

Abstract
Serpins are a widely distributed family of high molecular weight protein proteinase inhibitors that can inhibit both serine and cysteine proteinases by a remarkable mechanism-based kinetic trapping of an acyl or thioacyl enzyme intermediate, that involves massive conformational transformation. The trapping is based on distortion of the proteinase in the complex, with energy derived from the unique metastability of the active serpin. Serpins are the favored inhibitors for regulation of proteinases in complex proteolytic cascades, such as are involved in blood coagulation, fibrinolysis and complement activation, by virtue of the ability to modulate their specificity and reactivity. Given their prominence as inhibitors, much work has been carried out to understand not only the mechanism of inhibition, but how it is fine-tuned, both spatially and temporally. The metastability of the active state raises the question of how serpins fold, while the misfolding of some serpin variants that leads to polymerization and pathologies of liver disease, emphysema and dementia makes it clinically important to understand how such polymerization might occur. Finally, since binding of serpins and their proteinase complexes, particularly PAI-1, to the clearance and signaling receptor LRP1, may affect pathways linked to cell migration, angiogenesis, and tumor progression, it is important to understand the nature and specificity of binding. The current state of understanding of these areas is addressed here.
1. Introduction
The initial identification of a relationship that would grow into the serpin superfamily of proteins was made in 1980 by Hunt and Dayhoff [1] from a comparison of the complete sequence of chicken ovalbumin with partial sequences of two human proteinase inhibitors, antithrombin and α1-proteinase inhibitor (α1PI)1,2. Since then, the family has grown to thousands of proteins [2] that are found not only in mammals and other vertebrates, but in other animals, in plants [3], in viruses [4], in bacteria and in archaea [5–7]. Whereas the name “serpin” was coined by two of the pioneers in the field, Robin Carrell and Jim Travis, as a convenient shorthand for serine proteinase inhibitor [8], based on the ability of several of the first identified members of the family to inhibit serine proteinases, the name is somewhat misleading, since some serpins can inhibit cysteine proteinases, and quite a number of serpins are not proteinase inhibitors at all, though they do, in many cases, possess other important biological functions (Table 1). However, since most serpins are indeed inhibitors of serine proteinases, the name has stuck.
Table 1.
| Serpin | Synonyms | Size | RCL (P4-P4') | Chromosomal Location | Target Proteinases | Cofactors |
|---|---|---|---|---|---|---|
| CLADE A | ||||||
| SERPINA1 | α1proteinase inhibitor (α1PI), α1antitrypsin (α1AT) | 394 (418) | AIPM--SIPP | 14q32.1 | neutrophil elastase | |
| SERPINA2 | α1AT-related protein | 399 (420) | EKAW--SKYQ | 14q32.1 | ||
| SERPINA3 | α1antichymotrypsin (α1ACT) | 398 (423) | ITLL--SALV | 14q32.1 | cathepsin G, chymase prostate specific antigen |
|
| SERPINA4 | kallistatin, kallikrein inhibitor, PI4 | 401 (427) | IKFF--SAQT | 14q32.1 | tissue kallikrein | |
| SERPINA5 | protein C inhibitor (PCI), plasminogen activator inhibitor -3 (PAI-3) | 387 (406) | FTFR--SARL | 14q32.1 | activated protein C, uPA. plasma kallikrein acrosin |
heparin |
| SERPIN A6 | corticosteroid binding globulin (CBG) | 383 (405) | LNLT--SKPI | 14q32.1 | elastase substrate non-inhibitory |
. |
| SERPINA7 | thyroxin binding globulin | 395 (415) | LSDQ--PENT | Xq22.2 | non-inhibitory | . |
| SERPINA7P1 | Xq22.3 | |||||
| SERPINA8 | angiotensinogen (AGT) | 452 (485) | NKPE--VLEV | 1q42-q43 | renin substrate non-inhibitory |
|
| SERPINA9 | centerin | 434 | FIVR--SKDG | 14q32.13 | ||
| SERPINA10 | protein Z-dependent protease inhibitor (ZPI) | 423 (444) | ITAY--SMPP | 14q32.13 | factor Xa, factor XIa | protein Z, Ca2+, phospholipid , heparin |
| SERPINA11 | pseudogene | 14q32.13 | ||||
| SERPINA12 | vaspin | 395 (414) | TLPM--ETPL | 14q32.13 | kallikrein 7 | |
| SERPINA13P | pseudogene | |||||
| SERPINA15P | pseudogene | |||||
| CLADE B | ||||||
| SERPINB1 | leukocyte elastase inhibitor (LEI), monocyte/neutrophil elastase inhibitor (MNEI), PI2, EI, ELANH2 | 379 | ATFC--MLMP | 6p25 | neutrophil elastase, cathepsin G, proteinase 3, cathepsin L | |
| SERPINB2 | plasminogen activator inhibitor-2 (PAI-2), placental PAI, | 415 | MTGR--TGHG | 18q21.3 | uPA | . |
| SERPINB3 | squamous cell carcinoma antigen 1 (SCCA1) | 390 | GFGS--SPAS | 18q21.3 | cathepsin K, cathepsin L cathepsin S |
|
| SERPINB4 | squamous cell carcinoma antigen 2 (SCCA2), leupin | 390 | VVEL--SSPS | 18q21.3 | cathepsin G, chymase | |
| SERPINB5 | maspin, PI5 | 375 | ILQH--KDEL | 18q21.3 | non-inhibitory | |
| SERPINB6 | placental thrombin inhibitor (PTI), PI6, cytoplasmic antiprotease (CAP) | 376 | MMMR--CARF | 6p25 | thrombin plasmin chymotrypsin cathepsin G |
|
| SERPINB7 | megsin | 380 | IVEK--QLPQ | 18q21.3 | ||
| SERPINB8 | cytoplasmic antiprotease 2 (CAP2), PI8 | 374 | RNSR--CSRM | 18q21.3 | furin thrombin subtilisin A |
|
| SERPINB8P1 | pseudogene | 6p25 | ||||
| SERPINB9 | cytoplasmic antiprotease 3 (CAP3), PI9 | 376 | VVAE--CCME | 6p25 | granzyme B, caspase 1 (ICE), subtilisin A | |
| SERPINB9P | pseudogene | 6p25.2 | ||||
| SERPINB10 | bomapin, PI10 | 397 | IDIR--IRVP | 18q21.3 | thrombin | |
| SERPINB11 | epipin | 392 | IAVK--SLPM | 18q21.3 | ||
| SERPINB12 | yukopin | 405 | VSER--SLRS | 18q21.3 | trypsin, plasmin | |
| SERPINB13 | headpin, hurpin, PI13 | 391 | FTVT--SAPG | 18q21.3 | ||
| CLADE C | ||||||
| SERPINC1 | antithrombin (AT), antithrombin III (ATIII) | 432 (464) | IAGR--SLNP | 1q23-q25 | thrombin, factor Xa factor IXa |
heparin, heparan sulfate |
| CLADE D | ||||||
| SERPIND1 | heparin cofactor II (HCII), leuserpin 2 | 480 (499) | FMPL--STQV | 22q11 | thrombin | heparin, heparan sulfate, dermatan sulfate |
| CLADE E | ||||||
| SERPINE1 | plasminogen activator inhibitor-1 (PAI-1), endothelial PAI | 379 (402) | VSAR--MAPE | 7q21.3-q22 | uPA, tPA, thrombin, activated protein C | vitronectin |
| SERPINE2 | protease nexin 1 (PN1), glia derived nexin (GDN) | 379 (398) | LIAR--SSPP | 2q33-q35 | uPA, tPA, thrombin acrosin factor XIa |
heparin |
| SERPINE3 | 404 (424) | LLKR--SRIP | 13q14.3 | |||
| CLADE F | ||||||
| SERPINF1 | pigment epithelium-derived factor (PEDF), EPC-1 | 403 (418) | PAHL--TFPL | 17p13.3 | non-inhibitory | |
| SERPINF2 | α2antiplasmin (α2AP), A2AP | 452 (491) | AMSR--MSLS | 17pter-p12 | plasmin | |
| CLADE G | ||||||
| SERPING1 | C1-inhibitor (C1 INH) | 478 (500) | SVAR--TLLV | 11q11-q13.1 | C1r, C1s plasma kallikrein |
|
| CLADE H | ||||||
| SERPINH1 | heat shock protein 47 (HSP47), collagen-binding protein 1 (CBP1), colligin 1 | 400 (417) | EELR--SPKL | 11p13.5 | non-inhibitory | |
| SERPINH1P1 | 9p13.3 | |||||
| CLADE I | ||||||
| SERPINI1 | neuroserpin | 394 (410) | AISR--MAVL | 3q26 | tPA uPA plasmin |
|
| SERPINI2 | myoepithelium-derived serine proteinase inhibitor, MEPI, pancpin | 387 (405) | IPVI--MSLA | 3q26 |
2. Overview of Structure and Mechanism
2.i Serpin structure
The defining characteristic of a serpin is a primary structure that encompasses about 380 residues that fold into a single domain composed of three β-sheets, and 8 or more α-helices packed mostly on one side of the major β-sheet (sheet A). Critical for functioning as a proteinase inhibitor is an exposed loop, termed the reactive center loop (RCL), that is linked to s5A on the N-terminal end and s1C on the C-terminal end. The serpin structure is exemplified by the abundant human serpin α1-proteinase inhibitor, the principal inhibitor of neutrophil elastase (Fig 1). Enough x-ray structures of other serpins have been determined to confirm the ubiquity of this fold amongst serpins identified as such from their primary structures [9]. In humans 34 serpins and 5 serpin pseudogenes have been identified so far [10] (Table 1), with many of the genes found in 2 clusters on chromosomes 14 (most clade A serpins) and 18 (clade B serpins), reflecting the role of gene duplication and differentiation in their fanilial expansion.
Fig 1. Principal serpin conformations.

The native state (panel A, α1PI pdb code 1QLP [176]) and latent state (panel C, PAI-1 pdb code 1CG5 [177]) represent the metastable and most stable states respectively of the single-chain forms of the serpin. They differ principally in the location of the RCL (blue) as being exposed (native) or integrated into β-sheet A (latent) and of s1C as being part of β-sheet C (native) or an unstructured exposed linker (latent). Cleavage of the RCL permits the serpin to have both the RCL integrated into β-sheet A and s1C still as part of β-sheet C (panel B, α1PI pdb code 7API [178]). The remainder of β-sheet A is shown in red. β-sheet C is top left and β-sheet B is behind β-sheet C. Most helices pack on the “back” face of the serpin in this presentation, with the major exceptions of helix F (across the front of β-sheet A) and helices D and E (D above E) on the right edge. Adapted from ref [179]. Copyright Elsevier.
In addition to the core serpin domain, which corresponds to a mass of about 42kDa, some serpins have N- and/or C-terminal extensions, expected to be related to auxiliary functions, which can significantly increase their size. The most extreme case is C1 inhibitor (serpinG1), an inhibitor of the complement pathway serine proteinases C1s and C1r, where an N-terminal tail of about 100 residues is heavily glycosylated, bringing the total mass to ~76 kDa [11, 12]. Other important examples are heparin cofactor II (serpinD1), antithrombin (serpinC1) and protein Z-dependent proteinase inhibitor (ZPI, serpinA10), all with sizeable N-terminal extensions.
2.ii Serpin metastability
From a protein structural perspective, the most remarkable feature of serpins is that, unlike almost all other known proteins, they spontaneously fold into a metastable conformation, which is the normal, active conformation that, with the notable exception of PAI-1, has long-term stablility under physiological conditions. In this metastable state, the region of the protein that contains the recognition sequence for target proteinase, the RCL, is an exposed loop (Fig 1A). It is this loop that is energetically far from its most stable conformation. The more stable conformation, termed the latent state, involves insertion of the RCL into the center of β-sheet A as the fourth strand (s4A), thereby expanding β-sheet A to 6 strands of anti-parallel β-sheet, though with concomitant extraction of strand 1 from β-sheet C to permit the insertion of s4A into β-sheet A (Fig 1C). Energetically, the gain in stability from expansion of β-sheet A more than offsets the reduction in stability from loss of s1C, which is reflected in an increase of 17°C in the denaturation temperature in the case of human PAI-1 [13]. A third, very much more stable, conformation is obtained by cleavage of the RCL towards its C-terminus, where the proteinase recognition sequence lies. Cleavage of the polypeptide then allows s4A to insert into β-sheet A without the need to extract s1C from β-sheet C to again give a 6-stranded anti-parallel β-sheet (Fig 1B). The much greater increase in stability compared to the latent conformation is reflected in the very much higher Tm of >120°C [14], representing an increase over the native conformation of >60 deg C. This has been estimated to represent over 30kcal mol−1 of additional stabilization compared to the native state [15]. The highly favorable energetics for formation of the cleaved-like conformation of serpins from the metastable conformation underlie the unique mechanism of inhibition of both serine and cysteine proteinases. However, a negative consequence of the requirement to fold along a precise pathway that leads to the metastable state appears to be that certain mutations in serpins can lead to folding intermediates accumulating along the folding pathway, so that polymers can form in addition to the desired functional monomers [16]. The best example of this is the E342K mutation in α1PI (termed the Z variant), which is quite common in Northern European populations (~4%). In the ZZ homozygote this leads to formation of intracellular polymeric inclusions in both the liver [17] and the lung [18], with associated disease resulting both from reduced levels of functional proteinase inhibitor and from the toxic effects of the inclusions [18, 19].
2.iii Serpin inhibition mechanism
Early studies on the inhibition of serine proteinases by serpins suggested that they differ little from classical inhibitors, both in terms of formation of the initial encounter complex and of any subsequent reaction of the proteinase with the scissile peptide bond. It was suggested that one or two residues of the RCL, distal from the scissile bond, insert into β-sheet A, thereby rigidifying the remaining exposed loop and permitting at most formation of a tetrahedral intermediate between the proteinase active site and the scissile peptide bond carbonyl [20]. This was shown to be unlikely from the demonstration (i) that several serpins, including antithrombin [21] and C1 inhibitor [22] behave as branched pathway suicide substrate inhibitors towards their target proteinases, leading to either inhibited complex or cleaved serpin from a common intermediate, and (ii) from a study on an RCL variant of α1PI that, while still an inhibitor of proteinase, had an altered distribution of cleaved and complexed serpin, despite unaltered stability and reactivity of the starting conformation towards proteinases [23]. This led to a much more appealing proposal for the inhibition mechanism [24] that involved initial formation of a non-covalent complex analogous to those of classical inhibitors, but with subsequent successful cleavage of the peptide bond to form an acyl enzyme intermediate. Such cleavage would then allow the energetically favorable insertion of the RCL into β-sheet A. Since the proteinase would still be attached to the C-terminal end of the cleaved RCL, it would be transported from one end of the serpin to the distal end, where distortion of the active site might kinetically trap the acyl-enzyme intermediate (Fig 2). This proposal took into account the much greater stability of the cleaved conformation and evidence from SDS-PAGE of the formation of a covalent linkage between proteinase and serpin. This proposal has now been fully validated experimentally. Initial evidence that the serpin-proteinase complex contains an acyl enzyme linkage [25] was followed by demonstration that the serpin undergoes major conformational change upon complex formation with proteinase [26] that involves movement of over 70Å to the distal end of the serpin, with the concomitant requirement of full insertion of the RCL into β-sheet A [27]. The resulting conformation of the serpin moiety in the complex was shown to be like that of the cleaved conformation [28]. Finally, an x-ray structure of the complex between trypsin and α1PI not only confirmed the major translocation of the proteinase and the insertion of the RCL into β-sheet A, but showed both the covalent linkage between the active site serine of the proteinase and the carbonyl of the cleaved RCL, and the distortion of the active site [29], thus accounting for the kinetic stability of what would otherwise be a very short-lived intermediate. Subsequent fluorescence studies on covalent complexes of PAI-1 with cognate proteinases [30] and an x-ray structure of the covalent complex of α1PI with elastase [31] further supported this mechanism as the common one for inhibition of serine proteinases by serpins. An analogous study comparing the inhibition of the serine and cysteine proteinases, granzyme B and caspase-1, by the serpin crmA found evidence for the same mechanism with these different classes of proteinase [32]. In another study on the inhition of cysteine proteinases by crmA, it was shown for several caspases that a covalent intermediate is formed and that distortion of the proteinase active site also results in dissociation of the p10 caspase subunits, further helping to render the covalent intermediate kinetically stable [33], since p10:p10 intersubunit interactions within the heterotetramer are necessary for activity [34].
Fig 2. Suicide substrate branched pathway mechanism.

Serpin (I) and proteinase (E) initially associate non-covalently to form a Michaelis complex (EI) in which the proteinase active site recognizes and engages the P1-P1' residues and flanking residues and/or exosites. Upon formation of the acyl-enzyme intermediate through cleavage of the scissile bond (EI'), the cleaved RCL, with proteinase attached, translocates to the “bottom” of the serpin where compression and distortion of the proteinase lead to a kinetically-trapped covalent complex (E–I*). Should the proteinase complete the deacylation reaction prior to the inactivating distortion, cleaved serpin (I*) and regenerated free proteinase (E) are formed. Deacylation of E–I* can also slowly occur (k5) to give the same products. The ratio of (k3 + k4)/k4 gives the number of moles of serpin required to inhibit one mole of proteinase (if k5 is negligeable) and is called the stoichiometry of inhibition (SI). For many serpins and cognate proteinases this is close to 1. Figure from ref [179]. Copyright Elsevier.
