three low energy conformations of the isopropyl groups without affecting the core structure composed of the imine and the binaphtyl backbone. Therefore, in Figure 3a and in the further discussion, these isopropyl conformations are Boltzmann averaged but not depicted.
Figure 3.
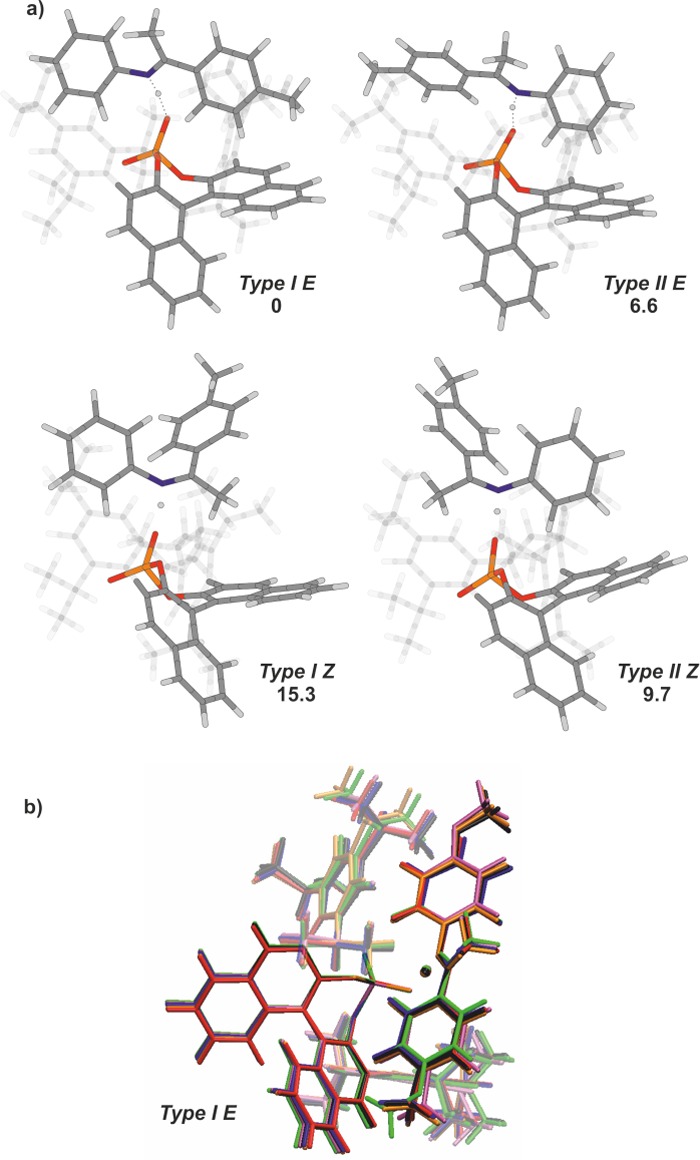
(a) Calculated structures of the 2a/1c complexes. Energies are shown in kJ mol–1 relative to Type I E.24 The bis(2,4,6-triisopropylphenyl) groups are shown transparent for the sake of clarity. (b) Overlay of global minima of investigated E-2a-c/1c and E-3a-c/1cType I E complexes. For the sake of clarity, a different orientation of the catalyst was chosen.
The structural analysis suggests that the methyl substituent in the para-position of the imine is situated above the binaphtyl moiety of the catalyst for conformation Type I E, which indicates a CH−π interaction. In contrast to structure Type I E, the imine in conformation Type II E is rotated by 180°. The aniline part of the imine is now located proximal to the binaphtyl moiety of the catalyst. The conformational analysis of E-isomer complexes suggests a preference (ca. 4.5–9.3 kJ/mol) for structure Type I E due to the CH−π stabilization rather than Type II E (Figure 3; E-2a/1c). This thermodynamic preference is conserved in the series of the investigated E-imine/1c complexes and seems to be independent of functional group induced electronic modulations, which is demonstrated by superimposed global minima in Figure 3b. Even the inverted electronic properties of the imine substituents in E-3a/1c, E-3b/1c, and E-3b/1c are not sufficient to alter the relative thermodynamic stability between Type I E and Type II E. The similar structures of the aromatic imine/catalyst complexes suggest that the electronic properties of the imine have very little influence on the structure of the binary complex.25
Structurally, the two complexes involving Z-imines exhibit either an interaction between the aniline part of the imine and the binaphthyl backbone of the catalyst (Type II Z) or an interaction between the α-methyl group and the catalyst backbone (Type I Z) (see Figure 3a). Here the conformation Type II Z is always thermodynamically favored (4.9–9.5 kJ/mol) over Type I Z in the series of the investigated Z-imine/1c complexes.
