

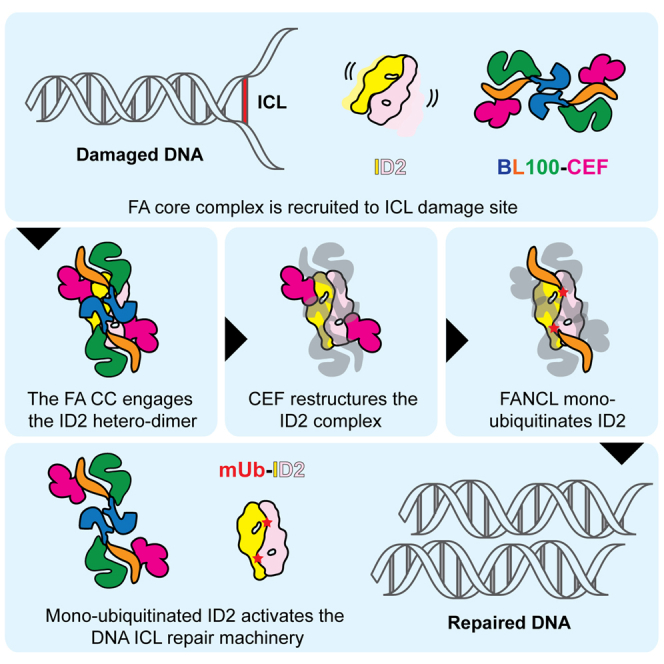
Summary
Activation of the main DNA interstrand crosslink repair pathway in higher eukaryotes requires mono-ubiquitination of FANCI and FANCD2 by FANCL, the E3 ligase subunit of the Fanconi anemia core complex. FANCI and FANCD2 form a stable complex; however, the molecular basis of their ubiquitination is ill defined. FANCD2 mono-ubiquitination by FANCL is stimulated by the presence of the FANCB and FAAP100 core complex components, through an unknown mechanism. How FANCI mono-ubiquitination is achieved remains unclear. Here, we use structural electron microscopy, combined with crosslink-coupled mass spectrometry, to find that FANCB, FANCL, and FAAP100 form a dimer of trimers, containing two FANCL molecules that are ideally poised to target both FANCI and FANCD2 for mono-ubiquitination. The FANCC-FANCE-FANCF subunits bridge between FANCB-FANCL-FAAP100 and the FANCI-FANCD2 substrate. A transient interaction with FANCC-FANCE-FANCF alters the FANCI-FANCD2 configuration, stabilizing the dimerization interface. Our data provide a model to explain how equivalent mono-ubiquitination of FANCI and FANCD2 occurs.
Keywords: interstrand crosslink, Fanconi anemia, ubiquitin ligase, DNA repair, cryo-electron microscopy, mono-ubiquitination
Graphical Abstract
Highlights
-
•
FANCB, FANCL, and FAAP100 form a symmetric dimer of trimers
-
•
FANCL is ideally poised for the symmetric mono-ubiquitination of FANCI-FANCD2
-
•
Two separate FANCC-FANCE-FANCF complexes bind to the opposing poles of FANCB-FANCL-FAAP100
-
•
FANCC-FANCE-FANCF stabilizes FANCI-FANCD2 for efficient mono-ubiquitination
Mono-ubiquitination of FANCI-FANCD2 by the Fanconi anemia core complex activates a major DNA interstrand-crosslink repair pathway important for genome stability maintenance. Here, Swuec et al. reveal the structural basis of this reaction by showing that the core complex exists as a dimeric catalytic module for the symmetric mono-ubiquitination of FANCI-FANCD2.
Introduction
Interstrand crosslinks (ICLs) are toxic DNA lesions that prevent the separation of the two filaments in the DNA double helix, impairing DNA replication and transcription. The Fanconi anemia (FA)/BRCA pathway is the major interstrand crosslink repair process in higher eukaryotes (Knipscheer et al., 2009, Kottemann and Smogorzewska, 2013). Activation of this pathway depends on the specific mono-ubiquitination of the FANCI and FANCD2 proteins by the FA core complex (Ceccaldi et al., 2016, Garcia-Higuera et al., 2001, Sims et al., 2007, Smogorzewska et al., 2007, Timmers et al., 2001). Mutations in the FA genes cause a rare genetic disorder characterized by hypersensitivity to crosslinking agents, chromosomal instability, and cancer predisposition (Deans and West, 2011).
Despite the enormous effort of several research groups, a mechanistic understanding of the activation of the FA ICL repair pathway is only starting to emerge (Walden and Deans, 2014). A key player in the recognition of ICLs is the FA anchor complex (consisting of the FANCM DNA translocase, FAAP24, and two histone-like factors named MHF1-2), which, in turn, recruits the FA core complex to the DNA damage site (Ciccia et al., 2007, Coulthard et al., 2013, Kim et al., 2008, Yan et al., 2010). The core complex is composed of three distinct protein assemblies with non-redundant functions: FANCB-FANCL-FAAP100 (hereinafter called BL100), FANCC-FANCE-FANCF (CEF), and FANCA-FANCG-FAAP20 (AG20) (Huang et al., 2014, Rajendra et al., 2014). While the AG20 and CEF play loosely defined ancillary roles (Wang and Smogorzewska, 2015), the BL100 complex is a three-member ubiquitin ligase, with FANCL as the minimal unit required for ubiquitination (Huang et al., 2014, Meetei et al., 2003, Rajendra et al., 2014). FANCL is an E3 ligase containing a RING domain that is essential for the interaction with the E2 enzyme, UBE2T/FANCT, and for the mono-ubiquitination of the FANCI and FANCD2 molecular targets (Cole et al., 2010, Hodson et al., 2011, Hodson et al., 2014, Machida et al., 2006). Although FANCL is active in isolation, its catalytic function is greatly stimulated by the interaction with FANCB and FAAP100 (Huang et al., 2014, Rajendra et al., 2014). Whether binding of these two factors causes an activating conformational switch in the FANCL catalytic center, or whether FANCB and FAAP100 play an architectural role important for mono-ubiquitination, is unknown.
