

Abstract
Donor-substituted diazo reagents, generated in situ from sulfonyl hydrazones in the presence of base, can serve as suitable radical precursors for Co(II)-based metalloradical catalysis (MRC). The cobalt(II) complex of D2-symmetric chiral porphyrin [Co(3,5-DitBu-Xu(2’-Naph)Phyrin)] is an efficient metalloradical catalyst that is capable of activating different N-arylsulfonyl hydrazones for asymmetric radical cyclopropanation of a broad range of alkenes, affording the corresponding cyclopropanes in high yields with effective control of both diastereoselectivity and enantioselectivity. This Co(II)-based metalloradical system represents the first catalytic protocol that can effectively utilize donor-type diazo reagents for asymmetric olefin cyclopropanation.
Graphical Abstract

There has been lasting interest in devising strategies to control stereoselectivity, especially enantioselectivity, of radical reactions for applications in organic synthesis.1 Among recent advances,1,2 metalloradical catalysis (MRC), which aims at the development of metalloradical-based systems for both catalytic initiation and selective control of radical processes, has led to the discovery of new catalytic pathways for stereoselective radical reactions.3,4 As stable metalloradicals, cobalt(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)] have emerged as effective catalysts for asymmetric radical transformations through catalytic generation of metal-stabilized organic radicals, such as the fundamentally new α-metalloalkyl radicals and α-metalloaminyl radicals, as the key intermediates.5 To date, Co(II)-based metalloradical catalysis (Co(II)-MRC) has exhibited capability of activating both acceptorand acceptor/acceptor-substituted diazo reagents to generate corresponding α-Co(III)-alkyl radicals (also known as Co(III)-carbene radicals) as key intermediates for various C-centered radical processes, including radical cyclopropanation of alkenes.6,7 Studies suggest that potential hydrogen-bonding interactions between carbonyl groups of these types of diazo reagents and the amide units of the porphyrin ligands in the resulting α-Co(III)-alkyl radicals play an important role in enhancing reactivity and controlling stereoselectivity. It was unclear if Co(II)-MRC could also be applicable to other types of diazo reagents that lack a carbonyl group, such as donor-substituted diazo reagents. Specifically, we wondered whether [Co(D2-Por*)] metalloradical catalysts were able to activate aryldiazomethanes to generate the corresponding α-Co(III)-benzyl radical intermediate I (Scheme 1). In the absence of carbonyl functionalities, what element of interaction could be explored for the effective control of enantioselectivity in the subsequent radical addition of the α-Co(III)-benzyl radical I to the olefin substrate? Furthermore, would the final step, 3-exo-tet radical cyclization of the resulting γ-Co(III)-alkyl radicals II, be diastereoselective? Additional questions arose from the intrinsic instability of donor-substituted diazomethanes, which are typically generated in situ from stable sulfonyl hydrazone precursors.8 Would Co(II)-MRC system be compatible with the basic conditions required for the in-situ generation protocol? If these questions could be answered positively, it would further expand the application of Co(II)-based radical cyclopropanation for the catalytic synthesis of enantioenriched 1,2-bisaryl and related cyclopropane compound 3.
Scheme 1.

Proposed Pathway for Asymmetric Radical Cyclopropanation with Donor-Substituted Diazo Reagents
Asymmetric olefin cyclopropanation with diazo reagents represents one of the most general methods for the construction of optically active three-membered carbocycles.9 While tremendous progress has been made with the use of several types of diazo reagents, asymmetric cyclopropanation with donor-substituted diazo reagents has been much less developed.10 As a novel alternative involving the further transfer of the initially formed electrophilic Rh2-carbenes into nucleophilic chiral sulfur ylides, Aggarwal and coworkers developed an indirect approach for asymmetric cyclopropanation with sulfonyl hydrazones as diazo precursors using the combination of dirhodium carboxylates and chiral sulfides as the catalyst that works specifically for electron-deficient olefins.8c,10c Nevertheless, it would be desirable to develop new catalytic systems that are capable of direct asymmetric cyclopropanation of diverse alkenes with donor-substituted diazo reagents.11 In view of the radical pathway of MRC (Scheme 1), we hypothesized that the radical cyclopropanation approach could potentially address the aforementioned issues with donor-substituted diazo reagents because a neutral radical process is generally less sensitive to electronic effects. Herein, we wish to report the development of the first asymmetric catalytic system that is highly effective for direct cyclopropanation of alkenes with α-aryl diazomethanes. Under mild conditions, the Co(II)-based metalloradical system is suitable for various alkenes, allowing for the efficient construction of cyclopropane rings with excellent control of both diastereoselectivity and enantioselectivity. The Co(II)-catalyzed cyclopropanation is further highlighted by its functional group tolerance, a feature that is closely related to the underlying radical mechanism.