Noncovalent Interaction (NCI) Analysis26,27
All investigated binary complexes feature a strong hydrogen bond independent of their structural arrangements,14 which is marked by the critical point (s(ρ) = 0) around sgn(λ2)ρ = −0.06 to −0.07 in the plot of density ρ vs reduced density gradient s(ρ) (Figure 4: NCI analysis of the global minima of complex E,Z-2a/1c). This is in full agreement with our current experimental results, which showed a downfield shifted proton in the 1H spectra around 15–19 ppm. The strong hydrogen bond is also depicted by the blue surface which exists in all investigated complexes (Figure 5; E,Z-2a/1c).
Figure 4.
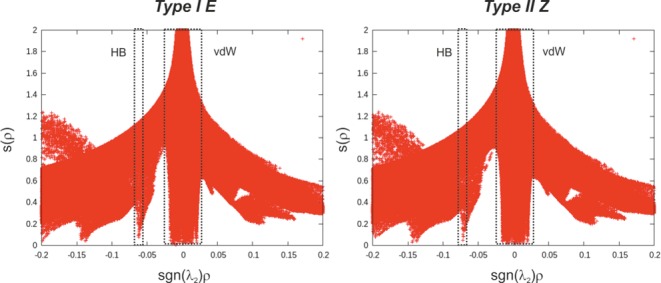
Plot of density (sgn(λ2)ρ) vs reduced density gradient (s(ρ)) showing a large area of van der Waals interaction and the strong hydrogen bond in the complex Type I E and Type II Z of E,Z-2a/1c.
Figure 5.
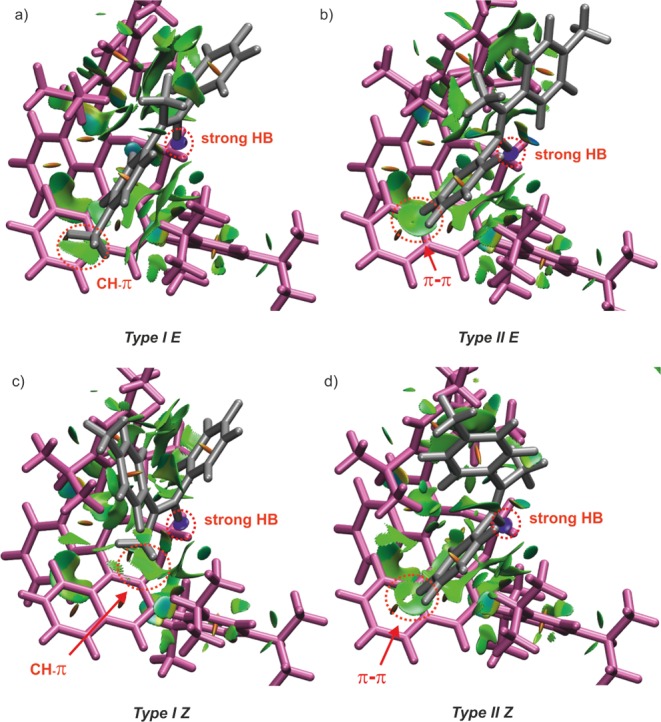
NCI analysis of the 2a/1c complexes (global minima) showing weak dispersive interactions (green surfaces) and an attractive strong hydrogen bridge between imine and catalyst (blue surfaces). (a) Type I E shows CH−π interaction between the p-methyl substituent of the imine and BINOL of the catalyst. (b) The imine is rotated 180° in Type II E and shows the aniline moiety lies on top of the BINOL aromatic surface and the p-methyl group faces outward from the catalyst. (c) ) In Type I Z, the imine is also rotated and exhibits a CH−π interaction between the α-methyl group and BINOL. (d) Similar to Type II E, Type II Z shows a π−π interaction between the aniline moiety and BINOL, as well as a CH−π interaction between the α-methyl group with the 2,4,6-triisopropylphenyl group of the catalyst; For comparison and clarity, a similar orientation as in Figure 3b was adopted. Isovalue 0.5 and a color range between −3.0 and 3.0 were chosen.