Multiple other architectural aspects of the mono-ubiquitination of FANCI and FANCD2 likewise remain poorly understood. First, crystallographic studies on mouse proteins indicate that the FANCI-FANCD2 (ID2) hetero-dimer forms a pseudo-2-fold symmetric arrangement. However, the mono-ubiquitination sites map at the dimerization interface within the ID2 complex (Joo et al., 2011), suggesting that a reconfiguration of this interface is needed at some point during the mono-ubiquitination reaction to allow for access of the E3 ligase assembly. Second, whether a stable ID2 complex is recruited to the DNA damage site prior to mono-ubiquitination is unclear, although recent cryoelectron microscopy (cryo-EM) studies on the unmodified human proteins support this notion (Liang et al., 2016). Remarkably the symmetric hetero-dimer captured by crystallography cannot be recognized in the electron microscopy (EM) structure, suggesting that the ID2 complex might visit different conformational states throughout the ubiquitination reaction. Finally, although both members of the ID2 dimer need to be mono-ubiquitinated for activation of the ICL repair pathway (Sims et al., 2007, Smogorzewska et al., 2007, Timmers et al., 2001), so far, biochemical reconstitution has mainly focused on the minimal ability of the BL100 E3 ligase to mono-ubiquitinate FANCD2, with little to no FANCI mono-ubiquitination observed (Rajendra et al.,2014). How equivalent mono-ubiquitination of both FANCI and FANCD2 is achieved and whether any of the ancillary proteins in the FA core complex are involved in this process remain unresolved questions.
To address these issues, we have used EM, combined with crosslinking and mass spectrometry (XL/MS), to analyze the architecture of the BL100 complex and its substrate ID2, either in isolation or with other components of the FA core complex. We found that BL100 forms a symmetric dimer of trimers, inviting a molecular model for the equivalent ubiquitination of the dimeric ID2 assembly. The CEF component of the FA core complex binds to the two extremities in the BL100 dimer, probably acting as a molecular matchmaker that mediates the BL100 and ID2 interaction. We also uncovered an unexpected structural function for CEF, which stabilizes the dimerization interface of the ID2 complex. We postulate that this function might have a direct role in the efficient mono-ubiquitination of the ID2 hetero-dimeric substrate. Our findings provide key insights into the structural basis for the mono-ubiquitination of the ID2 complex, the defining step in the activation of the FA/BRCA ICL repair pathway.
Results
BL100 Assembles in a 2-Fold Symmetric Dimer
Although the FANCL subunit of the core complex is an active E3 ligase in isolation (Alpi et al., 2008), its mono-ubiquitination function is stimulated by 6-fold in the presence of FANCB and FAAP100 (Rajendra et al., 2014). To understand whether the B100 association provides a structural framework for efficient mono-ubiquitination, or whether it causes a conformational change in FANCL that stimulates its activity, we have expressed and purified human BL100 from insect cells. Several steps of purification yielded a pure trimeric complex that eluted with a sharp peak under gel filtration and with an apparent molecular weight of 440 kDa (Figures 1A and S1). EM imaging confirmed that our preparation is highly homogeneous and monodisperse (Figures 1B and 1C). Two-dimensional classification of negative stain data revealed an elongated bow-tie structure containing apparent 2-fold symmetry (Figure 1D). The particle consists of a compact center and two globular modules, mapping at the two extremities of the assembly. Cryo-EM analysis yielded classes that were highly reminiscent of those obtained after negative stain imaging; however, no significant improvement in resolution could be detected, hinting at an inherent flexible nature in the BL100 complex (Figure 1E). To test our hypothesis that BL100 is mobile and can adopt different structural states, we have performed focused classification on the central core of the structure, followed by classification on the whole particle without further alignment. This approach revealed that the globular domains at the two poles in the structure move as rigid bodies, either tilting or stretching, relative to the central module (Figure S1; Movie S1). Despite this flexibility, the apparent 2-fold symmetry is maintained in the central core and at the two poles in the structure. From the data shown so far, it remains unclear whether BL100 is, indeed, a 2-fold symmetric assembly or, rather, contains a pseudo-2-fold symmetry axis, similar to ID2 (Figure 1F). To address this issue, we have repeated the two-dimensional analysis, using a FANCL derivative that contains maltose-binding protein (MBP) fused to the N terminus (Figures 2A and S2). MBP tags provide an electron-dense feature that can be averaged in negative stain EM, and they serve as pointers to localize individual proteins in a macromolecular assembly (Chen et al., 2008). We reasoned that a bona fide 2-fold symmetric complex should show two (FANCL-fused) MBP densities that are dyad related. Indeed, as evident from two-dimensional class averages, B-(MBP)L100 is decorated with two symmetry-related MBP densities, establishing that the three factors assemble into a homo-dimer (B:L:100 = 2:2:2 stoichiometry, with an expected molecular weight of 468 kDa, coherent with our analytical gel filtration profile) (Figures 2B and 2C). A dimeric nature of the FA core complex was unexpected and invites a mechanistic model for the ID2 mono-ubiquitination reaction that is further detailed in the Discussion section.
Figure 1.

FANCB-FANCL-FAAP100 under the Electron Microscope
(A) Coomassie-stained SDS-PAGE gel of the purified BL100 complex.
(B and C) Negative-stain (B) and cryo-EM (C) of the purified BL100 complex.
(D and E) Representative negative-stain (D) and cryo-EM (E) two-dimensional class averages. Box size is 458 Å × 458 Å.
(F) Top: BL100 forms an elongated, apparently 2-fold symmetric particle containing a dimerization core (blue) and two globular poles (red). Bottom: a cartoon representation of the assembly with overall particle dimensions.
See also Figure S1.
Figure 2.

Subunit Architecture of the FANCB-FANCL-FAAP100 Assembly Studied by Single-Particle Analysis
(A) Silver-stained SDS-PAGE gel of the assembly containing an MBP N-terminally fused to FANCL.
(B) Comparison between wild-type and MBP fused BL100 (negative-stain two-dimensional classes). Two MBP density features are marked with an orange arrowhead. Box size is 458 Å × 458 Å.
(C) Cartoon representation of the BL100 complex with MBP in orange.
(D) Coomassie-stained SDS-PAGE gel of the purified B100 subcomplex.
(E) Negative-stain two-dimensional classes of B100. Box size is 458 Å × 458 Å.
(F) Cartoon model of B100. In the absence of FANCL, the dimerization core remains unperturbed. The poles in the particle are more flexible.
(G) Coomassie-stained SDS-PAGE gel of the isolated FAAP100 subunit.