In forming such kinetically-compromised covalent acylenzyme intermediates, the energy required for translocating the proteinase and distorting the active site in the final state is expected to come from the much greater stability of the cleaved conformation of the serpin, which in turn derives, as explained above, from the metastability of the native state. Although the energy difference between native and cleaved conformations is more than enough to distort the enzyme active site in the covalent complex, the sequence in which the energy is used must be carefully choreographed during the transformation from initial non-covalent to final covalent state so that enough is available when needed rather than being spent prematurely [35]. A better understanding of the intermediate stages during the inhibition process has therefore been a continuing goal, that has been the focus of some more recent work that is included in this review [36].
2.iv Advantages of serpins for inhibition of proteinase
Such a complex mechanism of proteinase inhibition by serpins places great demands on the serpin if inhibition is to function correctly. First, the serpin must be able to fold efficiently into the metastable conformation and not continue too quickly to the latent state [13]. Second, the RCL must be just the right length to permit full translocation of the proteinase without excess slack, so that distortion by compression can take place [37, 38]. Third, the residues within the RCL, beyond those needed for proteinase recognition, must be compatible with integration into a β-sheet, with consequent burial into the hydrophobic interior of alternate side chains [39, 40]. Fourth, translocation must occur rapidly enough that significant deacylation does not occur prior to active site distortion [23, 41].
With such stringent demands, it might seem very strange at first that serpins would be the inhibitor of choice for regulating so many proteinases in so many different organisms, when much smaller canonical inhibitors might serve just as well. The key to understanding this paradox is to appreciate that an all-or-nothing inhibition, determined only by proteinase and inhibitor concentrations and the association constant, is not necessarily sufficient for the fine temporal and spatial regulation of proteinases that are often swiftly generated in large amounts but that then need to be quickly turned off once their job is done. Complex feedback loop proteolytic cascades, such as blood coagulation or fibrinolysis are examples where inhibition is needed only in certain places and for certain periods. In such situations serpins come into their own as regulatable inhibitors that exploit the complex conformational and kinetic requirements of their inhibition mechanism to give them a great advantage over the small, rigid canonical inhibitors. This may involve them acting alone or with cofactors. In addition, the mechanism-based mode of inhibition gives them additional scope to inhibit different structural classes of both serine and cysteine proteinases, an attribute not found with canonical inhibitors [42].
2.v Scope of the review
For inhibitory serpins, much of the basic mechanism of inhibition has been worked out. However, the area where serpins exploit their unique properties to advantage in regulating specificity or rate of reaction with proteinase, in modulating efficiency of inhibition over simple cleavage, or in use of cofactors to regulate their action or mediate their clearance and permit signaling remain areas where much still remains to be uncovered. The folding pathway to the metastable state and how that may lead to polymerization under certain circumstances also remain controversial and unsettled areas, with important consequences for the development of treatments for patients with pathological serpin variants. This review focuses primarily on recent studies, many from our own laboratories, that address some of these areas.
3. Folding and Polymerization
3.i How Serpins Fold
Since it was recognized over 40 years ago by Anfinsen that the active conformation of essentially all proteins is dictated only by the primary structure and represents the most stable state [43], the “protein folding problem” has been trying to understand how polypeptide chains can rapidly fold into this state. Serpins represent the most important exception to this paradigm. As discussed above, serpins fold into a kinetically-trapped metastable state that, in most instances, has a very long half-life, measured in days. This state represents the active proteinase inhibitory state by virtue not simply of having the scissile bond within the RCL exposed and accessible to attacking proteinase, but most importantly by having a large energy difference between it and the conformation of the serpin in the final serpin-proteinase complex. This energy difference is then exploited to effect the distortion of the proteinase active site that represents the kinetic trap of the mechanism. For serpins, the protein folding problem is therefore a very different question from other proteins and one that has two parts. The first part is how the serpin efficiently adopts the active metastable conformation without the need for a chaperone [44] and the second is what prevents the rapid transformation of this state (Fig 1A) into the most stable (of the single chain conformations) latent state (Fig 1C). Answers to both of these questions have now been found.
In 2008 Hirose and colleagues examined the outcome of denaturing the cleaved serpin ovalbumin in 8 M urea, in both wild type and R339T forms, and then diluting it 20-fold and examining whether and how the two chains could re-associate [45]. The choice of these two forms of ovalbumin for comparison was based on the following properties and allowed them not only to suggest a folding sequence for ovalbumin, but to propose what constrains the metastable state from rapidly converting into the most stable latent state. Thus, although not a proteinase inhibitor, wild type ovalbumin adopts the same conformation as other metastable inhibitory serpins [46]. The presence of arginine at position 339, at the hinge point for insertion of the RCL into β-sheet A, prevents ovalbumin from undergoing the favorable transition to the loop-inserted cleaved structure seen for other RCL-cleaved serpins [39, 47]. However, mutation of Arg339 to threonine allows this transition to occur, so that the R339T variant folds initially into the metastable state and can then transition to the most stable two-chain cleaved state upon reaction with proteinase. In this interaction insertion of the RCL into β-sheet A is still too slow to give proteinase inhibition [48], but does result in a structure for the cleaved ovalbumin that is like that of other cleaved inhibitory serpins [40].
For the study, the same cleavage site within the RCL was used in each ovalbumin, so that the resulting species each consisted of two chains representing residues 1–352 and 353–385. The light chain (353–385) consisted of residues that form the C-terminal end of the RCL (s1C, hI, s4B and s5B), while the heavy chain contained the remainder of the serpin. From various complementary spectroscopic, kinetic and thermodynamic measurements, the authors were able to show that both heavy chains refolded to form essentially the same initial intermediate in a rapid burst. This intermediate was proposed to have most secondary structure formed, and importantly, to have s5A that precedes the C-terminal light chain inserted into β-sheet A. Association of the light chain with this intermediate then led to the formation of native states with identical kinetics for each form of ovalbumin. However, the native state of the R339T ovalbumin was of the RCL-inserted state, while that of wt ovalbumin was of the metastable-like conformation. It was then shown that the cleaved state of the R339T variant is formed only after passing through the metastable conformation and thus that the metastable conformation of serpins is a necessary intermediate on the folding pathway to the relaxed states [45].
More recently, we extended these studies by examining the ability of various peptides that make up the full-length serpin α1PI to associate and form native-like species to further probe the folding pathway [49]. Unlike ovalbumin, α1PI is an inhibitory serpin and so provides a functional assay for protein that has correctly adopted the metastable state. The initial observation was that two chains consisting of residues 1–323 and 324–394 were able to reassociate after dilution from 6 M guanidine HCl to give fully functional α1PI. The break point of the two chains lies immediately prior to strand s5A, so that the resulting chains differ from those in the ovalbumin study by the light chain also having s5A and the full RCL (this becomes s4A in loop-inserted conformations). By examining the ability of heavy chains that contained additional secondary structure element-forming residues (s5A, or s5A + RCL) to associate with correspondingly shorter light chains, we independently formulated a folding mechanism that is in remarkable agreement with that proposed earlier for the non-inhibitory ovalbumin. Again, critically, the intermediate with which the C-terminal peptide that contains s1C, s4B and s5B associates must have s5A present, and presumably inserted into β-sheet A. If it is absent, the C-terminal peptide associates only very poorly. Furthermore, if the heavy chain contains both s5A and s4A, s4A can only insert into β-sheet A after the C-terminal peptide has associated to give the metastable conformation. This is equivalent to the finding in the ovalbumin study that the loop-inserted conformation of the R339T variant must first form the metastable conformation. Taken together, these two studies support the same folding pathway for serpins, and furthermore offer an explanation of why the most stable latent conformation forms so slowly from the metastable conformation.
This folding pathway is outlined in Fig 3. It does not attempt to identify the sequence of folding events leading up to formation of the critical intermediate species II, other than to propose that the last event is insertion of s5A into β-sheet A to transform species I into species II. The subsequent association of the C-terminus, containing the remaining elements of β-sheets B and C, requires the completion of β-sheet A. This is reasonable given the close interior packing of residues from β-sheet A against those of β-sheet B, so that, whether from a kinetic or thermodynamic perspective, β-sheet A must be complete to make the association favorable. Furthermore, using C-terminal peptides that either lacked or contained s1C, it was found that the presence of s1C greatly enhances association of the remainder of the C-terminus. Completion of β-sheet C may serve to position the hairpin of s4B and s5B appropriately for more facile insertion. Most importantly, the insertion of s4A (the RCL) into β-sheet A was found to only occur after the C-terminal peptide had associated. This may again result from favorable packing interactions between the expanded β-sheet A and underlying β-sheet B that can only occur once β-sheet B has been completed by insertion of the C-terminus. This requirement, however, underlies the preferred folding route to the metastable state and explains why the latent state only forms from the metastable state rather than directly by an alternative pathway. Thus, in Fig 3, species II cannot convert to species IV in which s4A has inserted into β-sheet A. If it could, the metastable state would be bypassed and species IV would lead directly to the most stable latent state (see Fig 1C) by subsequent insertion of the C-terminus. Instead, the latent state can only form from the first-formed metastable state (species III). The one possible fly in the ointment is an H/D exchange study carried out on the refolding of full length α1PI after dilution from 6 M guanidine HCl, which concluded that incorporation of the C-terminal residues into β-sheets B and C is largely complete before the center of β-sheet A (s5A) folds [50]. The authors, however, proposed a reconciliation by suggesting that s5A may be slow to form stable hydrogen bonds despite being correctly positioned. This might happen if helix F, which overlies β-sheet A, is the very last element to adopt stable structure and, until it does, can reversibly insert into β-sheet A. The ability of helix F to unfold and partially insert into β-sheet A has been observed in a variant of the serpin α1-antichymotrypsin [51] and in wild-type α1PI incubated at physiological temperature for long periods [52].
Fig 3. Serpin folding pathway.

Initial collapse and folding leads to species I, in which the whole C- terminal region from s5A (green), through the RCL (s4A, yellow) and s1C/s4B/s5B (cyan) have not yet associated. Insertion of s5A leads to completion of β-sheet A and formation of II. The remaining C-terminal region is now better positioned to associate and complete the B/C β-barrel. In addition completion of β-sheet A has presumably put in place necessary contacts for this C-terminus to now associate with favorable energy. Such association leads to the metastable state, III. With the Z mutation at the end of s5A it is likely that a species closely resembling II can build up and would thus represent the on-pathway polymerogenic intermediate. Insertion of the RCL into β-sheet A only becomes favorable once the B/C barrel has been completed and so, critically, species IV, and subsequently species V are not formed directly from II. Instead, V can only form from III. This ensures that the metastable state, with exposed RCL is the preferred species formed during folding, rather than the latent state, V. Figure from ref [49]. Copyright ASBMB.
The second part of the serpin folding problem, namely what slows the final step from the metastable to latent states, has also been addressed by several studies. Since cleavage within the RCL of an inhibitory serpin leads to rapid insertion of the RCL into β-sheet A, it seems likely that restraints at the C-terminal end of the RCL (s1C) prevent insertion in the absence of cleavage. In support, it was shown that insertion of an extra 30 residues into the RCL of α1PI led to direct formation of a folded form with properties of the cleaved serpin, implying that direct connection of the RCL to s1C prevents facile RCL insertion and formation of the latent state [15]. A further ingenious study from the same group using a circularly permuted form of α1PI, with s1C now at the C-terminus and s4B and s5B at the N-terminus, found that the polypeptide folded into the equivalent of the latent state. Since s4B and s5B are positioned close in space to the normal N-terminus in latent α1PI, this unusual chimera was thought not likely to affect the ability of s4B and s5B to complete β-sheet B. Accordingly, the formation of a latent conformation suggested that the restraint imposed by s1C being covalently anchored to the C-terminal s4B and s5B hinders the latency transition in the normal protein [53]. Furthermore, the stability of β-sheet C itself, and hence of s1C is important. Mutations at Val364 and Phe366 of s1C in α1PI and of residues that directly pack with them caused very large increases in the rate of conversion to the latent conformation that are best understood as reducing the activation energy for the transition [54], highlighting the importance of interactions within the B/C barrel in maintaining the kinetic trap.
3.ii Serpin Polymerization
3.ii.a Polymers formed during folding
It has been known for many years that the Z variant of α1PI, resulting from an E342K mutation at the C-terminal end of s5A, results both in greatly reduced secretion of functional serpin from the liver, as well as liver disease and emphysema [55]. In 1992 it was shown that the reduced secretion from hepatocytes results from formation of polymeric species in the endoplasmic reticulum [17]. Later it was established that polymer formation occurs as a result of much slower folding of the serpin and the accumulation of an aggregation-prone intermediate on the normal folding pathway [16]. Two variants (S49P and S52R) of another serpin, neuroserpin (serpinI1), also give rise to pathology resulting from formation of polymers [56], though in this instance they form in neurons and result in a dementia that has been given the acronym FENIB [57]. Despite the parallels between the pathologies in α1PI and neuroserpin, the mutations occur in very different parts of the secondary and tertiary structure of the serpin; at the C-terminal end of s5A for the E342K mutation in α1PI and within helix B (located on the back side of β-sheet A) for both S49P and S52R in neuroserpin. Although a mechanism to explain polymerization in both types of mutant serpin was put forward that started from the folded structure and postulated the insertion of the RCL of one molecule into β-sheet A of a second in an iterative manner [58], the demonstration that polymers of Z α1PI can form from a normal folding intermediate [16] suggests a very different route to intracellular polymer and consequently a different structure for such aggregates.
Considering the folding pathway outlined in Fig 3, together with the location of the E342K mutation (indicated in the figure), a plausible candidate for the accumulating, polymerogenic, on-pathway intermediate is one that resembles species II. The introduction of an unfavorable mutation at the top of s5A might be expected to slow down the last part of the insertion of s5A into β-sheet A and result in an accumulation of this species, whereas such accumulation would not occur for wild-type protein. When final insertion of the C-terminal end of s5A occurs, insertion of the C-terminal s1C and the hydrophobic β-hairpin s4B/s5B might occur in two ways, intramolecularly to give normally-folded protein (Fig 4A) or intermolecularly to give a heterodimer (Fig 4B). Such intermolecular association would be concentration-dependent and thus only compete well with intramolecular association when there is an elevated concentration of the intermediate (true for the Z variant). In the resulting heterodimer, the “acceptor” molecule has a completed serpin fold, though with a free C-terminus that also includes the RCL (s4A), while the “donor” molecule still lacks a completed B/C barrel. At this point it would be highly energetically favorable for the exposed and unconstrained RCL of the acceptor molecule to rapidly insert into its own β-sheet A [15] and be transformed into a conformation resembling the cleaved serpin, but again with an exposed C-terminus consisting of s1C, s4B and s5B (Fig 4C). This heterodimer could then add to another intermediate molecule, which would subsequently undergo the same native-to-cleaved-like transformation. In this way a polymer would be built up that would have internal molecules that are identical and be structurally indistinguishable from the highly stable cleaved conformation, while the end molecules would differ by also having a C-terminal tail for the acceptor end serpin and an incomplete B/C barrel for the donor end serpin (Fig 4D). Furthermore the free C-terminal peptide could potentially complete the serpin fold of the molecule at the donor end to give a circular polymer of identical cleaved-like serpin molecules. A recent SAXS study that examined short ring oligomers of Z α1PI formed intracellularly, and therefore presumably during folding, concluded that their extended structures were most consistent with such a C-terminal swap mechanism [59].
Fig 4. Polymer formation from unfolded state.

Based on the folding pathway of Fig 3, the intermediate II, can subsequently follow two paths, depending on concentration of intermediate. Pathway A leads to normally folded serpin, while pathway B, in which two molecules of type II associate, with the C-terminus of one completing the B/C barrel of the second, leads to a heterodimeric species, in which the acceptor molecule (cyan) has a native like fold, with B/C barrel completed by the C-terminus of the donor serpin (red), while the donor molecule still has an incomplete B/C barrel. In this state, with the B/C barrel of the cyan molecule complete, the RCL of the acceptor molecule is free to rapidly insert into its own β-sheet A (pathway C). This is energetically very favorable, being equivalent to the insertion that occurs during reaction with proteinase. The resulting new heterodimer has the acceptor serpin now in the extremely stable cleaved-like conformation, but with its own C-terminus still exposed and available to act as a donor to another species II serpin. After two additional rounds of addition and RCL insertion (steps B and C) a polymer of form D would be obtained. The central serpin moieties are identical, while the left and right molecules differ in either still having an incomplete B/C barrel or an available s1C/s4B/s5B extension to donate, respectively. If this extension is donated to the incomplete leftmost molecule a circular structure (here a tetramer) of identical cleaved-like molecules would form. This is the same type of structure as for the trimer in Fig 6B.