Additionally, the plot in Figure 4 exhibits also a large region of many weak interactions characteristically indicated by the critical points around sgn(λ2)ρ = −0.03 to −0.03, which is expected for complexes with many aromatic surfaces. Figure 5a illustrates the NCI analysis of the conformation Type I E in the complex E-2a/1c. The existence of the weak interactions, to which the CH−π interaction (red circle) also belongs, is indicated by the green surface. These numerous van der Waals interactions are contributing significantly to the geometry of the complex and conformational distribution. Therefore, the application of the dispersion corrected functional (D3)18 is indispensable in the simulation. Respectively, the chemical shift calculation of the protons of the methyl group predicted a typical shielding by the aromatic ring as discussed below. Compared to the conformation Type I E, the aniline part of the imine in the conformation Type II E is expected to interact with the binaphtyl moiety of the catalyst. Accordingly, a weak π–π interaction is predicted by the NCI analysis (Figure 5b). Moreover, the para-methyl group of the imine in the conformation Type II E faces outward from the catalyst. Therefore, no particular interaction and shielding is expected.
Similar to the situation in E-2a/1c complexes, several characteristic dispersive interactions can also be identified in Z-2a/1c complexes (Figure 5c,d). While in the conformation Type I Z the α-methyl substituent has a CH−π interaction with the binaphthyl moiety of the catalyst, the conformation Type II Z has a distinctive π–π interaction (T-shape) between the aniline part and the binaphthyl moiety of the catalyst. In this case, we expect that some of the protons of the aniline are shielded by the aromatic ring. Furthermore, it is also recognizable that the α-methyl substituent in Type II Z interacts weakly with the 2,4,6-triisopropylphenyl group of the catalyst (see the Supporting Information for a rotated picture). Therefore, it is expected that the protons of the α-methyl in both conformations Type II Z and Type I Z experience similar shielding effects.
In all conformations (Type I/II E/Z), the NCI analysis showed also many weak dispersive interactions between the 3,3′ substituent of the catalyst and the aniline/ketone part of the substrate. This demonstrates that these interactions, which often serve as a target for modulation to achieve high stereoselectivity in synthetic application, are already preformed and pronounced in the binary complexes. However, the NCI analysis as a whole suggests that modifications both of the 3,3′ substituent and of the binaphthyl backbone are important targets for catalyst design.
NMR Studies
At 300 K, the 1:1 mixtures of imine/1c complexes show only one averaged set of 1H signals for the Brønsted acid catalyst, whereas two sets of signals are detected for each imine, corresponding to their E- and Z-isomers with E/Z ratios ranging from 80:20 to 70:30 (for details, see the Supporting Information). By lowering the temperature to 180 K, two complete sets of signals, one of an E-imine/1c complex and one of a Z-imine/1c complex, are observed. Even at 180 K, an exchange between E-imine inside the E-imine/1c complex and E-imine outside the complex takes place. This exchange is slow on the NMR time scale and leads to an additional set of signals for the E-imine. An in depth investigation of the hydrogen bonds revealed for all imines a mainly ion-paired character for both E and Z complexes at 180 K and a fixed geometry of the hydrogen bond.14 Using various homo- and heteronuclear 2D spectra, 1H and 13C chemical shift assignments of the Z- and E-imines as well as a complete chemical shift assignment of the 1H signals of the imine/binaphthyl core structure were achieved (for details, see the Supporting Information).