(H) Comparison between FAAP100 two-dimensional classes and two-dimensional classes of B100, focused on one pole in the particle. Box size is 458 Å × 458 Å.
(I) Cartoon of the BL100 architecture derived from single-particle EM combined with subunit dropout experiments.
See also Figures S2 and S3.
FANCB Is the Sole Candidate for the Homo-dimerization Interface in the FA Core Complex
Previous reports indicate that RING domains can dimerize (Brzovic et al., 2001, Liew et al., 2010, Lorick et al., 1999, Pruneda et al., 2011), and our MBP analysis shows that the RING-containing FANCL subunit maps in proximity of the dimerization interface of BL100 (Figures 2B and 2C). To test whether the FA core complex dimerization requires FANCL, we have expressed and purified a subunit dropout assembly that only contains FANCB and FAAP100 for single-particle analysis (Figures 2D and S3). In the absence of FANCL, the dimerization interface remains intact, while the globular poles in the assembly become markedly flexible (Figures 2E and S3; Movie S2), revealing a structural role for FANCL in stabilizing the BL100 assembly (Figure 2F), in line with previous reports (Alpi et al., 2008). To identify the dimerization component in the BL100 complex, we have attempted to purify the isolated FANCB subunit, but we found that this factor is insoluble when expressed in the absence of FAAP100 (data not shown). Conversely, the FAAP100 subunit is soluble and monodisperse in the absence of FANCB, according to analytical gel filtration and single-particle EM (Figures 2G and S3). Comparison of FAAP100 with the B100 two-dimensional class averages, centered on the flexible globular pole component, allows us to recognize FAAP100 as the element mapping at the extremities of the B100 particle. No resemblance was detected between the isolated FAAP100 and the dimerization core in the assembly (Figures 2H and S3). From these data, we conclude that FANCB and FAAP100 provide a dimeric scaffold to position FANCL on opposite poles in the protein assembly (Figure 2I). This suggests that efficient substrate ubiquitination requires a 2-fold symmetric arrangement in the FA core complex. Furthermore, our subunit dropout experiments are incompatible with a homo-dimerization role for either FANCL or FAAP100, leaving the FANCB subunit as the sole candidate for the dimerization element of the BL100 complex.
XL/MS Analysis of the BL100 Assembly
Analytical biochemistry combined with single-particle EM analysis allowed us to propose a model for the molecular architecture of a dimeric BL100 assembly. Difference mapping suggests that FANCB is located at the homo-dimerization interface; however, we failed in our attempts to directly show that the isolated FANCB can self-associate. Therefore, we turned to XL/MS analysis, a powerful tool to study the architecture of macromolecular assemblies, especially when combined with structural EM (Bui et al., 2013) (Figure 3A; Table S1). We have used an amino-reactive bi-functional crosslinker (BS3) to covalently bridge between surface-exposed primary amines that map within a distance range of 26–30 Å between Cα of lysine residues. Samples were crosslinked and trypsinized, enriched for larger crosslinked peptides through gel filtration, and analyzed by nanoLC-MS (nano-liquid chromatography-mass spectrometry) using an LTQ Orbitrap Velos. We first validated the XL/MS data by comparing our results with the available crystal structure of the human FANCL central domain. All detected crosslinks in FANCL exist within solvent-exposed lysines that are spaced within a range compatible with the dimensions of the crosslinker spacing arm (Figure 3B). This control provided us with confidence in the interaction network built for the rest of the protein assembly. Remarkably, FANCB contains, by far, the highest frequency of intra-molecular crosslinks, followed by FANCL (Figures 3A and 3C). Only one FAAP100 intra-molecular crosslink was observed. Possible explanations for this deficiency include the unusually low number of lysines in this subunit (1.9% of the sequence, with 5.7% being the average in human proteins), their location in large peptides lacking trypsin-cleavage sites, or the absence of proximal lysines in the tertiary structure of FAAP100. Instead, the majority of crosslinked lysines observed for FAAP100 were involved in inter-molecular crosslinks with FANCB. Altogether, these data provide additional support to the notion that FANCB maps at the homo-dimerization interface in the core complex (Figures 2 and 3C). We note that one of the intra-FANCB crosslinks detected involves surface-exposed lysines (in position 35 and 39), which flank a known FA-associated mutation (L37S). Given the immediate proximity of Lys35 and Lys39, Leu37 is likely solvent exposed and could serve a role in interacting with other FA proteins. Further confidence in our XL/MS results is derived from the striking match between the inter-molecular crosslinks and our biochemical/EM analysis of the BL100 assembly. For example, four crosslinks connect FAAP100 with FANCB, in accord with our finding that FANCB-FAAP100 can exist as an isolated assembly (Figures 3A and 3C). FANCL and FANCB are connected by five crosslinks, which agrees with the observation that the MBP N-terminally fused to FANCL maps in close proximity to the central FANCB homo-dimer (Figure 2B). Conversely, FANCL and FAAP100 are not crosslinked, in agreement with our findings that (1) co-expression of FANCL and FAAP100 does not yield a L100 hetero-dimer (data not shown) and that (2) the two subunits only appear loosely associated in the globular pole in the BL100 particle (Figure 2I). In conclusion, XL/MS analysis, combined with our single-particle EM, provides a compelling architectural description of the dimeric BL100 catalytic assembly of the FA core complex.
Figure 3.

Subunit Architecture of the FANCB-FANCL-FAAP100 Assembly Studied by XL/MS
(A) Inter-subunit crosslinks between the six-protein BL100 complex. The lengths of the subunits correspond to the lengths of the colored rectangles that they represent: blue, FANCB; green, FAAP100; orange, FANCL. Red lines represent intra-molecular crosslinks. Blue lines represent inter-molecular crosslinks.
(B) Crystal structure of the human FANCL central domain with detected lysine residues highlighted in red (PDB: 3ZQS). All detected crosslinks in the FANCL region covered by the crystal structure map involve lysines that are surface exposed and are spaced within a range compatible with the spacer arm in the BS3 crosslinking agent.
(C) Cartoon representation integrating the crosslinking/mass spectrometry and single-particle analysis characterization of the BL100 architecture. The total number of detected inter-\intra-molecular crosslinks is indicated for each cartoon.
See also Table S1.