Such a mechanism of polymer formation might also, less obviously, explain why the neuroserpin variants with mutations in the B helix are polymerogenic. An x-ray structure of cleaved neuroserpin reveals that the side chain of Ser49 contacts the side chain of Leu383 [60], which is a residue within the C-terminal s4B/s5B β-hairpin. Mutation of this residue to proline, or a potentially more disruptive mutation of the nearby Ser52 to arginine (which is a buried residue) could slow down the insertion of the C-terminus and so, for quite different reasons than for the Z mutation, lead to a build up of an intermediate resembling species II in Fig 3. In keeping with this mode of modulating intermediate accumulation through influencing the ease of insertion of the C-terminal peptide, an F51L mutation has been shown to suppress the folding defect in Z α1PI [61]. This residue, which is structurally equivalent to the residue preceding Ser49 in neuroserpin, packs against the side chain of Met385 in strand s5B. The mutation to leucine may make insertion of the C-terminal hairpin easier and so partially compensate for the effects of the E342K mutation in slowing down this last step.
3.ii.b Polymers formed from folded serpin
While there is persuasive evidence that serpin polymers formed within the ER during folding have the kind of structure described above in Fig 4D, serpin polymers that form from folded native serpins may not use the same mechanism of polymerization. Although polymers formed from the folded state are typically studied because of experimental ease of generation, they might nevertheless still be of physiological and pathological importance, since it has been observed that polymers of Z α1PI also occur in circulation [62] and are even found to co-localize with neutrophils in lung alveoli [63], though not within the neutrophils [64]. Understanding whether such polymers use the same mechanism of formation as polymers formed intracellularly during folding is therefore of high significance.
Here it is instructive to examine an energy diagram for the folding pathway (Fig 5), and consider whether unfolding from the native state is likely to occur by an exact reversal of this pathway. The energy diagram was constructed assuming that incorporation of s1C, s4B and s5B results in a greater stabilization than does incorporation of s5A, and that there is a much higher activation energy for insertion of s1C/s4B/s5B into species I of Fig 3 than for insertion of s5A into the same species. These assumptions are based on the much greater stabilization expected from completion of both β-sheets B and C than from completion of just β-sheet A, together with the observed difference in kinetics of association of s5A compared with s1C/s4B/s5B during folding [49]. The result is that, while the forward folding pathway (shown in black) leads from insertion of s5A to insertion of s1C/s4B/s5B, unfolding from the native state would lead more favorably to removal of s5A followed subsequently by removal of s1C/s4B/s5B (shown in red). The structure of the first formed intermediate from such an unfolding would have s4A and s5A exposed and unstructured and consequently have an open center to β-sheet A. Polymers formed through engagement of the center of β-sheet A by one or both of s4A and s5A of a second molecule would thus be the ones most likely to be formed by incubating folded α1PI, whether native or Z variant, at elevated temperature or by chaotropic agents such as guanidine HCl or urea. Indeed, a structure of a dimer of antithrombin induced by low pH, as well as the properties of polymers of α1PI formed through incubation with 0.75 M guanidine HCl at physiological temperature, suggested a mechanism of polymerization that, in both cases, involved a swap of both s5A and s4A from one molecule to the next, and that could account for long chain linear polymers (Fig 6A) [65].
Fig 5. Possible folding/unfolding energy diagram for serpins.

From elucidation of the folding pathway of α1PI, the preferred next association with species I of Fig 3 is of s5A rather than s1C/s4B/s5B. This is reflected in the higher energy barrier (red) going from I to species IIIa (s5A and s4A exposed), than in going from I to II (black), even though the stability of IIIa might be greater than that of II. Once s5A has associated with I to form II, s1C/s4B/s5B rapidly and favorably forms the native state, III. However, starting from the native state, the activation energy barrier in going to II, with loss of s1C/s4B/s5B, is much higher than for loss of s5A to lead to IIIa. Accordingly, the preferred route for unfolding from the native state is the red pathway from III to IIIa rather than the black route to II. This explains how different types of polymerogenic intermediate can be formed (II vs IIIa) depending on whether the starting point is the unfolded or the folded state.
Fig 6. Observed and modeled serpin polymers formed by different methods.

Panel A, polymer modeled from an x-ray structure of an s4A/s5A-swapped antithrombin dimer [65]. This is the kind of swap that would be predicted by the red unfolding pathway of Fig 5, in which the first formed intermediate (IIIa) has s5A extracted from β-sheet A, and so both s4A and s5A available for donation to another molecule. Panel B, x-ray structure of a heat-induced trimer of α1PI in which the red pathway has been deliberately blocked by restraining s5A through disulfide linkage to s6A [69]. This only leaves the reversal of the black pathway from III to II, and so results in polymers that are identical to polymers formed during folding that occur from build up of species II from species I (Z or other adverse mutations). Panel C, modeled polymer formed from donation of part of the RCL (s4A) of one molecule to the bottom of β-sheet A of a second, thereby partially expanding the sheet A of the second molecule [72]. This would not be expected to result in as large an increase in stability as for formation of polymers such as in panel B or in Fig 4 panel D.
For the Z variant, the E342K mutation within s5A, might be expected to lower the energy barrier for its removal from the folded state and so promote polymerization by this second mechanism. This is supported by a recent unfolding study using fluorescent reporters. By detection of structural differences at the top of β-sheet A caused by the Z mutation [66] and by the greater ease with which RCL-derived peptides can anneal with native Z variant than with native wt serpin [67], the authors concluded that increased exposure of side chains within s5A and helix F occur as the first events upon exposure to increasing concentrations of guanidine HCl [68],
Given our proposal that serpin polymers formed during folding involve swapping of s1C/s4B/s5B and have the structure of Fig 4D, while those formed from the native state by unfolding involve intermolecular swapping of s4A/s5A and have the structure given in Fig 6A, it is initially surprising that an x-ray structure of a circular trimer of α1PI generated by heating the native protein (Fig 6B) [69] is identical to the structure predicted by us for the folding polymer (Fig 4D), if the two ends of the latter are joined. However, it should first be noted that heating close to the denaturation temperature has been found to result in overlapping polymer ladders [69], suggesting that, at this elevated temperature, both mechanisms of polymer formation might occur simultaneously. Realizing the possibility of dual mechanisms of polymer formation, the authors deliberately prevented the s4A/s5A swap by tying s5A to s6A through an engineered disulfide bond. If the result of blocking this pathway were to leave removal of s1C/s4B/s5B as the only remaining likely unfolding event, the intermediate formed would be that of species II of Fig 3 and the resulting polymer would have the structure expected for the folding polymer.
The formation of two structurally distinct sets of polymers by heating α1PI may also explain the observation that a monoclonal antibody that is specific for the pathogenic polymers of Z α1PI found in liver [70] stains with heat polymers on a Western blot of polymerized α1PI, but not with polymers induced by guanidine HCl. If the antibody only recognizes polymers of the form proposed above (Figs 4D and 6B), it would bind to pathogenic folding polymers and one subset of heat-induced polymers (the s1C/s4B/s5B subset), but not the other subset (s4A/s5A swap) or to guanidine HCl-induced polymers (again the s4A/s5A swap).
3.iii Conflict and Resolution
The two mechanisms of polymerization outlined above both lead to elongated polymers in which the structure of the serpin moiety closely resembles that of either the extremely stable cleaved serpin or the stable latent state. As seemingly well folded proteins, each would be expected to be resistant to targeting for removal by the cell’s unfolded protein response and so accumulate as stable, but pathological, intracellular deposits. The fundamental difference between their routes to formation is that one occurs during folding and the other during unfolding. The energy diagram in Fig 5 explains why these pathways might be different, with polymers formed during folding that lead to build-up of a species II-type intermediate (Z, S49P, S52R) having a swapped s1C/s4B/s5B structure and polymers that result from unfolding, with removal first of s5A, having a swapped s4A/s5A hairpin. These two types of structure might both occur, but under different physiological circumstances. While this leads to a possible pleasing reconciliation of apparently conflicting structures for physiological polymers, it still leaves the third long-standing structural proposal for polymers in major conflict.
David Lomas and colleagues have for many years championed a quite different mechanism of polymerization of Z α1PI for both intra- and extracellularly-formed polymers that involves insertion of the RCL of one molecule into β-sheet A of a second (Fig 6C) [58, 71, 72]. While conceptually this might seem to be a plausible mechanism, given the ability of β-sheet A to undergo an expansion from 5 strands to 6, there is no x-ray structure to support it, in contrast to the structures of polymers formed by either swapping of s1C/s4B/s5B or s4A/s5A discussed above. Furthermore, the extensive studies that have attempted to substantiate the mechanism are also consistent with one or other of the demonstrated polymerization mechanisms. For example, polymerization of the Ser53Phe variant of α1PI (Siiyama) has been held up as evidence in support of the RCL-sheet A polymerization mechanism [73]. However, this polymer reacts with the same monoclonal antibody, 2C1, as does the circular s1C/s4B/s5B swap trimer [69, 70]. Since it is claimed that this antibody uniquely recognizes polymers of the structure of physiological Z polymers [70], this suggests that they involve s1C/s4B/s5B swap and not insertion of the RCL into β-sheet A. Interestingly, another monoclonal antibody recently reported by the Lomas group, that prevents intracellular polymerization of Z α1PI has its epitope well removed from β-sheet A or the RCL, and is instead located on the “back” side of the molecule (see Fig 1) on helices A, C and G [74]. It is hard to understand on the RCL-sheet A insertion model why such an antibody might prevent polymer formation. However, for the s1C/s4B/s5B mechanism of polymerization during folding a much more obvious explanation is apparent. Thus, residues from helices A and G contact hydrophobic residues in the C-terminal β-hairpin of s4B-s5B and so may stabilize states with the C-terminus inserted. This may speed up the last step of the folding pathway for the Z variant (see Fig 3), perhaps by favoring the red pathway of folding from I to III via IIIa in Fig 5 and so prevent the build-up of the polymerogenic intermediate.
4. Energy Coupling in the Serpin Inhibition Mechanism
4. i. The necessity for energy coupling
As noted earlier, serpins employ a novel branched pathway suicide substrate mechanism to inactivate their target proteinases (Fig 2). The salient feature of this mechanism is the triggering of a massive serpin conformational change at the acyl-intermediate stage of proteinase cleavage of the serpin RCL as a regular substrate that results in the kinetic trapping of the acyl-intermediate. In the conformational change, the cleaved RCL, now released from its attachment to β-sheet C through s1C, inserts into β-sheet A as s4A to relieve the strain of the metastable serpin fold, causing the RCL-tethered proteinase to be translocated to the opposite end of β-sheet A and inactivated by conformational distortion. Competing with this conformational change is a normal deacylation of the acyl-intermediate that produces free cleaved serpin and regenerates proteinase. Efficient kinetic trapping of the acyl-intermediate thus requires that the cleaved RCL and tethered proteinase insert rapidly into β-sheet A and induce conformational distortion of the proteinase before deacylation can occur. Rapid kinetic studies of serpin-proteinase reactions support an extremely fast rate of kinetic trapping of the acyl-intermediate by the serpin conformational change that is limited by the rate at which the Michaelis complex is converted to the acyl-intermediate or, in the case of several target proteinases, by the slower rate of disengagement of the RCL-linked proteinase from exosite interactions with the serpin body that stabilize the Michaelis and acyl-intermediate complexes [75–78]. Conversely, slowing the rate of the serpin conformational change by natural or engineered substitution of residues in the RCL that insert into the hydrophobic core of sheet A [23, 41], or engineering proteinase reactive variants of noninhibitory serpins that can only slowly undergo the metastable to stable transformation [39, 79], results in the inability to trap the acyl-intermediate complex, as a result of this intermediate deacylating before the serpin conformational change is completed.
Consideration of the remarkable structural changes and energetics involved in transforming the Michaelis docking complex of serpin and proteinase to the kinetically trapped acyl-intermediate complex has suggested that it must involve intermediate stages and a means of coupling the energetically favorable serpin conformational change to the energetically unfavorable conformational distortion of the proteinase. This is because the energy released by the serpin conformational change superficially appears to be expended in the process of translocating the proteinase across β-sheet A before it can be utilized to distort the proteinase. Recognizing this problem and noting in the structure of the serpin-proteinase complex that helix F overlies β-sheet A and appears to obstruct the path of the inserting RCL and tethered proteinase, it was hypothesized that the energy released by the serpin conformational change could be partially stored by movement or unfolding of the F helix at an intermediate stage of inserting the RCL and translocating the proteinase across β-sheet A. Following full RCL insertion and proteinase translocation, this stored energy could then be recovered by the return of the F helix to its native state and coupled to conformationally distorting the proteinase [35]. This hypothesis thus implied that kinetic trapping of the acyl-intermediate by the serpin conformational change occurs in two stages, both of which are coupled to structural changes in helix F.
4.ii. Involvement of helix F
Early evidence supporting an important role of the F helix in the ability to kinetically trap the acyl-intermediate was the report that a PAI-1 variant in which the F helix was deleted adopted the serpin fold, but reacted with proteinases solely as a substrate; i.e. it generated only RCL-cleaved PAI-1 in which the RCL had inserted into β-sheet A, but the acyl-linked proteinase had completely deacylated [80]. This is unlikely to be the result of helix F deletion making it harder for the proteinase to translocate to the bottom of the serpin. In a related study, monoclonal antibodies that induce PAI-1 to react as a proteinase substrate were found to bind to an epitope exposed on helix F by the trapping serpin conformational change, consistent with structural alterations in helix F occurring during this conformational change that are critical for trapping the acyl-intermediate complex [81, 82]. Subsequent H/D exchange [83] and unfolding studies of the serpin α1PI [84] have shown that helix F is highly flexible in the metastable state, with the C-terminal end of the helix existing partly in an unfolded state, consistent with the predicted importance of movement or unfolding of the F helix to store the energy released by RCL insertion and translocation of the acyl-linked proteinase for later proteinase distortion. Mutagenesis of residues in helix F and the C-terminal loop connecting helix F with s3A has shown that residues that comprise the interface between helix F and β-sheet A are highly conserved in all serpins and are critical for inhibitory function, with their mutation decreasing inhibitory activity by enhancing flux through the substrate pathway [85]. By contrast, mutation of surface exposed residues in helix F and the C-terminal loop minimally affect inhibitory function. Such studies are consistent with the helix F-sheet A interface playing a role in coupling the serpin conformational change to the kinetic trapping of the acyl-intermediate complex in reactions with proteinase.
That structural changes in the helix F-sheet A interface play a role in the kinetic trapping of the acyl-intermediate by the serpin conformational change is supported by a study in which substitutions of α1PI Gly117 in the middle of strand 2A were made with nonpolar residues that fill a hydrophobic cavity in the interface with helix F [86]. Such substitutions were found to stabilize the native metastable state and enhance substrate reactivity with proteinase in a manner that correlated with the volume of the nonpolar sidechain substitution. Such findings implied that stabilizing helix F-sheet A interactions reduces helix F flexibility and blocks the ability of the serpin conformational change to distort the proteinase. A later H/D exchange study showed that the G117F mutation indeed reduces the flexibility of helix F in the native state [87], in keeping with the importance of this flexibility in allowing the efficient trapping of the acyl-intermediate complex.
4.iii. Evidence for complex intermediates
Kinetic studies that have followed the conformational changes in the proteinase during the transformation of the Michaelis docking complex to the trapped acyl-intermediate complex support a two-stage kinetic trapping of the acyl-intermediate complex by the serpin conformational change. An early study of the reaction of a Trp-less α1PI with proteinase thus observed biphasic changes in proteinase Trp fluorescence consistent with rapid conformational alteration of proteinase to form an intermediate complex, followed by a slower additional proteinase conformational change to form the final complex [88]. The nature of these complexes was clarified in a subsequent study of the reaction of a fluorescently silent 4-fluoro-Trp substituted PAI-1 variant with proteinase in which similar biphasic changes in proteinase Trp fluorescence were observed [89]. In this case the intermediate complex was shown to be formed with kinetics indistinguishable from those of the SDS-stable covalent acyl-intermediate complex, as measured by rapid quench methods, confirming a two-stage trapping of the acyl-intermediate complex involving two sets of proteinase conformational changes.
A recent study, using a combination of FRET and fluorescence perturbation changes accompanying the reactions of dansyl and NBD fluorophore-labeled α1PIs with trypsin, more clearly established the structural attributes of the intermediate and final complexes [36]. Following very rapid formation of the Michaelis docking complex, an intermediate complex was formed at a rate limited by acylation of the Michaelis complex (~50 s−1) in which the proteinase was fully translocated to one end of β-sheet A close to s2A. This intermediate was then transformed at rates of 1–2 s−1 to a final complex in which the proteinase moved ~10Å to reside close to s5A, in keeping with the final position of proteinase observed in X-ray structures of the complex. Such findings were consistent with the inactivating conformational changes in the proteinase occurring in two stages and involving significant movements of the proteinase at the end of β-sheet A (Fig 7). Significantly, the two proteinase conformational changes were correlated with biphasic perturbations of a reporter fluorophore in the helix F-sheet A interface, consistent with the movement of proteinase coinciding with structural alterations in the interface. These findings thus provide direct support for the hypothesized coupling of structural alterations in the helix F-sheet A interface with the inactivating conformational changes in the proteinase [35], as schematically depicted in Fig 7. Moreover, they suggest that the disabling of proteinase catalytic function occurs in two stages, since the observation of a transiently stable intermediate en route to proteinase distortion implies a diminished ability of the intermediate complex to deacylate and, in the case of the PAI-1-proteinase reaction, the kinetics of intermediate complex formation parallels the formation of a deacylation-resistant covalent complex.