The E-Imine/1c Complexes
NOE studies were conducted in order to determine the structure of the complexes present in solution. Considering the experimentally detected high percentage of E-imine complexes (70–80%) and the computationally calculated preference for structure Type I E, the NOE between the varying functional groups of imines 2a–c and 3a–b and the binaphthyl backbone of 1c should be most pronounced.
Selective 1D 1H,1H NOESY spectra of the p-methyl group of E-2a were used to identify the predicted NOEs unambiguously (see Figure 6). Selective NOE buildup curves of the E-2a/1c complex show deviations from linearity even below a 100 ms mixing time, similar to small proteins. Therefore, only qualitative analysis of intermolecular distances is possible. Selective 1D 1H,1H NOESY spectra with mixing times of 150 ms were further used for the detection of intermolecular NOEs between the catalyst and the imine, in order to gain NOE intensity with as little chemical exchange as possible. For complexes 2b/1c and 3a/1c, the same NOE pattern was found by exciting the functional groups of the acetophenone parts of the E-imines with selective NOESY spectra (for spectra, see the Supporting Information). For imines 2b and 3a, 1H,19F HOESY spectra with mixing times of 500 ms were successfully used to identify NOEs between the p-trifluoromethyl groups and the binaphthyl backbone of the catalyst. In conclusion, for every investigated E-imine/1c complex, the same NOE pattern can be observed between the different functional groups of the acetophenone moiety of the imine (p-Me, p-MeO, and p-CF3) and the binaphtyl backbone of 1c (for 1D 1H,1H NOESY spectra and 2D 1H,19F HOESY spectra of the other complexes, see the Supporting Information). Thus, independent of the electronic properties of the imines, all complexes show a strong population of structure Type I E.
Figure 6.
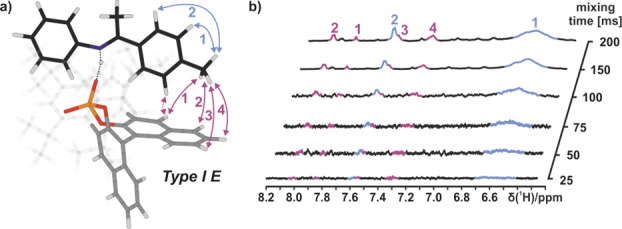
(a) Calculated structure Type I E of E-2a/1c corroborated by inter- (red arrows) and intramolecular NOEs (blue arrows). (b) Corresponding experimental data from 1D selective NOESY spectra at 180 K in CD2Cl2 (see the Supporting Information for additional data).
The identification of structure Type II E in complex 2a/1c, which showed an imine orientation similar to the crystal structure,3 is by far more complicated. According to the calculations, Type II E is less populated and the aniline signals providing the crucial NOE information show broad line widths. In addition, the expected NOE intensities between the aniline moiety and the binaphtyl backbone of the catalyst are further weakened by rotation. Therefore, imines 3a and 3b were used, which enable the measurement of selective 1D 1H,1H NOESY spectra by exciting the methoxy group of the anisidine moiety. The best signal dispersion was found in E-3a/1c (see Figure 7). The identified NOE pattern reveals a proximity of the anisidine moiety of the imine to the binaphtyl backbone of the catalyst (see Figure 7b). Since structures Type I E and Type II E are observed simultaneously, a fast exchange between both structures must occur, even at 180 K. In principle, the two structures are related by two potential exchange modes. Either the imine rotates by 180° under retention of the hydrogen bond or the imine switches the oxygen that constitutes the hydrogen bond and is thereby tilted inside the complex. Due to the extended shape of the E-imine and the steric configuration of the catalyst, the latter is more likely to occur. In comparison to structure Type I E, the NOE interaction in Type II E takes place on the other naphthyl half of the catalyst backbone (see Figure 7a). This experimentally proves that exchange occurs via the tilting mechanism (an exchange of Type I E and Type II E by rotation of the imine would lead to an interaction between both the acetophenone and anisidine moiety with the same naphthyl side of the catalyst). For the aniline derived imines 2a–c, even a weak NOE between the aniline moiety of the imines and the binaphthyl backbone of 1c was observed with the help of longer mixing times (300 ms) in 2D 1H,1H NOESY spectra. This proves the existence of Type II E for all investigated complexes (see the Supporting Information for spectra).24
Figure 7.