Two Copies of CEF Can Associate with the BL100 Dimer
We have, so far, established that the BL100 catalytic assembly of the FA core complex forms a homo-dimer. Whether this arrangement extends to the rest of the core complex, or whether the full assembly contains a symmetry mismatch element, remains to be established. Therefore, we decided to purify the CEF assembly and image it in isolation (as an MBP-FANCC fusion derivative to increase the overall molecular mass of the complex) (Figures 4A and S4). (MBP)CEF appears as an electron-dense, tri-lobed entity with no detectable 2-fold symmetry component (Figures 4B and S4). We have then reconstituted a BL100-(MBP)CEF co-assembly (Figures 4C and S4) and found that particles maintain the characteristic dimeric architecture of BL100 and are decorated with either one or two CEF additional densities, connected to the globular poles in the bow-tie structure (Figures 4D and S4). Therefore, binding of two copies of CEF to the BL100 complex is physically possible, indicating that the BL100 dimer acts as an organizing center for other core complex components. Whether two copies of AG20 are found in the full FA core complex remains to be established and will be the focus of future studies.
Figure 4.

The FANCC-FANCE-FANCF Complex and Its Interaction with FANCB-FANCL-FAAP100
(A) Coomassie-stained SDS-PAGE gel of the CEF complex, containing an MBP N-terminally fused to FANCC.
(B) Negative-stain two-dimensional classes of the of the MBP-CEF complex. Box size is 441 Å × 441 Å.
(C) Coomassie-stained SDS-PAGE gel of the reconstituted and purified MBP-CEF/BL100 assembly.
(D) Negative-stain two-dimensional classes of the reconstituted and purified (MBP)CEF-BL100 complex. Purple arrowheads point at the MBP-CEF density. Due to the flexible nature of the BL100-(MBP)CEF interaction, the MBP-CEF complex appears slightly smaller when BL100 is incorporated. A view of the isolated BL100 is shown, framed in white, for comparison. Box size is 580 Å × 580 Å.
(E) Cartoon representation of the BL100-CEF architecture.
See also Figure S4.
CEF-Co-expressed ID2 Particles Are Compact and Symmetric
In the BL100-CEF complex, CEF projects outward from the BL100 dimerization center and appears perfectly poised to latch onto the outer perimeter of the ID2 mono-ubiquitination substrate (Figure 4D). A direct contact between FANCE and FANCD2 has been previously reported (Gordon et al., 2005, Pace et al., 2002, Polito et al., 2014), and in an accompanying study, we show that human CEF can bind both FANCI and FANCD2 directly (van Twest et al., 2017). Reconstituting a CEF-ID2 assembly could, therefore, provide key insights into the mechanism of the FANCI-FANCD2 mono-ubiquitination by the FA core complex.
Xenopus laevis ID2 can be expressed and purified to homogeneity and efficiently processed in the mono-ubiquitination reaction by the human FA core complex (van Twest et al., 2017). To gain mechanistic insights into ID2 mono-ubiquitination by the human FA core complex, we chose to produce Xenopus ID2 for single-particle EM studies (Figure S5). In agreement with recently published work (Liang et al., 2016), we observed that ID2 forms an asymmetric, flexible, dimeric complex, which resists averaging and high-resolution structure determination (Figure S5). We hypothesized that CEF binding might stabilize the ID2 assembly, and we co-expressed the five proteins in the attempt to purify an ID2-CEF complex by affinity purification via a FLAG-tagged FANCI subunit. This purification strategy, including stringent ion exchange and size exclusion chromatography (SEC) steps, yielded an apparently stoichiometric ID2 assembly, according to Coomassie-stained SDS-PAGE gel analysis, but only trace amounts of the CEF assembly (Figure S6). As expected from inspection of the SDS-PAGE gel, the vast majority of ID2 particles show no sign of CEF co-complex, and no clear evidence for CEF association was detected by inspection of the two-dimensional classes. To our surprise, however, ID2 particles from the CEF co-expression experiments yielded neatly averaging two-dimensional classes (Figure S6). A pseudo-2-fold symmetric end-on view can be recognized in the assembly and perfectly matches the two-dimensional projections of an electron density map generated from the PDB coordinates of the mouse ID2 crystal structure (PDB: 3S4W) (Joo et al., 2011) (Figure 6A). To confirm this unexpected result, we repeated the co-expression experiment using a different CEF derivative, containing a Strep II tag on FANCC (Figure 5A). Single-particle analysis on the reconstituted complex confirms our initial observations (Figures 5B–5D and S6). To obtain a three-dimensional structure of the stabilized ID2 assembly, we generated an initial model using EMAN2, followed by three-dimensional classification, and auto-refined the most populated three-dimensional class to 18.3 Å resolution using RELION (Figure 6B; Figure S7). Rigid-body docking of the mouse atomic structure into the Xenopus ID2 map results in an excellent fit, with a striking match at the hetero-dimerization interface (Figure 6B). Conversely, minor adjustments would be needed in the C-terminal region of the two protomers to perfectly fit the atomic structure into the EM density (although we chose not to perform these local adjustments, given the limited resolution of our three-dimensional EM data). Coherent with a more flexible nature of the FANCI and FANCD2 carboxy-terminal modules, analysis of complex dynamics based on two-dimensional class averages of end-on views indicates that the peripheral lobes in the ID2 dimer are flexible (Movie S3).
Figure 6.

Three-Dimensional Reconstruction of the CEF-Co-expressed FANCI-FANCD2 Assembly
(A) Representative two-dimensional classes of the CEF-co-expressed ID2 complex matched to the two-dimensional projections of the electron density map generated from the ID2 crystallographic atomic coordinates (PDB: 3S4W). Box size is 350 Å × 350 Å.
(B) Three orientations and a cut-through view of the X. laevis ID2 EM structure with docked mouse ID2 crystal structure. The fit is excellent, in particular, at the dimerization interface. Lysines targeted for mono-ubiquitination are highlighted in red (residue numbering refers to the mouse proteins, and the corresponding lysines in the X. laevis ortholog are indicated in parentheses).
See also Figure S7.
Figure 5.

CEF Co-expression Stabilizes the FANCI-FANCD2 Interface
(A) Coomassie-stained SDS-PAGE gel of the ID2 complex, with trace amounts of CEF complex.
(B) Representative negative-stain micrograph of the ID2 assembly. Scale bar, 100 nm.