Our study began with cyclopropanation of styrene (2a) with benzaldehyde tosylhydrazone (1a), which is known to generate α-phenyldiazomethane under basic conditions (Table 1). Even simple metalloradical catalyst [Co(TPP)] (TPP = 5,10,15,20-tetraphenylporphyrin) could catalyze the reaction in the presence of Cs2CO3 in methanol, forming the desired 1,2-diphenyl cyclopropane 3aa in 46% yield (entry 1). This result signifies the compatibility of Co(II)-based metalloradical system with the use of both bases and polar solvents. The stereoselectivity of the reaction was then evaluated by using Co(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)]. When the first-generation catalyst [Co(P1)] (P1 = 3,5-DitBu-ChenPhyrin) was used,6f 3aa could form in higher yield with significant level of both enantioselectivity and diastereoselectivity (entry 2). To further improve the catalytic reaction, we then turned our attention to the second-generation Co(II)-based metalloradical catalysts.6b While the phenyl-substituted [Co(P2)] (P2 = 3,5-DitBu-QingPhyrin) was less selective, the naphthyl-substituted [Co(P3)] (P3 = 3,5-DitBu-Xu(2’-Naph)Phyrin)) could effectively catalyze the formation of 3aa with excellent diastereoselectivity but lower enantioselectivity (entry 4). It should be noted that comparable reactivity was observed with the use of preformed α-phenyldiazomethane (see Table S1 in SI). The ineffective asymmetric induction is likely attributed to the low-barrier rotation of the α-benzyl radical unit around the Co(III)–C σ bond in intermediate I (Scheme 1) that diminishes its approaching preference to the prochiral styrene substrate between the re and si faces. To restrict the rotation, we decided to use benzaldehyde tosylhydrazone derivative that contains an ortho-methoxy group (1b). Considering MeO group is known as good hydrogen bond acceptors,12 we postulated that the potential formation of N–H⋯O hydrogen bond between the amido group of the chiral ligand and the MeO group of tosylhydrazone 1b (Figure S1 in SI) might rigidify the resulting intermediate I, leading to improved stereoselectivity. Indeed, the corresponding cyclopropane 3ba was obtained in good yield (78%) with high diastereoselectivity (95:5 dr) and excellent enantioselectivity (99% ee) (entry 5; see Table S2 in SI for detailed solvent effect). Conversely, asymmetric induction of the cyclopropanation process was significantly reduced when the MeO group in 1b was replaced by a sterically comparable Et group despite the high reactivity (entry 6). Moreover, low enantioselectivity was observed when the MeO group in 1b was moved from ortho-to either meta- or para- positions of the phenyl ring although with similarly high diastereoselectivity (entries 7 and 8). These results seem support the hypothesized hydrogen-bonding interaction.
Table 1.
Asymmetric Cyclopropanation of Styrene with Tosylhydrazones by Metalloradical Catalysts [Co(D2-Por*)]a
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | catalyst | product | yield (%)b | dec | ee (%)d |
| 1 | H (1a) | 3aa | 46 | 88:12 | - | |
| 2 | H (1a) | 3aa | 64 | 89:11 | −42 | |
| 3 | H (1a) | 3aa | 65 | 78:22 | <5 | |
| 4 | H (1a) | 3aa | 67 | 95:5 | 26 | |
| 5 | 2-MeO (1b) | 3ba | 78 | 95:5 | 99 | |
| 6 | 2-Et (1c) | 3ca | 87 | 85:15 | 33 | |
| 7 | 4-MeO (1d) | 3da | 23 | 93:7 | 23 | |
| 8 | 3-MeO (1e) | 3ea | 69 | 95:5 | 23 | |
 | ||||||
Carried out with 1x (0.1 mmol) and 2a (1.5 equiv.) in the presence of Cs2CO3 (2 equiv.) in methanol (0.6 mL).