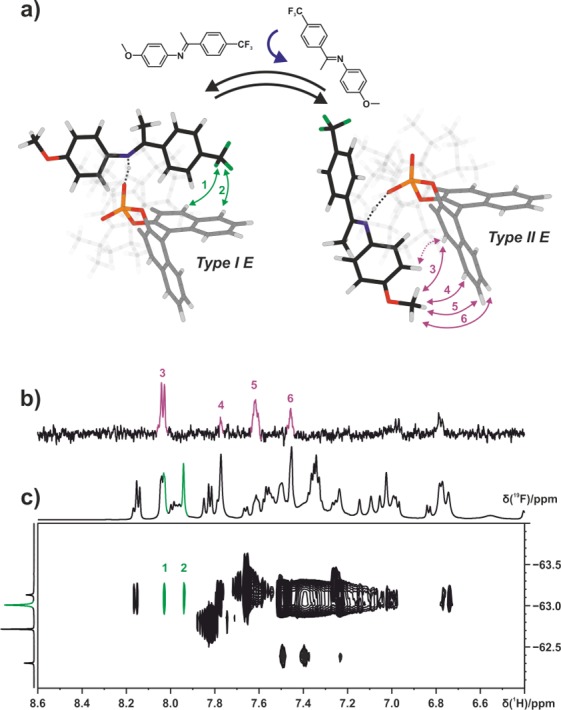
(a) Tilting of the imine E-3a in relation to the catalyst 1c. The observed NOE pattern (green and purple arrows) reveals the coexistence of complex structures Type I E and Type II E, which are in fast exchange on the NMR time scale at 180 K in CD2Cl2. (b) Section of a 1D selective NOESY of E-3a/1c at 180 K in CD2Cl2 at 600 MHz (excitation on the methoxy group of imine E-3a) showing intermolecular NOEs 3–6 corresponding to structure Type II E. (c) Section of a 1H,19F HOESY spectrum of E-3a/1c at 180 K in CD2Cl2 at 600 MHz showing NOEs 1 and 2 corresponding to structure Type I E.
In addition to the analysis of the NOE pattern, also the proton chemical shift pattern confirms the tilting process (switching the oxygen that forms the hydrogen bond) as the only exchange mode for E-imine/1c complexes. The two structures Type I E and Type II E exchange fast on the NMR time scale and produce only one set of chemical shifts. Within this assignment for any E-imine/1c combination, separated sets of chemical shifts are observed for the two naphthyl parts (Figure 8, blue and green colored peaks; for assignment and details, see the Supporting Information). In general, this chemical shift pattern can only be observed if one of the two described exchange processes (tilting or rotation) occurs exclusively. For the E complexes, tilting takes place (see above and Figure 7a). In cases where both exchange processes (tilting and rotation) are active, the complex would be symmetrized and an averaged set of signals for both halves of the binaphtyl backbone would be observed (see Z-imine/1c complexes and Figure 10 below).
Figure 8.
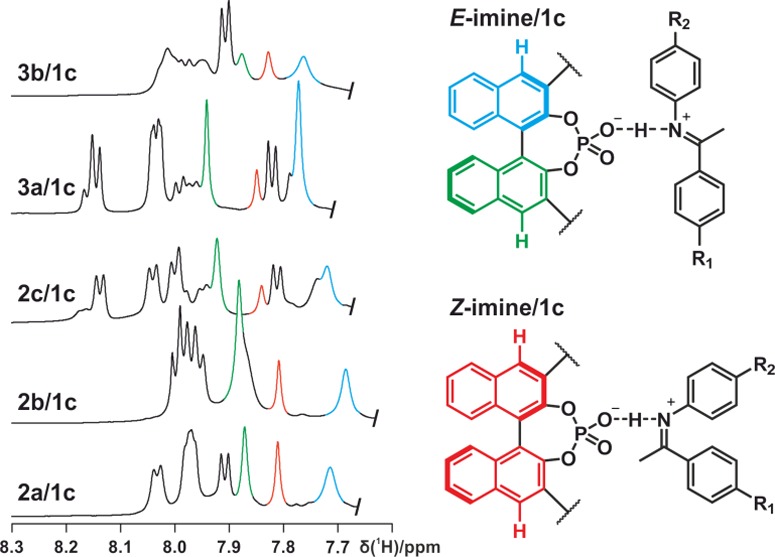
1H chemical shifts of the protons of the binaphthyl backbone of complexes 2a–c/1c and 3a–b/1c at 180 K in CD2Cl2 at 600 MHz. In every investigated sample, the asymmetry of the catalyst is preserved for the E-imine/1c complexes but broken for the Z-imine/1c complexes.