(C) Two-dimensional class averages of the CEF-stabilized ID2 assembly. Box size is 441 Å × 441 Å.
(D) CEF has a structural role in reshaping the ID2 interface, summarized in a cartoon.
See also Figures S5 and S6.
In summary, our characterization of the ID2-CEF interaction reveals that the two protein assemblies do not form a stoichiometric complex, at least in the experimental conditions tested. Remarkably, however, CEF co-expression alters the configuration of the ID2 dimer, stabilizing the dimerization interface and locking the assembly in a pseudo-2-fold symmetric structure (Figure 5D). This unexpected observation has key implications for the mechanism of ID2 mono-ubiquitination and suggests a role for the CEF assembly of the FA core complex.
Discussion
Because the two strands of DNA are antiparallel (Watson and Crick, 1953), the factors that spatially organize, cut, or open the double helix are often arranged as symmetric dimers. For example, a dyad element can be found in the protein core of the nucleosome (the building block of chromatin in eukaryotic cells) (Richmond et al., 1984), in DNA integrases (which concertedly cut the two DNA strands to insert foreign DNA) (Engelman and Cherepanov, 2014), or in DNA helicases that unwind the double helix (Li et al., 2015). This 2-fold symmetric architecture can then be propagated to a subsequent set of proteins that physically interact with the DNA-binding dimer. A paradigm for this concept is found in the eukaryotic origin of DNA replication, where two replisomes are built symmetrically around a dimer of ring-shaped helicases (Araki, 2016, Costa et al., 2014, Deegan et al., 2016) by a set of regulatory and chaperone proteins that are, in turn, symmetric dimers (Itou et al., 2015).
In our study, we report one additional example of how a 2-fold symmetric enzyme modifies a dimeric DNA interactor. In this case, a homo-dimeric regulator protein acts symmetrically on a hetero-dimeric assembly. In the FA/BRCA ICL repair pathway, the FANCI-FANCD2 dimer binds the double helix at the damaged site and acts as a landing platform that recruits a set of DNA-repair proteins for ICL processing (Ceccaldi et al., 2016). FANCI and FANCD2 are structural homologs that can arrange in a pseudo-2-fold-symmetric assembly (Joo et al., 2011). Recruitment of the DNA-repair machinery requires mono-ubiquitination of both factors (Smogorzewska et al., 2007). This function is provided by the FA core complex, an enzyme assembly whose architecture was previously unknown (Ceccaldi et al., 2016, Walden and Deans, 2014). Here, we showed that the trimeric center of the FA core complex (BL100) exists as a symmetric homo-dimer (Figure 2C). FANCB and FAAP100 play an architectural role, orienting the two E3 ligases in an ideal position to symmetrically target the FANCI and FANCD2 ubiquitination sites (Figure 2I). In particular, FANCB plays a key structural function, being the dimerization factor. We note that FANCB is the only protomer in the BL100 catalytic module that contains disease-associated missense mutations, emphasizing the key role of this factor in the assembly of a functional FA core complex (Chandrasekharappa et al., 2013) (http://databases.lovd.nl/shared/variants/FANCB/unique?search_var_status=%3D%22Marked%22|%3D%22Public%22). Inspection of EM two-dimensional class averages reveals that the BL100 particle is dynamic, with the two catalytic poles of the complex free to tilt and stretch with respect to the fixed FANCB dimerization center (Figure S1; Movie S1). Remarkably, this movement occurs symmetrically across the particle (when the upper lobe moves to the left, the lower lobe moves to the right, and vice versa). If the catalytic subunit FANCL is omitted, however, the two external lobes in the particle move independently of each other (Figure S3; Movie S2). Although the functional significance of this concerted movement is unclear, it is evident that each of the two catalytic subunits in the assembly must be able to sense the state of the symmetry mate in the assembly, and this allosteric communication must be mediated by the FANCB dimerization center. In summary, our study provides the first insights into the mechanics of this dimeric E3 ligase super-assembly and starts to explain how the FANCL mono-ubiquitination activity can be stimulated by the FANCB and FAAP100 architectural factors (Rajendra et al., 2014).
The BL100 assembly also functions as the organizing center of the FA core complex, providing a dimerization scaffold for the recruitment of a set of two CEF trimeric assemblies (Figures 4D and S4). The FANCE subunit of CEF has been previously reported to interact with the ID2 assembly (Gordon et al., 2005, Pace et al., 2002, Polito et al., 2014), and our structural data suggest that CEF might act as a molecular matchmaker to bridge between the ID2 mono-ubiquitination substrate and the BL100 E3 ligase machinery. Previous structural studies suggest that the ID2 dimer might exist in different configurations during the ubiquitination reaction. For example, crystallization of the murine complex captured a symmetric, compact state of the ID2 assembly, with the ubiquitination sites mapping at the dimerization interface, probably impairing access to the ubiquitination machinery (Joo et al., 2011). Conversely, a cryo-EM study of the human ID2 shows that the complex is more flexible (poorly averaged) in solution (Liang et al., 2016). In agreement with these recent findings, we show that the intact Xenopus ID2 complex expressed in isolation cannot be captured in discrete conformational states (two-dimensional images do not average) (Figure S5). In contrast, the co-expression of CEF with ID2 appears to stabilize a symmetric state of ID2, which is highly reminiscent of the form captured in the crystal structure (Figures 5C and 6; Movie S3). This hints at a possible role for CEF in manipulating the ID2 substrate to facilitate symmetric mono-ubiquitination of the two related protomers (Figure 7). Biochemical analysis of the reconstituted ID2 mono-ubiquitination reaction by the FA core complex supports our suggestion that the CEF assembly acts as a nexus between the BL100 enzyme and the ID2 substrate while also reconfiguring the ID2 structure (van Twest et al., 2017). Whereas the isolated BL100 assembly solely ubiquitinates the FANCD2 subunit, the presence of CEF significantly stimulates FANCI mono-ubiquitination, necessary for triggering initiation of the FA/BRCA ICL repair pathway. How BL100-CEF and ID2 interact and influence one another remains to be explained at a molecular level. We note that a disease-associated mutation on FANCE (R371W), predicted to destabilize the FANCE fold, abrogates the interaction with FANCD2 in a yeast two-hybrid study. The same Arg-to-Trp point mutation on FANCD2 (R302W), mapping within the outer perimeter of the N-terminal ID2 dimerization module, likewise abrogates the FANCE-D2 interaction (Joo et al., 2011, Nookala et al., 2007, Timmers et al., 2001). Such elements likely play an important role in the CEF-mediated stabilization of the ID2 substrate during the mono-ubiquitination reaction catalyzed by the homo-dimeric BL100 catalytic module. Notably, in addition to interacting with FANCE, the same R302 element in FANCD2 has been previously implicated in mediating a histone H3 contact (Sato et al., 2012). How the ID2 mono-ubiquitination reaction occurs in the context of chromatinized DNA is an exciting frontier topic.