Fig 7. Two-stage mechanism for helix F participation in kinetic trapping of the acyl-enzyme complex.

The scheme depicts the hypothesized two-stage mechanism by which the serpin and proteinase conformational changes triggered by proteinase cleavage of the serpin RCL in the acyl-enzyme complex are coupled to kinetically trap the acyl-enzyme complex following its formation from the initial Michaelis docking complex. In the scheme, the Michaelis docking complex (serpin colored cyan with P1 residue in blue spheres and proteinase colored green with active-site Ala195 residue in yellow spheres) undergoes transformation to an intermediate conformationally altered acyl-enzyme complex in which the cleaved RCL (blue) has fully inserted into β-sheet A, with displacement of helix F (red) from its native position to allow translocation of the RCL-linked proteinase to an intermediate position at the s2A end of β-sheet A. In the intermediate, the acyl-linkage between the serpin P1 residue and the proteinase catalytic Ser195 (yellow spheres) is partially distorted and the F helix is constrained in a higher energy state. The intermediate is then transformed to the final trapped complex by the relaxation of the F helix to its unconstrained position with the energy released by this relaxation driving the concomitant movement of the proteinase to the s5A side of sheet A and causing full inactivation of the proteinase by conformational distortion. The initial and final complexes are x- ray structures of Michaelis (1OPH [180]) and trapped acyl-enzyme (1EZX [29]) complexes of α1PI with trypsin, whereas the intermediate complex is modeled based on FRET and fluorescence perturbation changes accompanying the reaction of fluorophore-labeled α1PIs with trypsin as described in the text.
4. iv Multiple states of proteinase distortion
The nature of the disabling proteinase conformational changes in the intermediate are suggested by an earlier rapid kinetic study of the reaction of a P1 Trp α1PI variant with proteinase. This study observed the formation of a complex with kinetics resembling those of the intermediate complex in which the P1 Trp had undergone fluorescence changes indicative of its expulsion from the proteinase S1 pocket [90]. The second stage proteinase conformational changes may involve further distortion of the catalytic residues as well as one of the proteinase domains to account for the full inactivation of proteinase catalytic function and the observed susceptibility of the distorted proteinase to proteolysis in the kinetically trapped complex [28, 91, 92].
The FRET study discussed above also provided insight into an equilibrium between inactive and partially active forms of the proteinase in the serpin-proteinase complex that was earlier demonstrated by the finding that a proteinase cofactor, in this case calcium ions, could partially restore proteinase catalytic function in the complex [93]. Calcium binding to the proteinase in the stable α1PI-trypsin acyl-intermediate complex, observable by fluorescence perturbation of a reporter group at the end of β-sheet A, was found to shift the equilibrium to favor the partially active form of the complex. This partially active complex was found to be distinct from the intermediate complex formed in the two-stage inactivation of proteinase, since it was formed with the same two-stage kinetics of proteinase inactivation as the fully inactive complex [36]. The calcium ligand thus promotes the partial refolding of the distorted proteinase domain within the final complex and is not capable of recapitulating the movement and restoration of the partially disabled proteinase in the intermediate complex, presumably because this requires a coupled movement of the F helix to a constrained high energy state.
5 Use of Cofactors
While the above studies have advanced understanding of the intricate choreography of the serpin inhibition mechanism, they have also highlighted how demanding the mechanism is in terms of correct folding and precise timing and coordination of conformational changes upon reaction with proteinase. Thus, far from answering why serpins are often the inhibitor of choice, they may have made it seem even more puzzling that they are. This is where the use of cofactors plays such a critical and unique role in regulating the temporal and spatial activity and even tailoring the specificity of serpins, thereby giving an individual serpin a repertoire of reactivities that would be difficult or impossible to achieve with a canonical inhibitor. As examples, we consider below the use of glycosaminoglycans to regulate antithrombin and of protein Z to regulate ZPI.
5. i Use of glycosaminoglycans to regulate the localization and specificity of antithrombin action
Several serpins, most notably antithrombin, are known to utilize the negatively charged glycosaminoglycan, heparin, or related glycosaminoglycans (e.g., heparin sulfate and dermatan sulfate) to regulate their specificity and site of reaction with proteinases (Table 1). Antithrombin (serpinC1) is the most important inhibitor of blood coagulation proteinases, as judged from the increased risk of pathologic blood clots resulting from deficiency of the serpin in humans [94], and the embryonic lethality or coagulopathy associated with complete deficiency in mice [95] and zebrafish [96]. The activity of this serpin is regulated by heparin in a manner that enables the serpin to inhibit its target proteinases at the right time and place. Vascular sites of injury, where blood coagulation proteinases are rapidly activated, are thus abundant in the heparin sulfate glycosaminoglycans capable of activating the serpin, allowing antithrombin to rapidly inhibit proteinases that escape from an injury site and thus to prevent the dissemination of a blood clot beyond this site [97]. By contrast, the repressed activity of the serpin in the absence of heparin suffices to regulate the slow activation of these proteinases in the unperturbed vasculature.
The mechanism by which antithrombin is activated by heparin has been extensively studied and has undergone frequent revision, with recent clarification suggesting a mechanism that differs significantly from earlier proposals. While this serpin possesses a P1 Arg scissile bond that is compatible with the trypsin-like specificity of blood coagulation proteinases for cleaving at Arg-containing peptide bonds, the rate at which antithrombin inhibits these proteinases is relatively slow (102–104 M−1s−1) with half-lives of >30 s at plasma concentrations of the serpin. Heparin accelerates antithrombin inhibition of three coagulation proteinases, thrombin, factor Xa and factor IXa, by103–105-fold, to achieve rates of inhibition (107–108 M−1s−1) near the diffusion limit, suggesting that these proteinases are the main targets [98]. Conversely, the anticoagulant proteinase, activated protein C, which also prefers P1 Arg substrates, is inhibited extremely slowly by antithrombin even after heparin activation (0.1 M−1s−1). The mechanism of heparin acceleration of antithrombin involves two components: (i) an allosteric component in which a sequence-specific heparin pentasaccharide binds specifically and with high-affinity to a site on antithrombin centered on helix D [99–101] and induces an activating conformational change which enhances reactivity ~100-fold with factors Xa and IXa [99, 102]; and (ii) a bridging component in which the heparin chain that extends beyond the pentasaccharide binding site for antithrombin binds the proteinase in a conserved basic exosite to promote the Michaelis complex docking of serpin and proteinase [102–104]. The bridging mechanism augments antithrombin reactivity with factors Xa and IXa 100–1000-fold and is solely responsible for enhancing reactivity with thrombin ~10,000-fold.
5.i.a Early mechanisms
X-ray structures of antithrombin free and complexed with heparin pentasaccharide have illuminated the allosteric activating conformational changes induced in the serpin by the pentasaccharide and shown that these involve major changes in the heparin binding site in helix D, changes in the hydrophobic core and expulsion of the serpin RCL hinge from a stabilizing interaction with β-sheet A [100]. The changes in the heparin binding site produce a complementary fit of the heparin pentasaccharide that greatly enhances heparin affinity and account for the ability of the pentasaccharide to stabilize the energetically less favored activated state [105, 106]. Based on these structures it was initially proposed that the expulsion of the serpin RCL hinge from β-sheet A accounted for heparin allosteric activation of the serpin by increasing the accessibility of the RCL P1 Arg bait residue to proteinase [100, 107, 108]. However, later mutagenesis studies revealed that the determinants of the differential reactivities of repressed and allosterically-activated antithrombin were not contained in the RCL and instead resided in an exosite outside the RCL centered on strand 3 of β-sheet C [109–112]. Subsequent X-ray structures of Michaelis complexes of heparin-activated antithrombin with catalytically inactive factor Xa, factor IXa and thrombin (Fig 8) confirmed a critical interaction between Arg150 of factors Xa and IXa and an exosite binding pocket of antithrombin involving residues in sheet C that stabilize the Michaelis complexes [113, 114]. Thrombin, however, lacks the critical Arg150 residue and does not engage the exosite in the Michaelis complex. As compensation, the proteinase engages the extended chain of a bridging heparin through a basic exosite in a manner that positions the proteinase active-site for an optimal interaction with the RCL of the serpin [115, 116]. These structures thus rationalized why the allosteric activating effect of heparin on antithrombin was specific for factors Xa and IXa and did not affect thrombin. Moreover, these and other findings suggested that antithrombin utilizes an exosite to achieve specificity, because it had evolved an RCL sequence flanking the P1 Arg that disfavors the unwanted inhibition of activated protein C [117]. Based on these structures a new mechanism of allosteric activation was proposed in which the expulsion of the RCL from β-sheet A was suggested again to be the key activating structural change but in this case the change was deemed necessary to allow the RCL-bound proteinase to access the exosite to enhance antithrombin reactivity [113, 114].
Fig 8. Michaelis docking complexes of heparin-activated antithrombin (AT) with S195A variants of factor Xa, factor IXa and thrombin (IIa).

Shown are x-ray structures of Michaelis complexes of pentasaccharide heparin-activated antithrombin with factor Xa (H5-AT-FXa, left, pdb 2GD4 [113]) or with factor IXa (H5-AT-FIXa, middle, pdb 3KCG [114]) and of hexadecasaccharide heparin-activated antithrombin with thrombin (H16-AT-IIa, right, pdb 1TB6 [181]) in ribbon. Antithrombin is cyan with the RCL in blue and the P1Arg shown as spheres, the proteinases are green with Ala195 shown as yellow spheres and heparins depicted in black stick. Arg150 of factors Xa and IXa, depicted in yellow stick, interacts with an antithrombin exosite binding pocket, shown in red stick, that stabilizes these complexes. Thrombin lacks Arg150 and cannot interact with the antithrombin exosite but compensates by binding the extended heparin chain through a basic exosite region.
5.i.b Current understanding of the mechanism
This newer mechanism was subsequently challenged based on the observation that mutation of a key exosite residue in β-sheet C resulted in large losses in reactivity in both free and heparin pentasaccharide-activated antithrombin, but minimally affected heparin rate enhancement, implying that the exosite is critical for reactivity in both unactivated and heparin-activated states, and thus that RCL expulsion is not required to access the exosite [112, 118]. That RCL expulsion was not necessary for allosteric activation was supported by a later study of an antithrombin variant in which residues in the loop extending beyond helix D of the heparin binding site were substituted with helix promoting residues to mimic the helix extension that is induced upon heparin allosteric activation [119]. This variant was found to be essentially fully activated in its reactivity toward factors Xa and IXa in the native state without the need for heparin. Most notably, the variant exhibited a wild-type protein fluorescence and thus had not undergone the hallmark tryptophan fluorescence changes known to report RCL expulsion from β-sheet A [120]. Heparin pentasaccharide binding produced a further small activating effect of 3–4-fold on factor Xa and IXa reactivity and induced the tryptophan fluorescence changes signaling RCL expulsion from β-sheet A. Together, these findings suggested that RCL expulsion was not necessary for RCL-bound factors Xa and IXa to engage the β-sheet C exosite and that RCL expulsion produced at most a modest reactivity enhancement. Such studies complemented earlier findings that blocking helix D extension by deletion of residues in the helix D C-terminal loop or substitution of a Pro in this region resulted in the retention of a large heparin allosteric activating effect on antithrombin reactivity with factor Xa, despite the suppression of RCL expulsion, as indicated by loss of the hallmark tryptophan fluorescence changes [121, 122].
Based on these findings an alternative mechanism of heparin allosteric activation of antithrombin was proposed [118]. In this mechanism the interactions of RCL-bound proteinase with the exosite in the native repressed-reactivity state were suggested to be both positive and negative and heparin allosteric activation was proposed to enhance reactivity by alleviating the negative exosite interactions (Fig 9). This proposal received support from a study in which the six residues that comprise the exosite binding pocket in sheet C observed in the X-ray structures of the Michaelis complexes were mutated alone or in combination [123]. Whereas Tyr253 and His319 were both found to be key positive contributors to the exosite interaction with factors Xa and IXa in native and heparin activated states, Asn233, Arg235, Glu237 and Glu255 were found to make both negative and positive interactions with these proteinases that depended on the activation state and whether the critical Tyr253/His319 residues were mutated. Heparin activation was found to enhance the positive interactions and abrogate the negative interactions and account for the bulk of the allosteric activation effect. As expected, thrombin reactivity was minimally affected by the mutations. Significantly, mutation of Arg150 of factor Xa that binds in the antithrombin exosite pocket resulted in a loss of the exosite contribution to antithrombin reactivity in both native and heparin-activated states.
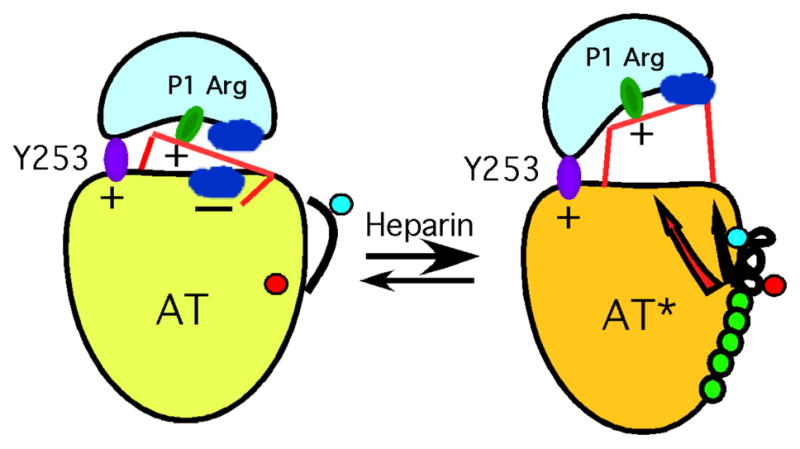
Fig 9. Heparin activation of antithrombin through mitigation of repulsion.
When proteinase engages the resting state of antithrombin (left) it recognizes the favorable P1 arginine side chain and binds to the exosite centered on Tyr253. The hinge of the RCL is also partially inserted into β-sheet A. In this state, unfavorable interactions at the interface between proteinase and serpin (shown as − ) lead to a relatively weak overall interaction and correspondingly slow rate of inhibition. Here, the favorable P1 and exosite interactions are partially offset by other unfavorable interactions. When heparin binds, to give AT*, the major consequence of the long-range conformational change induced in the body of antithrombin (open arrow) is a mitigation of the unfavorable interface interactions, such that favorable P1 and possibly better optimized exosite interactions are no longer opposed, thereby resulting in a greatly enhanced rate of inhibition. In addition, the expulsion of the hinge of the RCL, as a secondary, linked element of the conformational change (filled arrow), results in a small additional enhancement factor in the rate of inhibition. Most of the rate enhancement, however, comes from the more optimal surface-surface interactions. Adapted from ref [119]. Copyright ASBMB.
A more recent study clarified the role of the RCL hinge-sheet A interaction in the allosteric activation mechanism by characterizing fifteen antithrombin variants in which this native state interaction was perturbed by mutating the RCL hinge P14 residue [124]. The mutations produced the expected large changes in native state stability and these were correlated with corresponding major changes in the allosteric activation state of antithrombin, as judged from altered reactivities with factors Xa and IXa and heparin affinities, in agreement with limited earlier studies [125–127]. However, the minimal perturbation of wild-type fluorescence of the variants and inability of a two-state allosteric equilibrium to account for the correlations between native state stability and allosteric activation state implied a minimal three-state allosteric activation model involving an intermediate state in which changes in the heparin binding site and in the hydrophobic core that enhance heparin affinity and antithrombin reactivity have occurred, but RCL expulsion from β-sheet A has not (Fig 10). X-ray structures have demonstrated the existence of such an intermediate activated state [128, 129] and rapid kinetic studies support a three-state model of heparin allosteric activation involving an intermediate state in which the RCL-β-sheet A interaction is retained [130]. Heparin binding to the P14 variants was found to produce variable Trp fluorescence enhancements that were consistent with heparin shifting the preexisting allosteric equilibrium between native and intermediate states to favor an equilibrium between intermediate and fully-activated states in which RCL expulsion from β-sheet A has occurred only in the latter. Together, these studies thus support an allosteric activation mechanism in which the bulk of the allosteric activating effect that arises from the mitigation of repulsive interactions in the exosite has occurred in an intermediate state in which the RCL-sheet A interaction is intact. A further modest activating effect occurs in the fully activated state as a result of RCL expulsion from β-sheet A allowing the RCL-bound proteinase to move further away from the serpin body and minimize residual repulsive interactions. Collectively, the studies discussed above thus provide a unifying three-stage mechanism for heparin allosteric activation that rationalizes the accumulated evidence of many past studies.
Fig 10. Three-step mechanism of allosteric activation of antithrombin by the heparin pentasaccharide.