Isolated yields.
Determined by 1H NMR.
Determined by chiral HPLC for the major trans diastereomer.
Under the optimized conditions, the scope of [Co(P3)]/1b-based catalytic system was then evaluated by employing different alkenes (Table 2). Like styrene, its derivatives bearing substituents with different electronic properties, including electron-donating MeO and electron-withdrawing CF3 groups, could be cyclopropanated in high yields with excellent diastereoselectivity and enantioselectivity (entries 1–3). Halogenated styrenes such as those with a Br atom at various positions were also shown to be suitable substrates for the catalytic process (entries 4–6). The relative and absolute configurations of cyclopropane 3bd (entry 4) were established as trans and (1S,2S), respectively (see Figure S2 in SI). As demonstrated for 2-bromostyrene (2f) together with 2,4,6-trimethylstyrene (2g), sterically hindered styrene derivatives could also undergo productive cyclopropanation (entries 6 and 7). Furthermore, this system was also applicable for 1,1-disubstituted olefins as demonstrated with α-substituted styrene derivatives 2h, 2i and 2j, affording the desired cyclopropanes with enantioselective control of the newly-formed quaternary stereogenic centers, albeit with moderate diastereoselectivity (entries 8–10). Distinctly, the Co(II)-based radical process could tolerate functional groups as exemplified by the highly enantioselective cyclopropanation of 3-aminostyrene (entry 11). In addition to the expanded aromatic olefins such as 2-vinylnaphthalene (entry 12), conjugated dienes, including both aromatic diene 2m and aliphatic diene 2n, could also be employed as substrates but with relatively lower stereoselectivities (entries 13 and 14). It was notable that both electron-rich and -deficient nonaromatic olefins, such as vinyl ether 2o, acrylamide 2p and vinyl ketone 2q could be enantioselectively cyclopropanated as well (entries 15–17) with moderate control of stereoselectivities. The observed substrate scope and reactivity profile of [Co(P3)]-catalyzed cyclopropanation is in agreement with the underlying radical mechanism (Scheme 1). It was also noteworthy to mention that the one-pot protocol of the Co(II)-catalyzed cyclopropanation could be scaled up ten-fold as demonstrated with the high-yielding synthesis of cyclopropane 3ba on 1.0 mmol scale without affecting the excellent stereoselectivity (entry 1).
Table 2.
Asymmetric Cyclopropanation of Different Olefins with Tosylhydrazone 1b Catalyzed by [Co(P3)]a
 |
Carried out with 1b (0.1 mmol) and 2x (1.5 equiv.) in the presence of Cs2CO3 (2 equiv.) in methanol (0.6 mL); isolated yields; dr was determined by 1H NMR; ee of the major trans diastereomer was determined by chiral HPLC.
Performed on 1.0 mmol scale.
Absolute configuration was determined by X-ray as (1S, 2S).
5 mol % [Co(P3)].
5.0 equiv. 2q.
Guided by the postulated hydrogen-bonding interaction in the key intermediates I, we then sought to identify other benzaldehyde sulfonylhydrazone derivatives as effective radical precursors for asymmetric cyclopropanation (Table 3). Like arylhydrazone 1b (entry 1), 2,5-dimethoxybenzaldehyde-derived tosylhydrazone (1f) could be also effectively activated by [Co(P3)] for cyclopropanation of styrene, affording cyclopropane 3fa in 91% yield with 90% de and 94% ee (entry 2). Considering that fluorine atoms in organic fluorides have been demonstrated as a potential hydrogen bond acceptor,13 we explored the use of fluorine-containing arylhydrazones as diazo precursors. Remarkably, fluoroarene-based tosylhydrazones such as 1g–1j were found to be even more active than 1b, enabling the catalytic process even at room temperature (entries 3, 6, 8 and 10). The desired fluorine-containing bisaryl cyclopropanes 3ga–3ja were obtained in up to 90% yield with 88–92% de and 68–88% ee. Attempts to further improve enantioselectivity with 1g at lower temperature were unsuccessful due to ineffective generation of 2,6-difluorophenyl diazomethane at 0 °C (entry 4). Since the decomposition of the sterically bulky (2,4,6-triisopropylphenyl)sulfonyl hydrazone salt was shown to be considerably faster than tosylhydrazone salt,14 the low temperature asymmetric cyclopropanation was reexamined with the use of 2,6-difluorobenzaldehydederived (2,4,6-triisopropylphenyl)sulfonyl hydrazone 1g’. As expected, the reaction could proceed effectively at 0 °C, affording 3ga in 83% yield with considerably enhanced stereoselectivities (entry 5). Likewise, the reactions with related (2,4,6-triisopropylphenyl) sulfonyl hydrazones 1h’–1j’ could also be productively conducted at 0 °C, affording the cyclopropanes 3ha–3ja with significantly improved diastereoselectivity and enantioselectivity (entries 7, 9 and 11). Under the same conditions, while o-Cl-benzaldehyde-derived sulfonylhydrazone gave a similar result to o-F-benzaldehyde-derived sulfonylhydrazone, o-Br- and o-I-benzaldehyde-derived sulfonylhydrazones proved to be less effective radical precursors for the catalytic process (see Scheme S1 in SI).