Figure 7.

Molecular Model for the Mechanism of FANCI-FANCD2 Mono-ubiquitination by the FA Core Complex
Conclusions
Several outstanding questions will need to be addressed to understand the mechanism of activation of the FA/BRCA ICL repair pathway. For example: how do disease mutations affect the structure and function of the FA core complex? What is the molecular mechanism for the formation of the FA core complex in cells? How does DNA influence ID2 mono-ubiquitination? Our discovery that the FA core complex is a symmetric dimer changes the way we think about these problems.
Experimental Procedures
Construction of Baculoviruses
Baculoviruses were constructed as detailed in the Supplemental Information.
Protein Expression and Purification
For the expression of the FANCB-FANCL-FAAP100 complex and the isolated FAAP100, 1 L of Sf9 cells at a titer of 2.0 × 106 cells per milliliter were suspended in 4 L spinner flasks (Corning) and infected with baculovirus at an MOI of 2 for 72 hr at 27°C. Cells were collected post-infection by centrifugation (5,000 rpm, 4°C, 15 min), washed in PBS buffer supplemented with 5 mM MgCl2, and resuspended in lysis buffer (25 mM HEPES NaOH [pH 7.6], 15 mM NaCl, 0.02% Tween 20, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 2 mM MgCl2, 2 mM 2-BME, 0.4 mM PMSF, and Roche protease inhibitor cocktail). The suspension was lysed by Dounce homogenization (40 strokes, tight pestle), and NaCl was added to a final concentration of 250 mM. The lysate was cleared by centrifugation (15000 rpm, 4°C, 35 min), and the supernatant was incubated (3 hr, 4°C) with anti-FLAG M2 affinity resin (Sigma Aldrich) pre-equilibrated with FLAG washing buffer (25 mM HEPES NaOH [pH 8.0], 200 mM NaCl, 0.02% Tween 20, 10% glycerol, 1 mM EDTA, 1 mM EGTA, and Roche protease inhibitor cocktail). After wash, the sample was eluted with FLAG elution buffer (25 mM HEPES NaOH [pH 8.0], 100 mM NaCl, 0.02% Tween 20, 5% glycerol, 1 mM EDTA, 1 mM EGTA, 0.3 mg/mL FLAG peptide, and Roche protease inhibitor cocktail). Elution fractions were combined and loaded onto a Mono Q 5/50 GL for ion-exchange chromatography (GE Healthcare). The column was washed with Q-Wash buffer (25 mM HEPES NaOH [pH 8.0], 150 mM NaCl, 5% glycerol, 1 mM EDTA, 1 mM EGTA, 0.5 mM TCEP, and Roche protease inhibitor cocktail) and eluted over a 150-mM to 650-mM NaCl linear gradient in the same buffer. The peak fraction from cation-exchange chromatography was loaded onto a Superose 6 PC 3.2/30 column (GE Healthcare) operating on an ÄKTAmicro HPLC (high-performance liquid chromatography) system (GE Healthcare) and was equilibrated in 25 mM HEPES NaOH (pH 8.0), 250 mM NaCl, and 1 mM TCEP. The proteins were separated by SDS-PAGE, using 4%–12% Bis-Tris gels (Invitrogen) running for 50 min at 150 V in MOPS buffer or using 3%–8% Tris-acetate gels (Invitrogen) running for 60 min at 150 V in Tris-acetate buffer. Protein identities were confirmed by MS. Protein concentration was determined by measuring absorbance at 280 nm (A280nm) using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies).
Expression of the FANCB-FAAP100 and FANCB-(MBP)FANCL-FAAP100 complexes were performed using the FANCB-FANCL-FAAP100 protocol; however, infection was performed using an MOI of 1 for 72 hr at 27°C. Protein extraction, purification, and identification were performed as described earlier. Freshly purified samples (BL100 at 1 mg/mL, B100 at 0.25 mg/mL, and FAAP100 at 1.5 mg/mL) were immediately used for negative-stain EM or cryo-EM experiments.
Expression and purification of (MBP)FANCC-FANCE-FANCF was performed as follows. 1 L of Sf9 cells at a titer of 1.0 × 106 cells per milliliter was suspended in 4-L spinner flasks (Corning) and infected with baculovirus at an MOI of 1 for 72 hr at 27°C. Cells were collected post-infection by centrifugation (5,000 rpm, 4°C, 15 min), washed in PBS buffer supplemented with 5 mM MgCl2, and resuspended in lysis buffer (25 mM HEPES NaOH [pH 7.6], 15 mM NaCl, 0.02% Tween 20, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 2 mM MgCl2, 15 mM 2-BME, 0.6 mM PMSF, and Roche protease inhibitor cocktail). The suspension was lysed by Dounce homogenization (40 strokes, tight pestle), and NaCl was added to a final concentration of 200 mM. The lysate was cleared by centrifugation (15,000 rpm, 4°C, 35 min), and the supernatant was incubated (2 hr, 4°C) with Amylose resin (New England Biolabs) pre-equilibrated with Amylose wash buffer (25 mM HEPES NaOH [pH 8.0], 150 mM NaCl, 0.02% Tween 20, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 0.25 mM TCEP, and Roche protease inhibitor cocktail). After extensive wash, the sample was eluted by ligand competition using the Amylose wash buffer supplemented with 20 mM maltose (Sigma). Peak fractions were pooled and loaded onto a Mono Q 5/50 GL for ion-exchange chromatography (GE Healthcare). The column was washed with buffer Q-A (50 mM HEPES NaOH [pH 8.0], 150 mM NaCl, 5% glycerol, 1 mM EDTA, and 0.5 mM TCEP) and eluted over a linear salt gradient with buffer Q-B (50 mM HEPES NaOH [pH 8.0], 1 M NaCl, 5% glycerol, 1 mM EDTA, and 0.5 mM TCEP). Peak fraction was successively loaded onto a Superose 6 PC 3.2/30 column (GE Healthcare) operating on an ÄKTAmicro HPLC system (GE Healthcare) and separated by SEC in 25 mM HEPES NaOH (pH 8.0), 100 mM NaCl, and 2 mM DTT. Peak fraction was analyzed by SDS-PAGE. Instant Blue SafeStain-stained (Expedeon) bands were identified by MS. Sample concentration was measured by A280nm using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies) and the (MBP)CEF theoretical extinction coefficient. Freshly purified (MBP)CEF sample at a final concentration of 0.5 mg/mL was immediately used for preparation of negative-stain EM grids or reconstitution with freshly purified BL100 sample.