The top shows X-ray structures of native, intermediate-activated and fully-activated antithrombin depicting the key secondary structural elements undergoing activating conformational changes upon heparin pentasaccharide binding. Strands 2, 3, 5 and 6 of β-sheet A are in cyan ribbon, strands 2, 3, 4 and 5 of β-sheet B are in green ribbon, the RCL is depicted in blue ribbon, with the P14 RCL hinge residue shown as spheres, the helix D site of heparin binding is in red ribbon and the heparin pentasaccharide is shown in black stick. The bottom scheme shows the preexisting conformational equilibria between the energetically favored native antithrombin conformational state (AT) and the less favored intermediate-activated (AT*) and fully-activated (AT**) conformational states. Heparin pentasaccharide binding to native antithrombin shifts the equilibrium in favor of intermediate and fully-activated states with the latter energetically favored as a result of heparin binding with the highest affinity to this state. The native repressed activity state is characterized by the burial of the RCL hinge P14 residue into the hydrophobic core beneath sheet A. In the intermediate state conformational changes are induced in the heparin binding site that are transmitted to the hydrophobic core. In the latter changes, a subdomain consisting of strands 5A, 6A and β-sheet A move to partly close the gap between s3A and s5A and alter the packing with the subdomains containing s2A, s3A and helix D. These changes activate antithrombin reactivity with factors Xa and IXa by mitigating negative interactions between RCL-bound proteinase and an antithrombin exosite. In the fully activated state, additional conformational changes are induced including the extension of the C-terminal loop of helix D and the expulsion of the RCL hinge from sheet A and movement of the RCL away from the serpin body to further enhance reactivity with proteinase.
5.ii Protein Z as a cofactor for ZPI
As described above, the negatively charged polysaccharide heparin can play important and complex roles in fine-tuning the specificity of antithrombin towards solution phase thrombin, factor IXa, factor Xa and activated protein C. There nevertheless appears to be an additional physiological need to be able to selectively inhibit factor Xa when it is membrane associated, such as when it is in the enzyme complex prothrombinase and protected from inhibition by the antithrombin-heparin complex. For such inhibition a different serpin, ZPI, is used together with a protein cofactor, protein Z.
Protein Z-dependent proteinase inhibitor (ZPI or serpin A10) is a selective inhibitor of factor XIa and membrane-associated factor Xa [131, 132], both arginine-specific proteinases. In addition to the core serpin domain, it possesses a highly acidic 39 residue N-terminal tail. Given this specificity it is at first surprising that ZPI has a P1 tyrosine residue, where the more familiar inhibitor of factor Xa, antithrombin, has the expected P1 arginine. The presence of tyrosine rather than arginine reduces the rate of reaction 300-fold but also greatly decreases the SI, from 380 to 3 [133]. To compensate for such unfavorable kinetics of inhibition, ZPI in vivo recruits a protein cofactor, protein Z (PZ), together with phospholipid and Ca2+ for specific, rapid inhibition of membrane-associated factor Xa [131, 134, 135]. Together, these cofactors not only confer specificity for inhibition of membrane-associated factor Xa, but increase the rate of proteinase inhibition over 200-fold, from a modest 4 × 104 M−1 s−1 to a value similar to what would occur with P1 Arg in the absence of cofactors (107 M−1 s−1), though now with a more respectable SI of 3 rather than 380 [133–135].
The specificity for inhibition of membrane-associated factor Xa arises from the presence of membrane-anchoring N-terminal Ca2+-dependent Gla domains in both factor Xa and the ZPI cofactor protein PZ. In both proteins these are followed by two EGF domains and a proteinase or (for PZ) pseudoproteinase domain. Initial experiments on Gla-deleted or Gla swap variants of factor Xa suggested only a minor role for Gla-Gla domain interactions [136], but later Gla domain swap experiments from the same group found a specific interaction between the membrane-anchoring Gla domains of PZ and factor Xa that both promotes the formation of the membrane-localized ternary complex of PZ/ZPI and factor Xa and contributes ~6-fold to the rate of inhibition of factor Xa [137].
Whereas the Gla domains clearly contribute to specificity and to co-localization of the PZ/ZPI complex and factor Xa on the membrane surface, they account for only a small fraction of the observed rate enhancement of all of the cofactors. Recent x-ray structures of ZPI in complex with protein Z have provided insights into how this interaction promotes the specificity of the serpin for membrane-associated factor Xa [133, 138]. The first structure [138] was of full length ZPI in complex with protein Z that lacked the first 83 residues encompassing both the N-terminal Gla domain and the first EGF domain, while the second structure had a longer protein Z that included the first EGF domain [133]. Despite this difference, both structures showed the same extensive interface between the C-terminal pseudo-proteinase domain and the EGF2 domain of protein Z and 6 contact residues on ZPI from helices A and G. From a subsequent mutagenesis and kinetic study it was shown that two of these contact residues are most critical for binding of protein Z, with unitary binding energies of −8.4 kcal mol−1 for Tyr240 in the gate region loop and −10.4 kcal mol− for Asp293, located on helix G [139]. Tyr240 occupies a hydrophobic pocket between the EGF2 and pseudoproteinase domains of PZ, which explains the inability of EGF2 domain swap variants of PZ to bind to ZPI and the importance of interaction with both domains for high affinity PZ:ZPI complex formation [139]. The discovery of such a small number of hot spot residues that account for the majority of the interaction suggests a strategy for design of small molecule therapeutics to disrupt the interface and promote clotting in hemophilia. It was further shown that the affinity of protein Z for cleaved ZPI was ~6-fold lower than for the native serpin, thereby promoting dissociation of ZPI-factor Xa complex from protein Z, and helping protein Z, which is usually present in limiting amounts relative to ZPI [135, 139, 140], to function as a catalyst. It was later found that Lys239 of ZPI promotes such dissociation by increasing the rate of protein Z dissociation, possibly explaining the conservation of this residue in a diverse range of vertebrates [140].
Modeling of the PZ/ZPI/factor Xa Michaelis docking complex, based on the x-ray structures of PZ with ZPI [133, 138], together with demonstration that the Gla domains of PZ and factor Xa interact at the membrane surface [137], and that Arg143 of factor Xa is critical for binding to ZPI [136], gave a structure depicted in Fig 11A. Such a model nicely explains the requirements for the cofactors in tailoring the specificity of ZPI and resolves the conundrum of the P1 Tyr for inhibition of an arginine-specific proteinase. The presence of P1 Tyr ensures that, in the absence of cofactors, ZPI reacts extremely slowly with factor Xa or similar arginine-specific proteinases. Only when cofactors are present is the rate of reaction greatly increased, but in a way that is specific for membrane-associated factor Xa. The high affinity complex of PZ with ZPI localizes the ZPI close to the membrane surface. Interaction between the Gla domains of PZ and factor Xa, together with a salt bridge between Glu313 of ZPI and Arg143 of factor Xa “presents” the ZPI P1 Tyr to the factor Xa active site and thereby overcomes the unfavorability of tyrosine at this position (Fig 11B). It is also possible that other interactions between the RCL of ZPI and subsites on factor Xa help to partially offset the unfavorableness of the P1 tyrosine, although there is as yet no evidence for this. This may explain why, when P1 is mutated to arginine, the SI increases so much; namely that the Michaelis complex affinity becomes so tight as to slow down RCL loop insertion during complex formation or else that deacylation becomes more favorable. Either or both of these possibilities would then cause most of the ZPI to be cleaved unproductively. An analogous use of an unfavorable P1 residue that requires compensation by other specific interactions to correctly present the P1 to the proteinase active site and so achieve specificity for only a single proteinase is heparin cofactor II (HCII) [141–143]. This has a P1 leucine [144] and so reacts very slowly with arginine-specific proteinases. However, when heparin or heparan sulfate binds to HCII, the heparin chain and the displaced acidic tail of HCII serve to bridge the serpin and the specific target proteinase thrombin and give greatly accelerated rates of inhibition, but only for this proteinase.
Fig 11. Modeled ternary complex of ZPI, protein Z and membrane-associated factor Xa.

Panel A, model of the complex at the membrane surface, with interaction between the Gla domains of factor Xa and protein Z at the membrane surface, tight interaction between ZPI and the EGF2 and pseudoproteinase domains of protein Z, and orientation of factor Xa such that the P1 Tyr of ZPI aligns with the active site of the proteinase, while Arg 143 of the proteinase interacts with Glu 313 of ZPI. This is based on the x-ray structure of ZPI and protein Z [133] and the demonstrated importance of Gla-Gla interactions [137], of Arg 143 of factor Xa [136] and of Glu 313 of ZPI [133]. Panel B shows an expansion of the modeled active site-ZPI RCL interactions, showing the role of Glu383 and Glu 313 of ZPI and of Arg143 of factor Xa in correctly positioning the P1 residue in the proteinase active site. From ref [133]. Copyright ASBMB.
A major unanswered question is what role the highly acidic N-terminal tail of ZPI serves. Residues in this region are not visible in either x-ray structure, suggesting that the tail is largely unstructured and mobile. Although no published study has examined possible roles for this extension, there are unpublished data (X. Huang and S. Olson) that show that the tail does not affect the in vitro interaction with cofactors or efficiency of factor Xa inhibition. A possible role in vivo thus may be to promote the inhibition of factor Xa when it is membrane associated and in complex with its cofactor, factor Va in the prothrombinase complex.
Finally, although heparin does not seem to be involved in the inhibition of factor Xa by the PZ/ZPI complex, it does bind to ZPI and enhances the rate of inactivation of both factor Xa, independently of PZ binding, and of factor XIa [145]. This heparin interaction may thus be important as an adjunct to the heparin:antithrombin interaction to help inhibit free factor Xa, as well as factor XIa.
6. Serpin-Receptor binding
It has long been known that plasma serpins, particularly when in complex with proteinases, are rapidly cleared from circulation [146, 147]. This led to the search for a “serpin-enzyme complex” or SEC receptor. After a number of false starts, such a receptor was identified as being the same as the α2-macroglobulin receptor, which is a high molecular weight member of the LDL receptor family of mosaic protein receptors named LRP1, for low density lipoprotein receptor-related protein 1 [148, 149]. LRP1 is a multifunctional scavenging and signaling receptor that binds and clears not only certain serpin-proteinase complexes, but many other structurally unrelated proteins [150]. Understanding how LRP1 (and related family members) can bind such a large repertoire of different proteins has thus been of great interest, not only for those working with serpins, but more generally as a ligand specificity question. Despite this interest, there is relatively little hard structural information on any such receptor-protein ligand complexes, and none until recently for interactions of LRP1 with any serpin or its proteinase complexes.
6.i PAI-1 binding to LRP1
The serpins known to bind to LRP1 are α1PI [151, 152], neuroserpin [153], C1 inhibitor [154], protein C inhibitor [155], antithrombin [152, 155], heparin cofactor II [152, 155], proteinase nexin 1[155] and PAI-1[155, 156]. However, only proteinase complexes of protein C inhibitor, proteinase nexin 1 and PAI-1 bind with high affinity in the absence of any other cofactors [155]. Of these three serpin-proteinase complexes, those of PAI-1 binding to LRP1 have been most extensively studied. This is because of the importance of PAI-1 in regulating uPA activity, which in turn is a major player in generating the proteinase plasmin. Through the ability of plasmin to not only dissolve blood clots, but also to cleave matrix proteins such as laminin and fibronectin, it is involved in many normal and pathological processes, such as wound healing and angiogenesis, as well as cell migration and the growth and dissemination of tumors.
It was originally thought that only PAI-1 in complex with proteinase could bind tightly to LRP1, and that this resulted from a conformational change in the serpin that resulted in exposure of a cryptic epitope [157, 158]. However, the elucidation of structures of native, cleaved and complexed serpins, including of native and cleaved PAI-1, showed that the structure of the serpin moiety in the covalent acylenzyme proteinase complex is almost identical to that of cleaved serpin, thus making it unlikely that complex formation exposes a novel high affinity epitope for LRP1. More recently, we have shown using fragments of LRP1 that comprise the principal binding region for protein ligands, that native and cleaved forms of PAI-1 can bind to these fragments with indistinguishable very high affinity (Kd < 3 nM), and that complex formation, whether to trypsin or low molecular weight uPA results in only a small increase in affinity [159]. Importantly, the strength of this interaction recapitulates the affinity for intact LRP1, and so is likely to represent the full binding site. In addition, binding of LRP1 fragments to PAI-1 does not seem to cause any significant change in conformation of PAI-1. It thus appeared that there must be a unique high affinity binding site on PAI-1 that is present in all of its conformations and that is used for binding of PAI-1 and its proteinase complexes to LRP1. This binding site has now been identified by solution phase binding measurements on wild type and mutant PAI-1 species in which putative basic binding residues had been mutated to alanine, either singly or in pairs [160].
Arginine and lysine residues were chosen for mutation, based on previous findings that the complement-like domains (CR domains), that are linked in clusters in LRP1 and related LDL receptor family members and that represent the ligand binding regions, engage basic residues on the target proteins [161–167]. It was found that four basic residues (Lys69, Arg76, Lys80 and Lys88) form the binding site for three adjacent CR domains and that just the interactions of these residues are sufficient to account quantitatively for the binding interaction (Fig 12). This is in contrast to earlier studies where mutation of some of the same individual residues resulted in no more than 11 fold increase in Kd and mostly much less [157, 158, 161, 168]. Although several overlapping three-domain fragments of LRP1 all bind with high affinity to PAI-1, the fragment CR4-CR5-CR6 binds the tightest, with Kd of 1.5 nM estimated at physiological ionic strength [160]. This is a 20-fold lower Kd than for CR5-CR6-CR7. In the complex between PAI-1 and CR4-CR5-CR6, CR4 binds to Lys88 and contributes least to affinity, Arg76 together with Lys80 bind to CR5 and contribute the most, while CR6 binds strongly to Lys69 (Fig 12). As expected, the positions of the four residues do not change significantly between native, latent and cleaved conformations and are not expected to change in complex with uPA or other proteinase. In addition, their locations are all well removed from the expected position of the proteinase in the covalent complex. The absence of a need for an additional binding contribution from the proteinase is consistent with reports that the binding site on uPA for LRP1 is in the N-terminus and so is unlikely to be available when uPA is complexed through this N-terminal region to its cell-surface receptor uPAR, which is the form that is most likely inhibited in vivo by PAI-1[169–172]. The proteinase is therefore unlikely to either augment binding affinity further by altering the relative positions of the four basic residues, or to contribute directly to binding through engaging a fourth CR domain. However, the fact that the uPA encountered by PAI-1 is membrane-associated, through being in complex with its membrane-anchored receptor uPAR, will result in the uPA/PAI-1 complex being localized to the membrane surface, where it can much more readily “find” membrane-anchored LRP1 than can uncomplexed solution-phase PAI-1, and so account for an effectively higher affinity interaction than in solution.
Fig 12. The binding site for LRP1 on PAI-1.

Residues Lys 88, Arg 76, Lys 80, and Lys 69 form the very high affinity (~2 nM) binding site for fragment CR4-CR5-CR6 from a principal ligand binding region of the receptor LRP1, cluster II [160]. Arg76 and Lys 80 engage CR5, and contribute over half of the binding energy. Lys 69 engages CR6 and contributes a major portion of the binding energy, while CR4 engages Lys 88 and contributes modestly to the overall affinity. These critical basic residues are shown in the structure of PAI-1 (green ribbon) complexed with the somatomedin domain (red space filling) of vitronectin (pdb code 1OC0 [173]) to illustrate the close proximity, and likely overlap, of the binding sites on PAI-1 for LRP1 and vitronectin. Thus, the separation between Arg 76 and the closest point of the somatomedin domain is only ~15Å, while CR domains, which would be roughly centered on each set of basic contact residues, are about 25Å across [162, 163, 182–184].
It is noteworthy that the location of the binding site on PAI-1 for LRP1 is very close to the binding site for vitronectin, which occurs through vitronectin's somatomedin B domain (Fig 12) [173]. The large size of full-length vitronectin and the proximity of the vitronectin and LRP1 binding sites may thus explain the antagonism shown by vitronectin to LRP1-mediated signalling by PAI-1 [174]. A final point of interest from the study is that the closely-related serpin proteinase nexin 1 (serpinE2), whose complex with uPA also binds very tightly to LRP1, has highly conserved basic residues at structurally homologous sites to the four basic residues, Lys69, Arg76, Lys80 and Lys88, identified as the binding site for LRP1 on PAI-1. These homologous sites may therefore represent the high affinity binding site for LRP1 in proteinase nexin 1 [160].
Footnotes
α1-Proteinase inhibitor was originally given the name α1-antitrypsin, even though its principal proteinase target in vivo is neutrophil elastase. This alternative name is still frequently used. The systematic name is serpinA1 for the protein and SERPINA1 for the gene (see Table 1).
Abbreviations used: α1PI, α1-proteinase inhibitor; s5A, etc, nomenclature designating secondary structure elements in serpins, here strand 5 of β-sheet A; CR, complement-like repeat; LRP1, low density lipoprotein receptor-like protein 1; PAI-1, plasminogen activator inhibitor 1; P1, P14, etc, nomenclature of Schechter and Berger [175] to designate proteinase substrate subsites. Using this nomenclature, the scissile bond is P1-P1', with P1' being C-terminal to P1; SI, stoichiometry of inhibition; uPA, urokinase-type plasminogen activator; uPAR, uPA receptor; ZPI, protein Z-dependent proteinase inhibitor.