Figure 10.
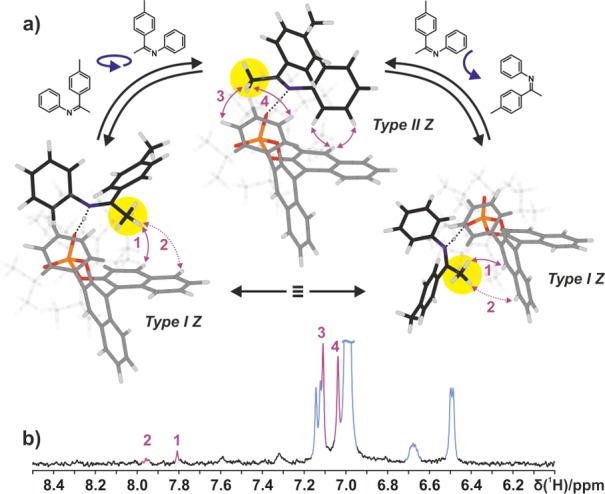
(a) NOE pattern (purple arrows) found for the investigated Z-2a/1c complexes. The intramolecular NOEs could not be assigned to a single structure Type II Z or Type I Z but instead show that an equilibrium of both structures exists in solution (see the Supporting Information for additional parameters). (b) Section of a selective 1D 1H,1H NOESY excited on the α-methyl group (yellow circle in part a) of Z-2a at 180 K in CD2Cl2 at 600 MHz.
Both the experimental and theoretical chemical shift analyses of the E-imine/1c complexes reveal strong high field shifts of the p-methyl group and the nearby aromatic protons again corroborating Type I E (see Figures 5a and 6a for structures and Figure 9 for spectra). In the structure of E-2a/1c, the p-methyl group is effectively shielded by the naphthyl backbone of the catalyst, while, with HBF4 as the noncoordinating protonating agent, no shielding can occur. This results in a high field shift of the p-methyl group in E-2a/1c compared to E-2a/HBF4 (Δδexp, 0.37 ppm; Δδcalc, 0.66 ppm) (see Figure 9 and the Supporting Information). Since the p-methyl group in Type II E is not shielded (see Figure 5b), the exchange between Type I E and Type II E leads to a reduction of the shielding effect caused by the binaphthyl moiety in Type I E. In the calculation of the chemical shielding analysis, only Type I E is considered, resulting in a larger chemical shift difference (Δδcalc: 0.66 ppm). Thus, the offset between calculated and experimentally determined Δδ corroborates the coexistence of Type I E and Type II E.
Figure 9.
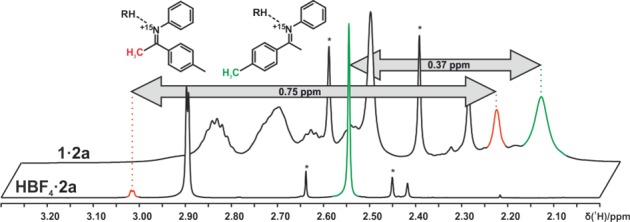
Shielding effects on methyl groups of 2a/1c due to CH−π interactions between the methyl groups and the extended aromatic systems of the catalyst. Free protonated imine was simulated with HBF4, and the chemical shifts were compared to the imine/1c complexes (180 K in CD2Cl2, 600 MHz).