Table 3.
Asymmetric Cyclopropanation of Styrene with Various Sulfonyl Hydrazones Catalyzed by [Co(P3)]a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry |
|
product | temp. (°C) |
yield (%)c |
drd | ee (%)e |
|
| 1 | Ts(1b) | 3ba | 40 | 78 | 95:5 | 99 | |
| 2 | Ts(1f) | 3fa | 40 | 91 | 95:5 | 94 | |
| 3 | 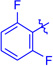 |
Ts(1g) | 3ga | RT | 90 | 94:6 | 86 |
| 4 | Ts (1g) | 3ga | 0 | <10 | - | - | |
| 5 | TPS (1g') | 3g'a | 0 | 83 | >99:1 | 93 | |
| 6 | Ts (1h) | 3ha | RT | 58 | 96:4 | 71 | |
| 7 | TPS (1h') | 3h'a | 0 | 75 | >99:1 | 76 | |
| 8 | 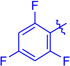 |
Ts (1i) | 3ia | RT | 85 | 96:4 | 88 |
| 9 | TPS (1i') | 3i'a | 0 | 85 | >99:1 | 93 | |
| 10 | 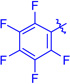 |
Ts (1j) | 3ja | RT | 82 | 95:5 | 68 |
| 11 | TPS (1j') | 3j'a | 0 | 81 | >99:1 | 89 | |
Carried out with 1x (0.1 mmol) and 2a (1.5 equiv.) in the presence of Cs2C O3 (2 equiv.) in methanol (0.6 mL).
Ts = 4-toluenesulfonyl; TPS = (2,4,6-triisopropyl) phenyl sulfonyl.
Isolated yields.
Determined by 1H NMR.
Determined by chiral HPLC for the major trans diastereomer.
To probe the underlying stepwise radical mechanism of the Co(II)-catalyzed cyclopropanation, both (E)- and (Z)-β-deuterostyrenes were employed as substrates (Scheme 2). While a concerted mechanism is usually stereospecific, a stepwise radical mechanism (Scheme 1) would result in the formation of four possible diastereomers from either (E)- or (Z)-β-deuterostyrene: two trans-isotopomers A & B and two cis-isotopomers C & D due to the rotation of β-C–C bond in the γ-Co(III)-alkyl radical intermediate II before ring closure (Schemes S2 and S3 in SI). Using catalyst [Co(P3)], cyclopropanation of (E)-β-deuterostyrene with tosylhydrazone 1g gave trans-3ga as the dominant product with a 93:7 ratio of isotopomers A and B. Under the same conditions, the reaction of (Z)-β- deuterostyrene resulted in the identical 93:7 ratio of isotopomeric distribution but in favoring B over A. The observation of trans-isotopomer B from (E)-β-deuterostyrene as well as trans-isotopomer A from (Z)-β-deuterostyrene is evidently a result of the rotation of β-C–C bond in intermediate II. When sterically less-hindered [Co(P4)] (P4 = 3,5-DitBu-IbuPhyrin) was used as the catalyst,15 a significantly different isotopomeric ratio of trans-isotopomers A & B (from 93:7 to 80:20) was observed, suggesting easier rotation of the β-C–C bond in a less-crowded ligand environment (Schemes S4 and S5 in SI). Together, these observations convincingly support the proposed stepwise radical mechanism of the Co(II)-based cyclopropanation with N-arylsulfonyl hydrazones. Furthermore experiments were performed for direct detection of the α-Co(III)-benzyl radical species I (Scheme 1) by high-resolution mass spectrometry (HRMS).16 The exposure of [Co(P4)] to 1g (10 equiv.) with base in methanol resulted in a mixture that was filtrated and analyzed by HRMS in the absence of any additives such as formic acids that commonly act as electron carriers for ESI ionization. As shown in Scheme S6 in SI, the obtained spectrum clearly revealed a signal corresponding to [Co(P4)(2,6-F2C6H3CH)]+ (m/z = 1361.6515), which resulted from the neutral α-Co(III)-benzyl radical I by the loss of one electron.