References
- 1.Hunt LT, Dayhoff MO. A surprising new protein superfamily containing ovalbumin, antithrombin III and α1-proteinase inhibitor. Biochem Biophys Res Commun. 1980;95:864–871. doi: 10.1016/0006-291x(80)90867-0. [DOI] [PubMed] [Google Scholar]
- 2.Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 2000;10:1845–1864. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- 3.Roberts TH, Hejgaard J. Serpins in plants and green algae. Functional and Integrative Genomics. 2008;8:1–27. doi: 10.1007/s10142-007-0059-2. [DOI] [PubMed] [Google Scholar]
- 4.Turner PC, Musy PY, Moyer RW. Poxvirus serpins. In: McFadden G, editor. Viroceptors, virokines and related immune modulators encoded by DNA viruses. R.G. Landes Co; Austin: 1994. pp. 67–88. [Google Scholar]
- 5.Irving JA, Steenbakkers PJM, Lesk AM, Op den Camp HJM, Pike RN, Whisstock JC. Serpins in prokaryotes. Mol Biol Evol. 2002;19:1881–1890. doi: 10.1093/oxfordjournals.molbev.a004012. [DOI] [PubMed] [Google Scholar]
- 6.Roberts TH, Hejgaard J, Saunders NF, Cavicchioli R, Curmi PM. Serpins in unicellular Eukarya, Archaea, and Bacteria: sequence analysis and evolution. J Mol Evol. 2004;59:437–447. doi: 10.1007/s00239-004-2635-6. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz WH, Zverlov VV. Protease inhibitors in bacteria: an emerging concept for the regulation of bacterial protein complexes. Mol Microbiol. 2006;60:1323–1326. doi: 10.1111/j.1365-2958.2006.05181.x. [DOI] [PubMed] [Google Scholar]
- 8.Carrell RW, Travis J. α1-Antitrypsin and the serpins. Variation and countervariation. Trends in Biological Sciences. 1985;10:20–24. [Google Scholar]
- 9.Dunstone MA, Whisstock JC. Crytallography of serpins and serpin complexes. Methods Enzymol. 2011;501:63–87. doi: 10.1016/B978-0-12-385950-1.00005-5. [DOI] [PubMed] [Google Scholar]
- 10.Heit C, Jackson BC, McAndrews M, Wright MW, Thompson DC, Silverman GA, Nebert DW, Vasiliou V. Update of the human and mouse SERPIN gene superfamily. Human Genomics. 2013;7:22. doi: 10.1186/1479-7364-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bock SC, Skriver K, Nielsen E, Thøgersen HC, Wiman B, Donaldson VH, Eddy RL, Marrinan J, Radziejewska E, Huber R, Shows TB, Magnusson S. Human C1 inhibitor: Primary structure, cDNA cloning, and chromosomal localization. Biochemistry. 1986;25:4292–4301. doi: 10.1021/bi00363a018. [DOI] [PubMed] [Google Scholar]
- 12.Perkins SJ, Smith KF, Amatayakul S, Ashford D, Rademacher TW, Dwek RA, Lachmann PJ, Harrison RA. Two-domain structure of the native and reactive center cleaved forms of C1 inhibitor of human complement by neutron scattering. J Mol Biol. 1990;214:751–763. doi: 10.1016/0022-2836(90)90290-3. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence DA, Olson ST, Palaniappan S, Ginsburg D. Engineering plasminogen activator inhibitor 1 mutants with increased functional stability. Biochemistry. 1994;33:3643–3648. doi: 10.1021/bi00178a022. [DOI] [PubMed] [Google Scholar]
- 14.Kaslik G, Kardos J, Szabó L, Závodszky P, Westler WM, Markley JL, Gráf L. Effects of serpin binding on the target proteinase: Global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry. 1997;36:5455–5474. doi: 10.1021/bi962931m. [DOI] [PubMed] [Google Scholar]
- 15.Im H, Ahn HY, Yu MH. Bypassing the kinetic trap of serpin protein folding by loop extension. Protein Sci. 2000;9:1497–1502. doi: 10.1110/ps.9.8.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu MH, Lee KN, Kim J. The Z type variation of human α1-antitrypsin causes a protein folding defect. Nat Struct Biol. 1995;2:363–367. doi: 10.1038/nsb0595-363. [DOI] [PubMed] [Google Scholar]
- 17.Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z α1-antitrypsin accumulation in the liver. Nature (London) 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- 18.Elliott PR, Bilton D, Lomas DA. Lung polymers in Z α1-antitrypsin deficiency-related emphysema. Am J Respir Cell Mol Biol. 1998;18:670–674. doi: 10.1165/ajrcmb.18.5.3065. [DOI] [PubMed] [Google Scholar]
- 19.Mahadeva R, Lomas DA. Alpha1-antitrypsin deficiency, cirrhosis and emphysema. Thorax. 1998;53:501–505. doi: 10.1136/thx.53.6.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matheson NR, van Halbeek H, Travis J. Evidence for a tetrahedral intermediate complex during serpin-proteinase interactions. J Biol Chem. 1991;266:13489–13491. [PubMed] [Google Scholar]
- 21.Olson ST. Heparin and ionic strength-dependent conversion of antithrombin III from an inhibitor to a substrate of α-thrombin. J Biol Chem. 1985;260:10153–10160. [PubMed] [Google Scholar]
- 22.Patston PA, Gettins P, Beechem J, Schapira M. Mechanism of serpin action: Evidence that C1 inhibitor functions as a suicide substrate. Biochemistry. 1991;30:8876–8882. doi: 10.1021/bi00100a022. [DOI] [PubMed] [Google Scholar]
- 23.Hood DB, Huntington JA, Gettins PGW. α1-Proteinase inhibitor variant T345R. Influence of P14 residue on substrate and inhibitory pathways. Biochemistry. 1994;33:8538–8547. doi: 10.1021/bi00194a020. [DOI] [PubMed] [Google Scholar]
- 24.Wright HT, Scarsdale JN. Structural basis for serpin inhibitor activity. Proteins. 1995;22:210–225. doi: 10.1002/prot.340220303. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence DA, Ginsburg D, Day DE, Berkenpas MB, Verhamme IM, Kvassman JO, Shore JD. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J Biol Chem. 1995;270:25309–25312. doi: 10.1074/jbc.270.43.25309. [DOI] [PubMed] [Google Scholar]
- 26.Stratikos E, Gettins PGW. Major proteinase movement upon covalent serpin-proteinase complex formation. Proc Natl Acad Sci U S A. 1997;94:453–458. doi: 10.1073/pnas.94.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratikos E, Gettins PGW. Formation of the covalent serpin-proteinase complex involves translocation of the proteinase by more than 70Å and full insertion of the reactive center loop into β-sheet A. Proc Natl Acad Sci U S A. 1999;96:4808–4813. doi: 10.1073/pnas.96.9.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterson FC, Gettins PGW. Insight into the mechanism of serpin-proteinase inhibition from 2D [1H-15N] NMR studies of the 69kDa α1-proteinase inhibitor Pittsburgh-trypsin covalent complex. Biochemistry. 2001;40:6284–6292. doi: 10.1021/bi010100x. [DOI] [PubMed] [Google Scholar]
- 29.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature (London) 2000;407:923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 30.Backovic M, Stratikos E, Lawrence DA, Gettins PGW. Structural similarity of the covalent complexes formed between the serpin plasminogen activator inhibitor-1 and the arginine-specific proteinases trypsin, LMW urokinase, HMW urokinase and tPA: Use of site-specific fluorescent probes of local environment. Protein Sci. 2002;11:1182–1191. doi: 10.1110/ps.4320102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dementiev A, Dobo J, Gettins PGW. Active site distortion is sufficient for proteinase inhibition by serpins. Structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J Biol Chem. 2006;281:3452–3457. doi: 10.1074/jbc.M510564200. [DOI] [PubMed] [Google Scholar]
- 32.Swanson R, Raghavendra MP, Zhang W, Froelich C, Gettins PGW, Olson ST. Serine and cysteine proteases are translocated to similar extents upon formation of covalent complexes with serpins. Fluorescence perturbation and fluorescence resonance energy transfer mapping of the protease binding site in CrmA complexes with granzyme B and caspase-1. J Biol Chem. 2007;282:2305–2313. doi: 10.1074/jbc.M609546200. [DOI] [PubMed] [Google Scholar]
- 33.Dobo J, Swanson R, Salvesen GS, Olson ST, Gettins PGW. Cytokine Response Modifier A Inhibition of Initiator Caspases Results in Covalent Complex Formation and Dissociation of the Caspase Tetramer. J Biol Chem. 2006;281:38781–38790. doi: 10.1074/jbc.M605151200. [DOI] [PubMed] [Google Scholar]
- 34.Boatright KM, Renatus M, Scott F, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 35.Gettins PGW. The F-helix of serpins plays an essential, active role in the proteinase inhibition mechanism. FEBS Lett. 2002;523:2–6. doi: 10.1016/s0014-5793(02)02924-1. [DOI] [PubMed] [Google Scholar]
- 36.Maddur AA, Swanson R, Izaguirre G, Gettins PGW, Olson ST. Kinetic intermediates en route to the final serpin-protease complex. Studes of complexes of alpha-1-protease inhibitor with trypsin. J Biol Chem. 2013;288:32020–32035. doi: 10.1074/jbc.M113.510990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou A, Carrell RW, Huntington JA. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J Biol Chem. 2001;276:27541–27547. doi: 10.1074/jbc.M102594200. [DOI] [PubMed] [Google Scholar]
- 38.Tesch LD, Raghavendra MP, Bedsted-Faarvang T, Gettins PGW, Olson ST. Specificity and reactive loop length requirements for CrmA inhibition of serine proteases. Protein Sci. 2005;14:533–542. doi: 10.1110/ps.041104905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huntington JA, Fan B, Karlsson KE, Deinum J, Lawrence DA, Gettins PGW. Serpin conformational change in ovalbumin. Enhanced reactive center loop insertion through hinge region mutations. Biochemistry. 1997;36:5432–5440. doi: 10.1021/bi9702142. [DOI] [PubMed] [Google Scholar]
- 40.Yamasaki M, Arii Y, Mikami B, Hirose M. Loop-inserted and thermostabilized structure of P1-P1' cleaved ovalbumin mutant R339T. J Mol Biol. 2002;315:113–120. doi: 10.1006/jmbi.2001.5056. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence DA, Olson ST, Muhammad S, Day DE, Kvassman JO, Ginsburg D, Shore JD. Partitioning of serpin-proteinase reactions between stable inhibition and substrate cleavage is regulated by the rate of serpin reactive center loop insertion into β-sheet A*. J Biol Chem. 2000;275:5839–5844. doi: 10.1074/jbc.275.8.5839. [DOI] [PubMed] [Google Scholar]
- 42.Gettins PGW. Serpin structure, mechanism and function. Chem Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 43.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 44.Hekman CM, Loskutoff DJ. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J Biol Chem. 1985;260:11581–11587. [PubMed] [Google Scholar]
- 45.Onda M, Nakatani K, Takahara S, Nishiyama M, Takahashi N, Hirose M. Cleaved serpin refolds into the relaxed state via a stressed conformer. J Biol Chem. 2008;283:17568–17578. doi: 10.1074/jbc.M709262200. [DOI] [PubMed] [Google Scholar]
- 46.Stein PE, Leslie AGW, Finch JT, Carrell RW. Crystal structure of uncleaved ovalbumin at 1.95Å resolution. J Mol Biol. 1991;221:941–959. doi: 10.1016/0022-2836(91)80185-w. [DOI] [PubMed] [Google Scholar]
- 47.Wright HT, Qian HX, Huber R. Crystal structure of plakalbumin, a proteolytically nicked form of ovalbumin. Its relationship to the structure of cleaved α-1-proteinase inhibitor. J Mol Biol. 1990;213:513–528. doi: 10.1016/s0022-2836(05)80212-8. [DOI] [PubMed] [Google Scholar]
- 48.Arii Y, Hirose M. Probing the serpin structural-transition mechanism in ovalbumi mutant R339T by proteolytic-cleavage kinetics of the reactive centre loop. Biochem J. 2002;363:403–409. doi: 10.1042/0264-6021:3630403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dolmer K, Gettins PGW. How the serpin α1 proteinase inhibitor folds. J Biol Chem. 2012;287:12425–12432. doi: 10.1074/jbc.M111.315465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsutsui Y, Dela Cruz R, Wintrode PL. Folding mechanism of the metastable serpin α1-antitrypsin. Proc Natl Acad Sci U S A. 2012;109:4467–4472. doi: 10.1073/pnas.1109125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gooptu B, Chang WSW, Dafforn TR, Carrell RW, Read RJ. Inactive conformation of the serpin α1-antichymotrypsin indicates two-stage insertion of the reactive center loop: implications for inhibitroy function and conformational disease. Proc Natl Acad Sci U S A. 2000;97:67–72. doi: 10.1073/pnas.97.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearce MC, Powers GA, Feil SC, Hansen G, Parker MW, Bottomley SP. Identification and characterization of a misfolded serpin formed at physiological temperature. J Mol Biol. 2010:493. doi: 10.1016/j.jmb.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Lee C, Seo EJ, Yu MH. Role of the connectivity of secondary structure segments in the folding of α1-antitrypsin. Biochem Biophys Res Commun. 2001;287:636–641. doi: 10.1006/bbrc.2001.5638. [DOI] [PubMed] [Google Scholar]
- 54.Im H, Woo MS, Hwang KY, Yu MH. Interactions causing the kinetic trap in serpin protein folding. J Biol Chem. 2002;277:46347–46354. doi: 10.1074/jbc.M207682200. [DOI] [PubMed] [Google Scholar]
- 55.Carrell RW. α1-Antitrypsin: Molecular pathology, leukocytes, and tissue damage. J Clin Invest. 1986;78:1427–1431. doi: 10.1172/JCI112731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis RL, Shrimpton AE, Holohan PD, Bradshaw C, Feiglin D, Collins GH, Sonderegger P, Kinter J, Becker LM, Lacbawan F, Krasnewich D, Muenke M, Lawrence DA, Yerby MS, Shaw CM, Gooptu B, Elliott PR, Finch JT, Carrell RW, Lomas DA. Familial dementia caused by polymerization of mutant neuroserpin. Nature (London) 1999;401:376–379. doi: 10.1038/43894. [DOI] [PubMed] [Google Scholar]
- 57.Miranda E, Römisch K, Lomas DA. Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J Biol Chem. 2004;279:28283–28291. doi: 10.1074/jbc.M313166200. [DOI] [PubMed] [Google Scholar]
- 58.Elliott PR, Lomas DA, Carrell RW, Abrahams JP. Inhibitory conformation of the reactive loop of α1-antitrypsin. Nat Struct Biol. 1996;3:676–681. doi: 10.1038/nsb0896-676. [DOI] [PubMed] [Google Scholar]
- 59.Behrens MA, Sendall TJ, Pedersen JS, Kjeldgaard M, Huntington JA, Jensen JK. The shapes of Z-α1-antitrypsin polymers in solution support the C-terminal domain-swap mechanism of polymerization. Biophys J. 2014;107:1905–1912. doi: 10.1016/j.bpj.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ricagno S, Caccia S, Sorrentino G, Antonini G, Bolognesi M. Human neuroserpin: structure and time-dependent inhibition. J Mol Biol. 2009;388:109–121. doi: 10.1016/j.jmb.2009.02.056. [DOI] [PubMed] [Google Scholar]
- 61.Kim J, Lee KN, Yi GS, Yu MH. A thermostable mutation located at the hydrophobic core of α1-antitrypsin suppresses the folding defect of the Z-type variant. J Biol Chem. 1995;270:8597–8601. doi: 10.1074/jbc.270.15.8597. [DOI] [PubMed] [Google Scholar]
- 62.Tan L, Dickens JA, Demeo DL, Miranda E, Perez J, Rashid ST, Day J, Ordoñez A, Marciniak SJ, Haq I, Barker AF, Campbell EJ, Eden E, McElvaney NG, Rennard SI, Sandhaus RA, Stocks JM, Stoller JK, Strange C, Turino G, Rouhani FN, Brantly M, Lomas DA. Circulating polymers in α1-antitrypsin deficiency. Eur Respir J. 2014;43:1501–1504. doi: 10.1183/09031936.00111213. [DOI] [PubMed] [Google Scholar]
- 63.Mahadeva R, Atkinson C, Li Z, Stewart S, Janciauskiene S, Kelley DG, Parmar J, Pitman R, Shapiro SD, Lomas DA. Polymers of Z α1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. 2005;166:377–386. doi: 10.1016/s0002-9440(10)62261-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van't Wout EFA, van Schadewijk A, Lomas DA, Stolk J, Marciniak SJ, Hiemstra PS. Function of monocytes and monocyte-derived macrophages in α1-antitrypsin deficiency. Eur Respir J. 2015;45:365–376. doi: 10.1183/09031936.00046114. [DOI] [PubMed] [Google Scholar]
- 65.Yamasaki M, Li W, Johnson DJD, Huntington JA. Crystal stucture of a stable dimer reveals the molecular basis of serpin polymerization. Nature. 2008;455:1255–1259. doi: 10.1038/nature07394. [DOI] [PubMed] [Google Scholar]
- 66.Knaupp AS, Bottomley SP. Structural change in β-sheet A of Z α1-antitrypsin is responsible for accelerated polymerization and disease. J Mol Biol. 2011;413:888–898. doi: 10.1016/j.jmb.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 67.Mahadeva R, Dafforn TR, Carrell RW, Lomas DA. 6-mer peptide selectively anneals to a pathogenic serpin conformation and blocks polymerization. Implications for the prevention of Z α1-antitrypsin-related cirrhosis. J Biol Chem. 2002;277:6771–6774. doi: 10.1074/jbc.C100722200. [DOI] [PubMed] [Google Scholar]