The Z-Imine/1c Complexes
The calculated energy differences between the two structures Type II Z and Type I Z ranging from 4.9 to 9.5 kJ/mol are similar to those between Type I E and Type II E. Again, a preference for Type II Z is expected. Similar to the E-imine/1c complex, the two structures Type II Z and Type I Z are in principle related by rotation of the imine by 180° without breaking the hydrogen bond, or by tilting the imine inside the complex and thereby switching the oxygen that forms the hydrogen bond. In comparison to Type I E and Type II E, only one averaged set of signals can be observed for both naphthyl halves of the catalyst backbone (see Figure 8). By exciting the α-methyl group of Z-2a in the 2a/1c complex in a selective 1D 1H,1H NOESY spectrum, structures Type II Z and Type I Z were identified simultaneously (see Figure 10b). Due to the reduced steric hindrance of the Z-imine compared to the E-imine, an exchange between the two complex structures Type II Z and Type I Z is possible by rotation, as well as tilting of the imine (see Figure 10a). This explains the broken asymmetry of the catalyst in the Z-imine/1c complexes (see Figure 8). With the help of selective 1D 1H,1H NOESY spectra, the exact same situation was identified for all investigated Z-imine/1c complexes (see the Supporting Information for spectra).
Moreover, the comparison of experimental and theoretical chemical shifts of Z-2a/1c fits to the Boltzmann weighted average of Type II Z and Type I Z. Both structures show a close proximity of the α-methyl group to either the 2,4,6-triisopropylphenyl moiety (Type II Z) or the binaphthyl backbone of the catalyst (Type I Z), which leads to a strong high field shift for the respective methyl group due to CH−π interactions (Δδexp, 0.75 ppm; Δδcalc, 0.74 ppm; see Figure 9).
Conclusion
By means of NMR and theoretical calculations, we were able to identify the structure of binary complexes, consisting of the chiral phosphoric acid 1c and E-/Z-isomers of aromatic imines. All E-imine/catalyst complexes possess two structures, which are in fast exchange on the NMR time scale even at 180 K. Due to the extended steric properties of the E-imine, this exchange occurs by tilting the imine inside the complex and thereby switching the oxygen that constitutes the hydrogen bond. This preserves the asymmetry of the E-imine/catalyst complexes. Additionally, the structure of the Z-imine/catalyst complexes was determined for the first time. For all Z-imine/catalyst complexes, a similar exchange process between two structures was observed. However, the compact structure of the Z-imine enables an additional exchange pathway by rotation, symmetrizing the catalyst. For all investigated imine/R-TRIP (3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate) complexes, dispersive interactions between substrate and catalyst were identified not only at the 3,3′ substituents of the catalyst but at the binaphthyl backbone as well, revealing the importance of modifications at this moiety for catalyst design. The four presented core structures are highly preserved; i.e., these core structures were found to be independent of electron density and substituent modulations of the aromatic imines. This invariance of the structures across all imine substrates reflects the strength of this class of catalysts. Extended dispersive interactions between catalyst and substrate, which are postulated to provide high stereoselectivities in the transition states, are preformed in the binary complex, while the strong hydrogen bond allows for a high substrate tolerance. For example, for phosphoramidites, it is known that variations in the electron density of the substrate often have to be compensated by changes in the ligand structure.28 The structural investigations presented here show that in Brønsted acid catalysis the strength of the hydrogen bond overrides the variations in the CH−π and π–π interactions caused by different functional groups of the substrates. Assuming that these structural features of the binary complexes are preserved in the transition states, Brønsted acid catalysis seems to combine both high stereoinduction due to dispersive interactions and structural invariance caused by the hydrogen bond.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b09244.
Assignments of all complexes in CD2Cl2; NOESY spectra used for identification of structures; NMR parameters and sample preparation procedure (PDF)
Financial Support was provided by the European Research Council (ERC-CoG 614182 – IonPairsAtCatalysis).
The authors declare no competing financial interest.