Expression and purification of the FANCI-FANCD2 complex was performed as follows. Briefly, 1 L of Sf9 cells at a titer of 2.0 × 106 cells per milliliter was suspended in 4-L spinner flasks (Corning) and co-infected with the two baculoviruses, each at MOI of 1, for 72 hr at 27°C. Following the co-infection, cells were collected by centrifugation (5,000 rpm, 4°C, 15 min) and washed in PBS buffer supplemented with 5 mM MgCl2. Cells were resuspended in lysis buffer (25 mM HEPES NaOH [pH 7.6], 15 mM NaCl, 0.02% Tween 20, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 2 mM MgCl2, 2 mM 2-BME, 0.4 mM PMSF, and Roche protease inhibitor cocktail) and lysed by Dounce homogenization (40 strokes, tight pestle). To preserve complex integrity, the lysate was added with NaCl to a final concentration of 150 mM and cleared by centrifugation (15,000 rpm, 4°C, 35 min). The supernatant was collected and incubated (3 hr, 4°C) with anti-FLAG M2 affinity resin (Sigma-Aldrich) pre-equilibrated in FLAG washing buffer (25 mM HEPES NaOH [pH 8.0], 120 mM NaCl, 0.02% Tween 20, 10% glycerol, 1 mM EDTA, 1 mM EGTA, and Roche protease inhibitor cocktail). After extensive wash, the proteins were eluted by incubation of the resin in FLAG elution buffer (25 mM HEPES NaOH [pH 8.0], 120 mM NaCl, 1 mM EDTA, 0.3 mg/mL FLAG peptide, and Roche protease inhibitor cocktail). Peak fractions were combined and loaded onto a Mono Q 5/50 GL(GE Healthcare) column running in buffer Q-A (25 mM HEPES NaOH [pH 8.0], 100 mM NaCl, 5% glycerol, 1 mM EDTA, and 0.25 mM TCEP). Elution of the sample was performed over a linear salt gradient with buffer Q-B (25 mM HEPES NaOH pH 8.0], 750 mM NaCl, 5% glycerol, 1 mM EDTA, and 0.5 mM TCEP). To increase sample homogeneity, the complex was further purified by pull-down of the Strep-tag II on FANCD2. Peak fraction from ion-exchange chromatography was incubated for 2 hr at 4°C with Strep-tactin Superflow plus resin (QIAGEN). The resin was washed in Strep-wash buffer (25 mM HEPES NaOH [pH 8.0], 120 mM NaCl, 10% glycerol, 1 mM EDTA and 0.25 mM TCEP) and eluted by incubation with Strep-wash buffer supplemented with 25 mM desthiobiotin. Fractions containing stoichiometric I-D2 complex were pooled and concentrated on Amicon Ultra MWCO 10K centrifugal filters (Millipore). Concentrated sample was separated by size exclusion on a Superdex 200 PC 3.2/30 column (GE Healthcare) operating on an ÄKTAmicro HPLC system (GE Healthcare) running in 25 mM HEPES NaOH (pH 8.0), 120 mM NaCl, and 1 mM TCEP. Elution peak was analyzed by SDS-PAGE followed by Instant Blue SafeStain staining (Expedeon). Band identity was confirmed by MS. Freshly purified ID2 sample at a final concentration of 0.25 mg/mL was immediately used for preparation of negative-stain EM grids.
Co-expression of FANCI-FANCD2-FANCC-FANCE-FANCF was obtained by infecting Sf9 cells with baculoviruses encoding for FANCI, FANCD2, and either MBP-tagged or StrepII-tagged CEF. Briefly, 1 L Sf9 cells at a titer of 2.0 × 106 cells per milliliter were co-infected with baculoviruses encoding for FLAG-FANCI, StrepII-FANCD2, and (MBP)CEF (or (StrepII)CEF), each at a MOI of 1. Cells were incubated for 72 hr at 27°C. Protein extraction, purification, and identification were carried out as previously described for the ID2 complex. The ID2-(StrepII)CEF sample purified from ion-exchange chromatography and the ID2-(MBP)CEF sample purified from SEC were used for preparation of EM grids by negative stain immediately after purification.
Reconstitution of the BL100-(MBP)CEF Complex
The reconstitution of the FANCB-FANCL-FAAP100-(MBP)FANCC-FANCE-FANCF complex was performed by mixing equal volumes of a freshly purified 1-μM solution of BL100 and a 3-μM solution of CEF (resulting in a molar ratio of 1:3) and loaded on a 30K MWCO Slide-a-Lyzer dialysis cassette (Thermo Scientific). The protein mix was first dialyzed for 1 hr at 4°C in Buffer 500 (25 mM Tris-HCl [pH 8.0], 500 mM NaCl, 5% glycerol, and 1 mM DTT), then for 2 hr in Buffer 350 (25 mM Tris-HCl [pH 8.0], 350 mM NaCl, 5% glycerol, and 1 mM DTT), and finally overnight in Buffer 100 (25 mM Tris-HCl [pH 8.0], 100 mM NaCl, 5% glycerol, and 1 mM DTT). The sample was recovered from the dialysis cassette and loaded onto a Superose 6 PC 3.2/30 column (GE Healthcare) operating on a ÄKTAmicro FPLC (fast protein liquid chromatography) system (GE Healthcare) equilibrated in 25 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 2 mM DTT. In control experiments, isolated dialyzed BL100 and (MBP)CEF complexes were also subject to SEC as described for the reconstituted samples. Samples were analyzed by SDS-PAGE on 3%–8% Criterion XT Tris-Acetate gels (Bio-Rad) followed by Instant Blue SafeStain staining (Expedeon). Freshly purified samples were immediately used for the preparation of EM grids.