- 68.Krishnan B, Gierasch LM. Dynamic local unfolding in the serpin α-1 antitrypsin provides a mechanism for loop insertion and polymerization. Nature Structural and Molecular Biology. 2011;18:222–226. doi: 10.1038/nsmb.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamasaki M, Sendall TJ, Pearce MC, Whisstock JC, Huntington JA. Molecular basis of α1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO reports. 2011;12:1011–1107. doi: 10.1038/embor.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miranda E, Perez J, Ekeowa UI, Hadzic N, Kalsheker N, Gooptu B, Portmann B, Belorgey D, Hill M, Chambers S, Teckman J, Alexander GJ, Marciniak SJ, Lomas DA. A novel monoclonal antibody to characterize pathgenic polymers in liver disease associated with α1-antitrypsin deficiency. Hepatology. 2010;52:1078–1088. doi: 10.1002/hep.23760. [DOI] [PubMed] [Google Scholar]
- 71.Sivasothy P, Dafforn TR, Gettins PGW, Lomas DA. Pathogenic α1-antitrypsin polymers are formed by reactive loop-α-sheet A linkage. J Biol Chem. 2000:275. doi: 10.1074/jbc.M004054200. [DOI] [PubMed] [Google Scholar]
- 72.Ekeowa UI, Freeke J, Miranda E, Gooptu B, Bush MF, Perez J, Teckman J, Robinson CV, Lomas DA. Defining the mechanism of polymerization in the serpinopathies. Proc Natl Acad Sci U S A. 2010;107:17146–17151. doi: 10.1073/pnas.1004785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. α1-Antitrypsin Siiyama (Ser53ÆPhe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem. 1993;268:15333–15335. [PubMed] [Google Scholar]
- 74.Ordonez A, Perez J, Tan J, Dickens JA, Motamedi-Shad N, Irving JA, Haq I, Ekeowa UI, Marciniak SJ, Miranda E, Lomas DA. A single-chain variable fragment intrabody prevents intracellular polymerization of Z α1-antitrypsin while allowing its antiprotease activity. FASEB J. 2015;29:2667–2678. doi: 10.1096/fj.14-267351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kvassman JO, Verhamme I, Shore JD. Inhibitory mechanism of serpins: Loop insertion forces acylation of plasminogen activator by plasminogen activator inhibitor-1. Biochemistry. 1998;37:15491–15502. doi: 10.1021/bi9814787. [DOI] [PubMed] [Google Scholar]
- 76.Olson ST, Swanson R, Day D, Verhamme I, Kvassman J, Shore JD. Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor-1, with tissue plasminogen activator and trypsin. Biochemistry. 2001;40:11742–11756. doi: 10.1021/bi0107290. [DOI] [PubMed] [Google Scholar]
- 77.Izaguirre G, Swanson R, Raja SM, Rezaie AR, Olson ST. Mechanism by which exosites promote the inhibition of blood coagulation proteases by heparin-activated antithrombin. J Biol Chem. 2007;282:33609–33622. doi: 10.1074/jbc.M702462200. [DOI] [PubMed] [Google Scholar]
- 78.Blouse GE, Perron MJ, Kvassman J, Yunus S, Thompson JH, Betts RL, Lutter LC, Shore JD. Mutation of the conserved tryptophan in the serpin breach region alters the inhibitory mechanism of plasminogen acticvator inhibitor-1. Biochemistry. 2003;42:12260–12272. doi: 10.1021/bi034737n. [DOI] [PubMed] [Google Scholar]
- 79.Takahashi N, Terakado K, Nakamura G, Soekmadji C, masuoka T, Yamasakio M, Hirose M. Dynamic mechanism for the serpin loop insertion as revealed by quantitative kinetics. J Mol Biol. 2005;348:409–418. doi: 10.1016/j.jmb.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 80.Vleugels N, Gils A, Bijnens AP, Knockaert I, Declerck P. The importance of helix F in plasminogen activator inhibitor-1. Biochim Biophys Acta. 2000;1476:20–26. doi: 10.1016/s0167-4838(99)00224-1. [DOI] [PubMed] [Google Scholar]
- 81.Bijnens AP, Gils A, Knockaert I, Stassen JM, Declerck PJ. Importance of the hinge region between α-helix F and the main part of serpins, based upon identification of the epitope of plasminogen activator inhibitor type 1 neutralizing antibodies. J Biol Chem. 2000;275:6375–6380. doi: 10.1074/jbc.275.9.6375. [DOI] [PubMed] [Google Scholar]
- 82.Komissarov AA, Zhou A, Declerck PJ. Modulation of serpin reaction through stabilization of transient intermediate by ligands bound through α-helix F. J Biol Chem. 2007;282:26306–26315. doi: 10.1074/jbc.M702089200. [DOI] [PubMed] [Google Scholar]
- 83.Tsutsui Y, Liu L, Gershenson A, Wintrode PL. The conformational dynamics of a metastable serpin studied by hydrogen exchange and mass spectrometry. Biochemistry. 2006;45:6561–6569. doi: 10.1021/bi060431f. [DOI] [PubMed] [Google Scholar]
- 84.Cabrita LD, Whisstock JC, Bottomley SP. Probing the role of the F-helix in serpin stability through a single tryptophan substitution. Biochemistry. 2002;41:4575–4581. doi: 10.1021/bi0158932. [DOI] [PubMed] [Google Scholar]
- 85.Wind T, Jensen JK, Dupont DM, Kulig P, Andreasen PA. Mutational analysis of plasminogen activator inhibitor-1. Interactions of α-helix F and its neighbouring structural elements regulates the activity and the rate of latency transition. Eur J Biochem. 2003;270:1680–1688. doi: 10.1046/j.1432-1033.2003.03524.x. [DOI] [PubMed] [Google Scholar]
- 86.Lee C, Park SH, Lee MY, Yu MH. Regulation of protein function by native metastability. Proc Natl Acad Sci U S A. 2000;97:7727–7731. doi: 10.1073/pnas.97.14.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sengupta T, Tsutsui Y, Wintrode PL. Local and global effects of a cavity filling mutation in a metastable serpin. Biochemistry. 2009;48:8233–8240. doi: 10.1021/bi900342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tew DJ, Bottomley SP. Intrinsic fluorescence changes and rapid kinetics of proteinase deformation during serpin inhibition. FEBS Lett. 2001;494:30–33. doi: 10.1016/s0014-5793(01)02305-5. [DOI] [PubMed] [Google Scholar]
- 89.Perron MJ, Blouse GE, Shore JD. Distortion of the catalytic domain of tissue-type plasminogen activator by plasminogen activator inhibitor-1 coincides with the formation of stable serpin-proteinase complexes. J Biol Chem. 2003;278:48197–48203. doi: 10.1074/jbc.M306184200. [DOI] [PubMed] [Google Scholar]
- 90.Futamura A, Stratikos E, Olson ST, Gettins PGW. Change in environment of the P1 side chain upon progression from the Michaelis complex to the covalent serpin-proteinase complex. Biochemistry. 1998;37:13110–13119. doi: 10.1021/bi981234m. [DOI] [PubMed] [Google Scholar]
- 91.Bock PE, Olson ST, Björk I. Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite 1. J Biol Chem. 1997;272:19837–19845. doi: 10.1074/jbc.272.32.19837. [DOI] [PubMed] [Google Scholar]
- 92.Stavridi ES, O'Malley K, Lukacs CM, Moore WT, Lambris JD, Christianson DW, Rubin H, Cooperman BS. Structural change in α-chymotrypsin induced by complexation with α1-antichymotrypsin as seen by enhanced sensitivity to proteolysis. Biochemistry. 1996;35:10608–10615. doi: 10.1021/bi9605806. [DOI] [PubMed] [Google Scholar]
- 93.Calugaru SV, Swanson R, Olson ST. The pH dependence of serpin-proteinase complex dissociation reveals a mechanism of complex stabilization involving inactive and active conformational states of the proteinase which are perturbable by calcium. J Biol Chem. 2001;276:32446–32455. doi: 10.1074/jbc.M104731200. [DOI] [PubMed] [Google Scholar]
- 94.Van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997;34:188–204. [PubMed] [Google Scholar]
- 95.Ishiguro K, Kojima T, Kadomatsu K, Nakayama Y, Takagi A, Suzuki M, Takeda N, Ito M, Yamamoto K, Matsushita T, Kusugami K, Muramatsu T, Saito H. Complete antithrombin deficiency in mice results in embryonic lethality. J Clin Invest. 2000;106:873–878. doi: 10.1172/JCI10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Y, Kretz CA, Maeder ML, Richter CE, Tsao P, Vo AH, Huarng MC, Rode T, Hu Z, Mehra R, Olson ST, Joung JK, Shavit JA. Targeted mutagenesis of zebrafish antithrombin III triggers disseminated intravascular coagulation anf thrombosis, revealing insight into function. Blood. 2014;124:142–150. doi: 10.1182/blood-2014-03-561027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Agostini A, Watkins SC, Slayter HS, Youssoufian H, Rosenberg RD. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: Antithrombin binding on cultured endothelial cells and perfused rat aorta. J Cell Biol. 1990;111:1293–1304. doi: 10.1083/jcb.111.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Hérault JM, Björk I. Accelerating ability of synthetic oligosacccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Thromb Haemost. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 99.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J Biol Chem. 1992;267:12528–12538. [PubMed] [Google Scholar]
- 100.Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci U S A. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petitou M, Casu B, Lindahl U. 1976–1983, a critical preiod in the history of heparin: the discovery of the antithrombin binding site. Biochimie. 2003;85:83–89. doi: 10.1016/s0300-9084(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 102.Bedsted T, Swanson R, Chuang YJ, Bock PE, Björk I, Olson ST. Heparin and calcium ions dramatically enhance antithrombin reactvity with factor IXa by generating new interaction exosites. Biochemistry. 2003;42:8143–8152. doi: 10.1021/bi034363y. [DOI] [PubMed] [Google Scholar]
- 103.Olson ST, Björk I. Predominant contribution of surface approximation to the mechanism of heparin acceleration of the antithrombin-thrombin reaction. Elucidation from salt concentration effects. J Biol Chem. 1991;266:6353–6364. [PubMed] [Google Scholar]
- 104.Rezaie AR. Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism. Evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J Biol Chem. 1998;273:16824–16827. doi: 10.1074/jbc.273.27.16824. [DOI] [PubMed] [Google Scholar]
- 105.Desai UR, Petitou M, Björk I, Olson ST. Mechanism of heparin activation of antithrombin. Role of individual residues of the pentasaccharide in the recognition of native and activated states of antithrombin. J Biol Chem. 1998;273:7478–7487. doi: 10.1074/jbc.273.13.7478. [DOI] [PubMed] [Google Scholar]
- 106.Desai UR, Petitou M, Björk I, Olson ST. Mechanism of heparin activation of antithrombin: Evidence for an induced-fit model of allosteric activation involving two interaction subsites. Biochemistry. 1998;37:13033–13041. doi: 10.1021/bi981426h. [DOI] [PubMed] [Google Scholar]
- 107.Gettins PGW, Fan B, Crews BC, Turko IV, Olson ST, Streusand VJ. Transmission of conformational change from the heparin binding site to the reactive center of antithrombin. Biochemistry. 1993;32:8385–8389. doi: 10.1021/bi00084a001. [DOI] [PubMed] [Google Scholar]
- 108.Pike RN, Potempa J, Skinner R, Fitton HL, McGraw WT, Travis J, Owen M, Jin L, Carrell RW. Heparin-dependent modification of the reactive center arginine of antithrombin and consequent increase in heparin binding affinity. J Biol Chem. 1997;272:19652–19655. doi: 10.1074/jbc.272.32.19652. [DOI] [PubMed] [Google Scholar]
- 109.Chuang YJ, Swanson R, Raja SM, Olson ST. Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence. Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin. J Biol Chem. 2001;276:14961–14971. doi: 10.1074/jbc.M011550200. [DOI] [PubMed] [Google Scholar]
- 110.Manithody C, Yang L, Rezaie AR. Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry. 2002;41:6780–6788. doi: 10.1021/bi0255367. [DOI] [PubMed] [Google Scholar]
- 111.Izaguirre G, Zhang W, Swanson R, Bedsted T, Olson ST. Localization of an antithrombin exosite that promotes rapid inhibition of factors Xa and IXa on heparin activation of the serpin. J Biol Chem. 2003;279:51433–51440. doi: 10.1074/jbc.M309266200. [DOI] [PubMed] [Google Scholar]
- 112.Izaguirre G, Olson ST. Residues Tyr253 and Glu255 in strand 3 of β-sheet C of antithrombin are key determinants of an exosite made accessible by heparin activation to promote rapid inhibition of factors Xa and IXa. J Biol Chem. 2006;281:13424–13432. doi: 10.1074/jbc.M600415200. [DOI] [PubMed] [Google Scholar]
- 113.Johnson DJD, Li W, Adams TE, Huntington JA. Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 2006;25:2029–2037. doi: 10.1038/sj.emboj.7601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Johnson DJD, Langdown J, Huntington JA. Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7Å structure of the ternary complex. Proc Natl Acad Sci U S A. 2010;107:645–650. doi: 10.1073/pnas.0910144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dementiev A, Petitou M, Herbert JM, Gettins PGW. The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nature Structural and Molecular Biology. 2004;11:863–867. doi: 10.1038/nsmb810. [DOI] [PubMed] [Google Scholar]
- 116.Li W, Johnson DJ, Esmon CT, Huntington JA. Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nature Structural and Molecular Biology. 2004;11:857–862. doi: 10.1038/nsmb811. [DOI] [PubMed] [Google Scholar]
- 117.Hopkins PCR, Pike RN, Stone SR. Evolution of serpin specificity: cooperative interactions in the reactive-site loop sequence of antithrombin specifically restrict the inhibition of activated protein C. J Mol Evol. 2000;51:507–515. doi: 10.1007/s002390010114. [DOI] [PubMed] [Google Scholar]
- 118.Gettins PGW, Olson ST. Activation of antithrombin as a factor IXa and Xa inhibitor involves mitigation of repression rather than positive enhancement. FEBS Lett. 2009;583:3397–3400. doi: 10.1016/j.febslet.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dementiev A, Swanson R, Roth R, Isetti G, Izahuirre G, Olson ST, Gettins PGW. The allosteric mechanism of activation of antithrombin as an inhibitor of factor IXa and Factor Xa. Heparin-independent full activatio though mutations adjacent to helix D. J Biol Chem. 2013;288:22611–33619. doi: 10.1074/jbc.M113.510727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meagher JL, Beechem JM, Olson ST, Gettins PGW. Deconvolution of the fluorescence emission spectrum of human antithrombin and identification of the tryptophan residues that are responsive to heparin binding. J Biol Chem. 1998;273:23283–23289. doi: 10.1074/jbc.273.36.23283. [DOI] [PubMed] [Google Scholar]
- 121.Meagher JL, Olson ST, Gettins PGW. Critical role of the linker region between helix D and strand 2A in heparin activation of antithrombin. J Biol Chem. 2000;275:2698–2704. doi: 10.1074/jbc.275.4.2698. [DOI] [PubMed] [Google Scholar]
- 122.Belzar KJ, Zhou A, Carrell RW, Gettins PGW, Huntington JA. Helix D elongation and allosteric activation of antithrombin. J Biol Chem. 2002;277:8551–8558. doi: 10.1074/jbc.M110807200. [DOI] [PubMed] [Google Scholar]
- 123.Izaguirre G, Aguila S, Qi L, Swanson R, Roth R, Rezaie AR, Gettins PGW, Olson ST. Conformational activation of antithrombin by heparin involves altered exosite interaction with protease. J Biol Chem. 2014;289:34049–34064. doi: 10.1074/jbc.M114.611707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roth R, Swanson R, Izaguirre G, Bock SC, Gettins PGW, Olson ST. Saturation mutagenesis of the antithrombin reactive center loop P14 residue supports a three-step mechanism of heparin allosteric activation involving intermediate and fully activated states. J Biol Chem. 2015;290:28020–28036. doi: 10.1074/jbc.M115.678839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huntington JA, Olson ST, Fan B, Gettins PGW. Mechanism of heparin activation of antithrombin. Evidence for reactive center loop pre-insertion with expulsion upon heparin binding. Biochemistry. 1996;35:8495–8503. doi: 10.1021/bi9604643. [DOI] [PubMed] [Google Scholar]
- 126.Huntington JA, Gettins PGW. Conformational conversion of antithrombin to a fully activated substrate of factor Xa without need for heparin. Biochemistry. 1998;37:3272–3277. doi: 10.1021/bi972182o. [DOI] [PubMed] [Google Scholar]