Supplementary Material
References
- Akiyama T.; Itoh J.; Yokota K.; Fuchibe K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]
- Uraguchi D.; Terada M. J. Am. Chem. Soc. 2004, 126, 5356–5357. 10.1021/ja0491533. [DOI] [PubMed] [Google Scholar]
- Storer R. I.; Carrera D. E.; Ni Y.; MacMillan D. W. C. J. Am. Chem. Soc. 2006, 128, 84–86. 10.1021/ja057222n. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Sugiono E.; Azap C.; Theissmann T. Org. Lett. 2005, 7, 3781–3783. 10.1021/ol0515964. [DOI] [PubMed] [Google Scholar]
- Hoffmann S.; Seayad A. M.; List B. Angew. Chem., Int. Ed. 2005, 44, 7424–7427. 10.1002/anie.200503062. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Sugiono E.; Azap C. Angew. Chem., Int. Ed. 2006, 45, 2617–2619. 10.1002/anie.200504344. [DOI] [PubMed] [Google Scholar]
- Mahlau M.; List B. Angew. Chem. 2013, 125, 540–556. 10.1002/ange.201205343. [DOI] [PubMed] [Google Scholar]
- Marcelli T.; Hammar P.; Himo F. Chem. - Eur. J. 2008, 14, 8562–8571. 10.1002/chem.200800890. [DOI] [PubMed] [Google Scholar]
- Simón L.; Goodman J. M. J. Am. Chem. Soc. 2008, 130, 8741–8747. 10.1021/ja800793t. [DOI] [PubMed] [Google Scholar]
- Tang W.; Johnston S.; Iggo J. a; Berry N. G.; Phelan M.; Lian L.; Bacsa J.; Xiao J. Angew. Chem., Int. Ed. 2013, 52, 1668–1672. 10.1002/anie.201208774. [DOI] [PubMed] [Google Scholar]
- Kim H.; Sugiono E.; Nagata Y.; Wagner M.; Bonn M.; Rueping M.; Hunger J. ACS Catal. 2015, 5, 6630–6633. 10.1021/acscatal.5b01694. [DOI] [Google Scholar]
- Merten C.; Pollok C. H.; Liao S.; List B. Angew. Chem., Int. Ed. 2015, 54, 8841–8845. 10.1002/anie.201501271. [DOI] [PubMed] [Google Scholar]
- Fleischmann M.; Drettwan D.; Sugiono E.; Rueping M.; Gschwind R. M. Angew. Chem. 2011, 123, 6488–6493. 10.1002/ange.201101385. [DOI] [PubMed] [Google Scholar]
- Sorgenfrei N.; Hioe J.; Greindl J.; Rothermel K.; Morana F.; Lokesh N.; Gschwind R. M. J. Am. Chem. Soc. 2016, 10.1021/jacs.6b09243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J. S.; Anet F. A. L. J. Org. Chem. 1988, 53, 2629–2630. 10.1021/jo00246a046. [DOI] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Marenich A. V; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Furche F.; Ahlrichs R. J. Chem. Phys. 2003, 119, 12753–12762. 10.1063/1.1627293. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Grimme S. J. Chem. Phys. 2003, 118, 9095–9102. 10.1063/1.1569242. [DOI] [Google Scholar]
- Neese F. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- The simulation at 180 K showed a Boltzmann population of Z-imine/catalyst complexes less than 1% relative to the E-imine/catalyst complexes. This is in agreement with the experiment which proved that no Z-imine/catalyst complexes were found when the complex was synthesized at 180 K.
- A quantitative analysis to determine the experimental ratio between Type I E and Type II E is currently not possible due to long mixing times and the inaccuracy of the distance measurements.
- Johnson E. R.; Keinan S.; Mori-Sánchez P.; Contreras-García J.; Cohen A. J.; Yang W. J. Am. Chem. Soc. 2010, 132, 6498–6506. 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-García J.; Johnson E. R.; Keinan S.; Chaudret R.; Piquemal J. P.; Beratan D. N.; Yang W. J. Chem. Theory Comput. 2011, 7, 625–632. 10.1021/ct100641a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann E.; Hammer M. M.; Gschwind R. M. Chem. - Eur. J. 2013, 19, 10551–10562. 10.1002/chem.201300095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.