Scheme 2.

Cyclopropanation of (E)- and (Z)-β-Deuterostyrenes to Probe Radical Reaction Mechanism.
In summary, Co(II)-based metalloradical catalysis (MRC) has, for the first time, been successfully applied to donor-substituted diazo reagents, generated from N-arylsulfonyl hydrazones in the presence of base, for olefin cyclopropanation. Chiral metalloradical complex [Co(P3)] has been shown to be an effective catalyst for asymmetric radical cyclopropanation with α-aryldiazomethane precursors. The Co(II)-based cyclopropanation is applicable to a broad combination of N-arylsulfonyl hydrazones and alkenes, affording the cyclopropane derivatives in high yields with excellent diastereoselectivity and enantioselectivity. In view of the underdevelopment of donor-substituted diazo reagents in asymmetric cyclopropanation, the successful development of this catalytic radical process will encourage further efforts in applying sulfonyl hydrazones as stable precursors for various asymmetric catalytic processes.
Supplementary Material
Acknowledgments
We are grateful for financial support by NIH (R01-GM102554).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental details and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.”
REFERENCES
- 1.For reviews on stereoselectivity of free radical transformations see: Yang Y-H, Sibi MP. In: Encyclopedia of Radicals in Chemistry, Biology and Materials. Chatgilialoglu C, Studer A, editors. Wiley: Weinheim; 2012. p. 655. Curran DP, Porter NA, Giese B. Stereochemistry of radical reactions: concepts, guidelines, and synthetic applications. John Wiley & Sons; 2008. Zimmerman J, Sibi MP. Top. Curr. Chem. 2006;263:107. Bar G, Parsons AF. Chem. Soc. Rev. 2003;32:251. doi: 10.1039/b111414j. Zard SZ. Radical reactions in organic synthesis. Oxford: Oxford University Press; 2003.
- (2).For select examples of stereoselective radical processes via photoredox catalysis see: Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681. doi: 10.1126/science.aad8313. Brimioulle R, Lenhart D, Maturi MM, Bach T. Angew. Chem. Int. Ed. 2015;54:3872. doi: 10.1002/anie.201411409. Huo HH, Shen XD, Wang CY, Zhang LL, Rose P, Chen LA, Harms K, Marsch M, Hilt G, Meggers E. Nature. 2014;515:100. doi: 10.1038/nature13892. Du JN, Skubi KL, Schultz DM, Yoon TP. Science. 2014;344:392. doi: 10.1126/science.1251511.
- (3).For reviews and highlights on Co(II)-MRC: Studer A, Curran DP. Angew. Chem. Int. Ed. 2016;55:58. doi: 10.1002/anie.201505090. Pellissier H, Clavier H. Chem. Rev. 2014;114:2775. doi: 10.1021/cr4004055. Degennaro L, Trinchera P, Luisi R. Chem. Rev. 2014;114:7881. doi: 10.1021/cr400553c. Olivos Suarez AI, Lyaskovskyy V, Reek JN, van der Vlugt JI, de Bruin B. Angew. Chem. Int. Ed. 2013;52:12510. doi: 10.1002/anie.201301487. Doyle MP. Angew. Chem. Int. Ed. 2009;48:850. doi: 10.1002/anie.200804940.