- 127.Futamura A, Gettins PGW. Serine 380 (P14) to glutamate mutation activates antithrombin as an inhibitor of factor Xa. J Biol Chem. 2000;275:4092–4098. doi: 10.1074/jbc.275.6.4092. [DOI] [PubMed] [Google Scholar]
- 128.Johnson DJD, Huntington JA. Crystal structure of antithrombin in a heparin-bound intermediate state. Biochemistry. 2003;42:8712–8719. doi: 10.1021/bi034524y. [DOI] [PubMed] [Google Scholar]
- 129.Langdown J, Belzar KJ, Savory WJ, Baglin TP, Huntington JA. The critical role of hinge region expulsion in the induced-fit heparin binding mechanism of antithrombin. J Mol Biol. 2009;386:1278–1289. doi: 10.1016/j.jmb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schedin-Weiss S, Richard B, Olson ST. Kinetic evidence that allosteric activation of antithrombin by heparin is mediated by two sequential conformational changes. Arch Biochem Biophys. 2010;504:169–176. doi: 10.1016/j.abb.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Han X, Fiehler R, Broze GJ., Jr Isolation of a protein Z-dependent plasma protease inhibitor. Proc Natl Acad Sci U S A. 1998;95:9250–9255. doi: 10.1073/pnas.95.16.9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Han X, Huang ZF, Fiehler R, Broze GJ., Jr The protein Z-dependent protease inhibitor is a serpin. Biochemistry. 1999;38:11073–11078. doi: 10.1021/bi990641a. [DOI] [PubMed] [Google Scholar]
- 133.Huang X, Dementiev A, Olson ST, Gettins PGW. Basis for the specificity and activation of the serpin protein Z-dependent proteinases inhibitor as an inhibitor of membrane-associated factor Xa. J Biol Chem. 2010:285. doi: 10.1074/jbc.M110.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Han X, Fiehler R, Broze GJ., Jr Characterization of the protein Z-dependent protease inhibitor. Blood. 2000;96:3049–3055. [PubMed] [Google Scholar]
- 135.Huang X, Swanson R, Broze GJ, Jr, Olson ST. Kinetic characterization of the protein Z-dependent protease inhibitor (ZPI) reaction with blood coagulation factor Xa. J Biol Chem. 2008:29770–29783. doi: 10.1074/jbc.M805214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rezaie AR, Manithody C, Yang L. Identification of factor Xa residues critical for interaction with protein Z-dependent protease inhibitor. Both active site and exosite interactions are required for inhibition. J Biol Chem. 2005;280:32722–32728. doi: 10.1074/jbc.M505517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rezaie AR, Bae JS, Manithody C, Qureshi SH, Yang L. Protein Z-dependent protease inhibitor binds to the C-terminal domain of protein Z. J Biol Chem. 2008;283:19922–19926. doi: 10.1074/jbc.M802639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wei Z, Yan Y, Carrell RW, Zhou A. Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood. 2009;114:366203667. doi: 10.1182/blood-2009-04-210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang X, Yan Y, Tu Y, Gatti J, Broze G, Jr, Zhou A, Olson ST. Structural basis for catalytic activation of protein Z-dependent protease inhibitor. Blood. 2012;120:1726–1733. doi: 10.1182/blood-2012-03-419598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang X, Zhou J, Zhou A, Olson ST. Thermodynamic and kinetic characterization of the protein Z-dependent protease inhibitor (ZPI)-protein Z interaction reveals an unexpected role for ZPI Lys-239. J Biol Chem. 2015;290:9906–9918. doi: 10.1074/jbc.M114.633479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Van Deerlin VMD, Tollefsen DM. The N-terminal acidic domain of heparin cofactor II mediates the inhibition of α-thrombin in the presence of glycosaminoglycans. J Biol Chem. 1991;266:20223–20231. [PubMed] [Google Scholar]
- 142.Baglin TP, Carrell RW, Church FC, Esmon CT, Huntington JA. Crystal structure of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc Natl Acad Sci U S A. 2002;99:11079–11084. doi: 10.1073/pnas.162232399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gettins PGW, Olson ST. Exosite determinants of serpin specificity. J Biol Chem. 2009;284:20441–20445. doi: 10.1074/jbc.R800064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Griffith MJ, Noyes CM, Tyndall JA, Church FC. Structural evidence for leucine at the reactive site of heparin cofactor II. Biochemistry. 1985;24:6777–6782. doi: 10.1021/bi00345a008. [DOI] [PubMed] [Google Scholar]
- 145.Huang X, Rezaie AR, Broze G, Jr, Olson ST. Heparin is a major activator of the anticoagulant serpin protein Z-dependent protease inhibitor. J Biol Chem. 2011;286:8740–8751. doi: 10.1074/jbc.M110.188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: Comparison of α1-proteinase inhibitor, α1-antichymotrypsin, antithrombin III, α2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry. 1991;30:1723–1730. doi: 10.1021/bi00220a039. [DOI] [PubMed] [Google Scholar]
- 147.Ohlsson K, Laurell CB. The disappearance of enzyme-inhibitor complexes from the circulation of man. Clin Sci Mol Med. 1976;51:87–92. doi: 10.1042/cs0510087. [DOI] [PubMed] [Google Scholar]
- 148.Orth K, Madison EL, Gething MJ, Sambrook JF, Herz J. Complexes of tissue-type plasminogen activator and its serpin inhibitor plasminogen activator inhibitor type 1 are internalized by means of the low density lipoprotein-related protein/α2-macroglobulin receptor. Proc Natl Acad Sci U S A. 1992;89:7422–7426. doi: 10.1073/pnas.89.16.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS. Sequence identity between the α2-macroglobulin receptor and low density lipoprotein receptor- related protein suggests that this molecule is a multifunctional receptor. J Biol Chem. 1990;265:17401–17404. [PubMed] [Google Scholar]
- 150.Strickland DK, Kounnas MZ, Argraves WS. LDL receptor-related protein: A multiligand receptor for lipoprotein and proteinase catabolism. FASEB J. 1995;9:890–898. doi: 10.1096/fasebj.9.10.7615159. [DOI] [PubMed] [Google Scholar]
- 151.Poller W, Willnow TE, Hilpert J, Herz J. Differential recognition of α1-antitrypsin- elastase and α1-antichymotrypsin-cathepsin G complexes by the low density lipoprotein receptor-related protein. J Biol Chem. 1995;270:2841–2845. doi: 10.1074/jbc.270.6.2841. [DOI] [PubMed] [Google Scholar]
- 152.Kounnas MZ, Church FC, Argraves WS, Strickland DK. Cellular internalization and degradation of antithrombin III-thrombin, heparin cofactor II-thrombin, and α1-antitrypsin-trypsin complexes is mediated by the low density lipoprotein receptor-related protein. J Biol Chem. 1996;271:6523–6529. doi: 10.1074/jbc.271.11.6523. [DOI] [PubMed] [Google Scholar]
- 153.Makarova A, Mikhailenko I, Bugge TH, List K, Lawrence DA, Strickland DK. The low density lipoprotein receptor-related protein modulates protease activity in the brain by mediating the cellular internalization of both neuroserpin and neuroserpin-tissue-type plasminogen activator complexes. J Biol Chem. 2003;278:50250–50258. doi: 10.1074/jbc.M309150200. [DOI] [PubMed] [Google Scholar]
- 154.Storm D, Herz J, Trinder P, Loos M. C1 Inhibitor-C(1)over-bar-s complexes are internalized and degraded by the low density lipoprotein receptor-related protein. J Biol Chem. 1997;272:31043–31050. doi: 10.1074/jbc.272.49.31043. [DOI] [PubMed] [Google Scholar]
- 155.Kasza A, Petersen HH, Heegaard CW, Oka K, Christensen A, Dubin A, Chan L, Andreasen PA. Specificity of serine proteinase serpin complex binding to very-low-density lipoprotein receptor and α2-macroglobulin receptor low-density-lipoprotein-receptor-related protein. Eur J Biochem. 1997;248:270–281. doi: 10.1111/j.1432-1033.1997.00270.x. [DOI] [PubMed] [Google Scholar]
- 156.Stefansson S, Lawrence DA, Argraves WS. Plasminogen activator inhibitor-1 and vitronectin promote the cellular clearance of thrombin by low density lipoprotein receptor-related proteins 1 and 2. J Biol Chem. 1996;271:8215–8220. doi: 10.1074/jbc.271.14.8215. [DOI] [PubMed] [Google Scholar]
- 157.Stefansson S, Muhammad S, Cheng XF, Battey FD, Strickland DK, Lawrence DA. Plasminogen activator inhibitor-1 contains a cryptic high affinity binding site for the low density lipoprotein receptor-related protein. J Biol Chem. 1998;273:6358–6366. doi: 10.1074/jbc.273.11.6358. [DOI] [PubMed] [Google Scholar]
- 158.Horn IR, Van den Berg BMM, Moestrup SK, Pannekoek H, van Zonneveld AJ. Plasminogen activator inhibitor 1 contains a cryptic high affinity receptor binding site that is exposed upon complex formation with tissue-type plasminogen activator. Thromb Haemost. 1998;80:822–828. [PubMed] [Google Scholar]
- 159.Jensen JK, Dolmer K, Gettins PGW. Specificity of binding of the low density lipoprotein receptor-related protein (LRP) to different conformational states of the clade E serpins PAI-1 and PN-1. J Biol Chem. 2009;284:17989–17997. doi: 10.1074/jbc.M109.009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gettins PGW, Dolmer K. The high affinity binding site on plasminogen activator inhibior-1 (PAI-1) for the low density lipoprotein receptor-related protein (LRP1) is composed of four basic residues. J Biol Chem. 2016;291:800–812. doi: 10.1074/jbc.M115.688820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rodenburg KW, Kjoller L, Petersen HH, Andreasen PA. Binding of urokinase-type plasminogen activator plasminogen activator inhibitor-1 complex to the endocytosis receptors α2-macroglobulin receptor low-density lipoprotein receptor-related protein and very-low-density lipoprotein receptor involves basic residues in the inhibitor. Biochem J. 1998;329:55–63. doi: 10.1042/bj3290055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fass D, Blacklow S, Kim PS, Berger JM. Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature (London) 1997;388:691–693. doi: 10.1038/41798. [DOI] [PubMed] [Google Scholar]
- 163.Fisher C, Beglova N, Blacklow SC. Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol Cell. 2006;22:277–283. doi: 10.1016/j.molcel.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 164.Rudenko G, Henry L, Henderson K, Ichtchenko K, Brown MS, Goldstein JL, Deisenhofer J. Structure of the LDL receptor extracellular domain at endosomal pH. Science. 2002;298:2353–2358. doi: 10.1126/science.1078124. [DOI] [PubMed] [Google Scholar]
- 165.Yasui N, Nogi T, Kiatao T, Nakano Y, Hattori M, Takagi J. Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc Natl Acad Sci U S A. 2007;104:9988–9993. doi: 10.1073/pnas.0700438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dolmer K, Campos A, Gettins PGW. Quantitative dissection of the binding contributions of ligand lysines of the receptor-associated protein (RAP) to the low density lipoprotein receptor-related protein (LRP1) J Biol Chem. 2013;288:24081–24090. doi: 10.1074/jbc.M113.473728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gettins PGW, Dolmer K. A proximal pair of positive charges provides the dominant ligand-binding contribution to complement-like domains from the LRP (low density lipoprotein-related receptor) Biochem J. 2012;443:65–73. doi: 10.1042/BJ20111867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Skeldal S, Larsen JV, Pedersen KE, Egelund R, Christensen A, Jensen J, Andreasen PA. Binding areas of urokinase-type plasminogen activator-plasminogen activator inhibitor-1 complex for endocytosis receptors of the low density lipoprotein receptor family, determined by site-directed mutagenesis. FEBS Journal. 2006;273:5143–5159. doi: 10.1111/j.1742-4658.2006.05511.x. [DOI] [PubMed] [Google Scholar]
- 169.Nykjaer A, Petersen CM, Moller BK, Jensen PH, Moestrup SK, Holtet TL, Etzerodt M, Thøgersen HC, Munch M, Andreasen AM. Purified α2-macroglobulin receptor/LDL receptor related protein binds urokinase plasminogen activator inhibitor-1 complex. Evidence that the α 2-macroglobulin receptor mediates cellular degradation of urokinase receptor-bound complexes. J Biol Chem. 1992;267:14543–14546. [PubMed] [Google Scholar]
- 170.Nykjær A, Kjoller L, Cohen RL, Lawrence DA, Garni-Wagner BA, Todd RF, III, van Zonneveld A-J, Gliemann J, Andreasen PA. Regions involved in binding of urokinase-type-1 inhibitor complex and pro-urokinase to the endocytic α2-macroglobulin receptor/low density lipoprotein receptor-related protein. Evidence that the urokinase receptor protects pro-urokinase against binding to the endocytic receptor. J Biol Chem. 1994;269:25668–25676. [PubMed] [Google Scholar]
- 171.Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J, Li Y, Yuan C, Bian C, Chen L, Furie B, Furie BC, Cines DB, Huang M. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656–659. doi: 10.1126/science.1121143. [DOI] [PubMed] [Google Scholar]
- 172.Xu X, Gårdsvoll H, Yuan C, Lin LF, Ploug M, Huang M. Crystal structure of the urokinase receptor in a ligand-free form. J Mol Biol. 2012;416:629–641. doi: 10.1016/j.jmb.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 173.Zhou A, Huntington JA, Pannu NS, Carrell RW, Read RJ. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol. 2003;10:541–544. doi: 10.1038/nsb943. [DOI] [PubMed] [Google Scholar]
- 174.Kamikubo Y, Neels JG, Degryse B. Vitronectin inhibits plasminogen activator inibitor-1-induced signalling and chemotaxis by blocking plasminogen activator inhibitor-1 binding to the low-density lipoprotein receptor-related protein. Int J Biochem Cell Biol. 2008;41:578–585. doi: 10.1016/j.biocel.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 175.Schechter I, Berger A. On the size of the active site of proteases. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 176.Elliott PR, Pei XY, Dafforn TR, Lomas DA. Topography of a 2.0Å structure of alpha-1-antitrypsin revelas targets for rational drug design to prevent conformational disease. Protein Sci. 2000;9:1274–1281. doi: 10.1110/ps.9.7.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tucker HM, Mottonen J, Goldsmith EJ, Gerard RD. Engineering of plasminogen activator inhibitor-1 to reduce the rate of latency transition. Nat Struct Biol. 1995;2:442–445. doi: 10.1038/nsb0695-442. [DOI] [PubMed] [Google Scholar]
- 178.Engh R, Löbermann H, Schneider M, Wiegand G, Huber R, Laurell CB. The S variant of human alpha-1-antitrypsin-activity, structure and implication for function and metabolism. Protein Eng. 1989;2:407–415. doi: 10.1093/protein/2.6.407. [DOI] [PubMed] [Google Scholar]
- 179.Olson ST, Gettins PGW. Regulation of proteases by protein inhibitors of the serpin superfamily. Progress in Molecular Biology and Translational Science. 2011;99:185–240. doi: 10.1016/B978-0-12-385504-6.00005-1. [DOI] [PubMed] [Google Scholar]
- 180.Dementiev A, Simonovic M, Volz K, Gettins PGW. Canonical inhibitor-like interactions explain reactivity of α1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J Biol Chem. 2003;278:37881–37887. doi: 10.1074/jbc.M305195200. [DOI] [PubMed] [Google Scholar]
- 181.Li W, Johnson DJD, Esmon CT, Huntington JA. 2.5Å structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nature Structural and Molecular Biology. 2004:11. doi: 10.1038/nsmb811. [DOI] [PubMed] [Google Scholar]
- 182.Huang W, Dolmer K, Gettins PGW. NMR solution structure of complement-like repeat CR8 from the low density lipoprotein receptor-related protein (LRP) J Biol Chem. 1999;274:14130–14136. doi: 10.1074/jbc.274.20.14130. [DOI] [PubMed] [Google Scholar]
- 183.Dolmer K, Huang W, Gettins PGW. NMR solution structure of complement-like repeat CR3 from the low density lipoprotein receptor-related protein (LRP). Evidence for specific binding to the receptor binding domain of human α2-macroglobulin. J Biol Chem. 2000;275:3264–3271. doi: 10.1074/jbc.275.5.3264. [DOI] [PubMed] [Google Scholar]
- 184.Simonovic M, Dolmer K, Huang W, Strickland DK, Volz K, Gettins PGW. Calcium coordination and pH dependence of the calcium affinity of ligand-binding repeat CR7 from the LRP. Comparison with related domains from the LRP and the LDL receptor. Biochemistry. 2001;40:15127–15134. doi: 10.1021/bi015688m. [DOI] [PubMed] [Google Scholar]