- (4).For Ti(III)-based MRC see: Gansäuer A, Hildebrandt S, Vogelsang E, Flowers Ii RA. Dalton Trans. 2016;45:448. doi: 10.1039/c5dt03891j. RajanBabu TV, Nugent WA. J. Am. Chem. Soc. 1994;116:986. Gansäuer A, Hildebrandt S, Michelmann A, Dahmen T, von Laufenberg D, Kube C, Fianu GD, Flowers RA. Angew. Chem. Int. Ed. 2015;54:7003. doi: 10.1002/anie.201501955. Gansäuer A, Fleckhaus A, Lafont MA, Okkel A, Kotsis K, Anoop A, Neese F. J. Am. Chem. Soc. 2009;131:16989. doi: 10.1021/ja907817y. Gansäuer A, Fan C-A, Keller F, Keil J. J. Am. Chem. Soc. 2007;129:3484. doi: 10.1021/ja0686211. Gansäuer A, Rinker B, Pierobon M, Grimme S, Gerenkamp M, Mück-Lichtenfeld C. Angew. Chem. Int. Ed. 2003;42:3687. doi: 10.1002/anie.200351240.
- (5).For detailed experimental and theoretical studies on the radical mechanism of Co(II)-MRC: Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B. J. Am. Chem. Soc. 2010;132:10891. doi: 10.1021/ja103768r. Belof JL, Cioce CR, Xu X, Zhang XP, Space B, Woodcock HL. Organometallics. 2011;30:2739. doi: 10.1021/om2001348. Lu HJ, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP. J. Am. Chem. Soc. 2011;133:8518. doi: 10.1021/ja203434c.
- (6).(a) Xu X, Zhu SF, Cui X, Wojtas L, Zhang XP. Angew. Chem. Int. Ed. 2013;52:11857. doi: 10.1002/anie.201305883. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu X, Lu HJ, Ruppel JV, Cui X, de Mesa SL, Wojtas L, Zhang XP. J. Am. Chem. Soc. 2011;133:15292. doi: 10.1021/ja2062506. [DOI] [PubMed] [Google Scholar]; (c) Zhu SF, Xu X, Perman JA, Zhang XP. J. Am. Chem. Soc. 2010;132:12796. doi: 10.1021/ja1056246. [DOI] [PubMed] [Google Scholar]; (d) Zhu SF, Ruppel JV, Lu HJ, Wojtas L, Zhang XP. J. Am. Chem. Soc. 2008;130:5042. doi: 10.1021/ja7106838. [DOI] [PubMed] [Google Scholar]; (e) Chen Y, Ruppel JV, Zhang XP. J. Am. Chem. Soc. 2007;129:12074. doi: 10.1021/ja074613o. [DOI] [PubMed] [Google Scholar]; (f) Chen Y, Fields KB, Zhang XP. J. Am. Chem. Soc. 2004;126:14718. doi: 10.1021/ja044889l. [DOI] [PubMed] [Google Scholar]; (g) Fantauzzi S, Gallo E, Rose E, Raoul N, Caselli A, Issa S, Ragaini F, Cenini S. Organometallics. 2008;27:6143. [Google Scholar]
- (7).For other catalytic processes involving α-Co(III)-alkyl radicals as intermediates see: Das BG, Chirila A, Tromp M, Reek JNH, Bruin Bd. J. Am. Chem. Soc. 2016;138:8968. doi: 10.1021/jacs.6b05434. Zhang J, Jiang J, Xu D, Luo Q, Wang H, Chen J, Li H, Wang Y, Wan X. Angew. Chem. Int. Ed. 2015;54:1231. doi: 10.1002/anie.201408874.
- (8).(a) Xiao Q, Zhang Y, Wang J. Acc. Chem. Res. 2013;46:236. doi: 10.1021/ar300101k. [DOI] [PubMed] [Google Scholar]; (b) Barluenga J, Valdés C. Angew. Chem. Int. Ed. 2011;50:7486. doi: 10.1002/anie.201007961. [DOI] [PubMed] [Google Scholar]; (c) Fulton JR, Aggarwal VK, de Vicente J. Eur. J. Org. Chem. 2005:1479. [Google Scholar]
- (9).(a) Intrieri D, Carminati DM, Gallo E. Dalton Trans. 2016;45:15746. doi: 10.1039/c6dt02094a. [DOI] [PubMed] [Google Scholar]; (b) Bartoli G, Bencivenni G, Dalpozzo R. Synthesis. 2014;46:979. [Google Scholar]; (d) Intrieri D, Caselli A, Gallo E. Eur. J. Inorg. Chem. 2011:5071. [Google Scholar]; (e) Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; (f) Davies HML, Antoulinakis EG. Org. React. 2001;57:1. [Google Scholar]; (g) Doyle MP, Forbes DC. Chem. Rev. 1998;98:911. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- (10).For asymmetric cyclopropanation reactions with donor-type diazo reagents see: Goudreau SR, Charette AB. J. Am. Chem. Soc. 2009;131:15633. doi: 10.1021/ja9074776. Berkessel A, Kaiser P, Lex J. Chem. Eur. J. 2003;9:4746. doi: 10.1002/chem.200305045. Aggarwal VK, Alonso E, Fang G, Ferrara M, Hynd G, Porcelloni M. Angew. Chem. Int. Ed. 2001;40:1433. Aggarwal VK, Smith HW, Jones RVH, Fieldhouse R. Chem. Commun. 1997:1785. For non-asymmetric cyclopropanation reactions with donor-type diazo reagents see: Verdecchia M, Tubaro C, Biffis A. Tetrahedron Lett. 2011;52:1136. Dai X, Warren TH. J. Am. Chem. Soc. 2004;126:10085. doi: 10.1021/ja047935q. Maas G, Seitz J. Tetrahedron Lett. 2001;42:6137. Li Y, Huang J-S, Zhou Z-Y, Che C-M. J. Am. Chem. Soc. 2001;123:4843. doi: 10.1021/ja003184q. Hamaker CG, Djukic J-P, Smith DA, Woo LK. Organometallics. 2001;20:5189.
- (11).For non-asymmetric cyclopropanation using tosylhydrazones as donor- type diazo precursors see: Cyr P, Cote-Raiche A, Bronner SM. Org. Lett. 2016;18:6448. doi: 10.1021/acs.orglett.6b03345. Reddy AR, Hao F, Wu K, Zhou C-Y, Che C-M. Angew. Chem. Int. Ed. 2016;55:1810. doi: 10.1002/anie.201506418. Barluenga J, Quiñones N, Tomás-Gamasa M, Cabal M-P. Eur. J. Org. Chem. 2012:2312. Zhang J-L, Hong Chan PW, Che C-M. Tetrahedron Lett. 2003;44:8733. Adams LA, Aggarwal VK, Bonnert RV, Bressel B, Cox RJ, Shepherd J, de Vicente J, Walter M, Whittingham WG, Winn CL. J. Org. Chem. 2003;68:9433. doi: 10.1021/jo035060c. Aggarwal VK, de Vicente J, Bonnert RV. Org. Lett. 2001;3:2785. doi: 10.1021/ol0164177. For intramolecular C–H alkylation using hydrazones as donor-type diazo precursors see: Soldi C, Lamb KN, Squitieri RA, Gonzalez-Lopez M, Di Maso MJ, Shaw JT. J. Am. Chem. Soc. 2014;136:15142. doi: 10.1021/ja508586t. Reddy AR, Zhou CY, Guo Z, Wei J, Che CM. Angew. Chem. Int. Ed. 2014;53:14175. doi: 10.1002/anie.201408102.
- (12).(a) Palusiak M, Grabowski SJ. J. Mol. Struct. 2002;642:97. [Google Scholar]; (b) Grabowski SJ. J. Phys. Org. Chem. 2004;17:18. [Google Scholar]
- (13).Champagne PA, Desroches J, Paquin JF. Synthesis. 2015;47:306. [Google Scholar]
- (14).(a) Chamberlin AR, Bond FT. J. Org. Chem. 1978;43:154. [Google Scholar]; (b) Cusack NJ, Reese CB, Risius AC, Roozepeikar B. Tetrahedron. 1976;32:2157. [Google Scholar]
- (15).Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org. Lett. 2008;10:1995. doi: 10.1021/ol800588p. [DOI] [PubMed] [Google Scholar]
- (16).Schäfer M, Drayß M, Springer A, Zacharias P, Meerholz K. Eur. J. Org. Chem. 2007:5162. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.