Negative-Stain EM Sample Preparation
A 2- to 6-nm-thick carbon layer was evaporated onto freshly cleaved mica sheets (EMS) using a Q150TE carbon coater (Quorum Technologies). Carbon was incubated overnight at 50°C before floating on 600-mesh copper grids (Agar Scientific) or Quantifoil R1.2/1.3 300-mesh copper grids. Prior to sample incubation, the carbon-coated grid was glow discharged for 30 s at 45 mA using a 100× glow discharger (EMS). Serial dilutions of each protein sample were performed to achieve optimal particle concentration on the EM grid (final concentrations ranged from 30 to 5 μg/mL). A 4-μL drop of the freshly purified sample solution was applied onto the glow-discharged grid and incubated for 1 min. Approximately 2 μL of sample solution was absorbed by gentle side blotting, and the grid was immediately stained with four subsequent applications onto 4× 50-μL drops of a 2% (wt/vol) uranyl formate solution with gentle stirring for 8 s. After staining, the grid was blotted dry and stored at room temperature prior to imaging.
Negative-Stain EM Data Collection
Particles of reconstituted BL100-(MBP)CEF, isolated (MBP)CEF, isolated ID2, and ID2 co-expressed with (MBP)CEF were imaged on a Tecnai G2 Spirit LaB6 transmission electron microscope (TEM; FEI) operating at 120 keV (Electron Microscopy Science Technology Platform, The Francis Crick Institute). Images were manually acquired on a UltraScan 1000 2k × 2k charge-coupled device (CCD) camera (Gatan) at a nominal magnification of 30,000× (corresponding to a pixel size of 3.45 Å at the specimen level). Particles of BL100, B100, B-(MBP)L100, isolated FAAP100, and ID2 co-expressed with (StrepII)CEF samples were imaged using a JEM-2100 LaB6 electron microscope (JEOL) operated at 120 kV. Images were recorded on a UltraScan 4k × 4k CCD camera (Gatan) at a nominal magnification of 42,000× corresponding to a pixel size of 2.73 Å at the specimen level. On both microscopes, images were collected in low-dose mode (electron dose of 35 e−/Å2) and with defocus values in the range of 0.5 μm to 2.0 μm. For the BL100 and FAAP100 samples, the micrographs were automatically recorded using the JEOL Automated Data Acquisition System (JADAS) with same electron dose and defocus value as the manual collection. A total of 567 images were collected for the BL100 sample, 250 images for the B100 sample, 412 images for the B-(MBP)L100 sample, 540 images for the FAAP100 sample, 277 images for the (MBP)CEF sample, 455 images for the BL100-(MBP)CEF sample,712 images for the ID2 sample, 468 images for the ID2-(MBP)CEF sample, and 447 images for the ID2-(StrepII)CEF sample.
Cryo-EM Sample Preparation and Data Collection
Ultrathin carbon-coated Lacey 300-mesh copper grids (Ted Pella) were glow discharged for 30 s at 45 mA using a 100× glow discharger (EMS) and mounted onto a Vitrobot Mark IV (FEI). A 4-μL drop of freshly purified BL100 at a concentration of 20 μg/mL was applied to the glow-discharged grid. The sample was incubated on the grid for 1 min in 100% humidity at 21°C. After incubation, the grid was double-side blotted for 2.0 s and rapidly plunged in liquid ethane. Cryo-EM data for the BL100 sample were collected on a Titan Krios TEM (FEI) operated at 300 keV and equipped with a back-thinned Falcon II (FEI) direct electron detector at a nominal magnification of 47,000×, resulting in a pixel size of 1.77 Å at the specimen level. A total of 2,798 images were recorded at defocus values in the range of 2.5–4.5 μm and with a cumulative electron dose of 56 e−/Å2 distributed over 26 frames. Beam-induced motion was corrected on each image using the Motioncorr program (Li et al., 2013). Micrographs showing contamination drift, or out-of-range defocus values were discarded.
Single-Particle Image Processing
For each dataset, particles were semi-automatically picked using the EMAN2 package, version 2.12 (Tang et al., 2007). From the negative-stain EM data collection, a total of 63,543 individual particles were extracted from the BL100 dataset, 63,365 particles from the B100 dataset, 51,941 particles from the B-(MBP)L100 dataset, 177,106 particles from the FAAP100 dataset, 36,768 particles from BL100-(MBP)CEF dataset, 47,898 particles from the (MBP)CEF dataset, 95,553 particles from the ID2 dataset, 27,084 particles from the ID2-(MBP)CEF dataset, and 71,295 particles from the ID2-(StrepII)CEF dataset. From the BL100 cryo-EM dataset, a total of 536,754 individual particles were extracted from selected micrographs. The parameters of the contrast transfer function (CTF) were estimated on each micrograph using CTFFIND3 (Mindell and Grigorieff, 2003) for negative-stain data and CTFFIND4 (Rohou and Grigorieff, 2015) for cryo-EM data. Reference-free two-dimensional alignment and averaging was performed using RELION, version 1.4 (Scheres, 2012). For three-dimensional reconstruction of the ID2 complex co-expressed with (StrepII)CEF, an initial three-dimensional volume was generated using the e2inimodel.py program in the EMAN2 package (Tang et al., 2007). The initial volume was used as a reference model for three-dimensional classification and refinement in RELION 1.4 (Scheres, 2012). The atomic model of the FANCI-FANCD2 hetero-dimer (PDB: 3S4W) (Joo et al., 2011) was used for rigid-body fitting into the EM map using UCSF (University of California, San Francisco) Chimera (Pettersen et al., 2004). The refined EM map of the ID2 hetero-dimer has been deposited in the three-dimensional-EM database (http://www.emdatabank.org) with accession number: EMD-3476.
XL/MS
All chemicals were purchased from Sigma at the highest grade possible, unless otherwise stated. BS3 d0 and BS3 d4 were purchased from ThermoFisher Scientific. nano-LC-MS solvents and additives were purchased as LC-MS grade from Fisher Scientific. The XL/MS workflow was conducted as previously described (Leitner et al., 2014). Briefly, BL100 dimer, at a concentration of 0.5 mg/mL in 25 mM HEPES (pH 8), 300 mM NaCl, 10% glycerol, and 0.25 mM tris(2-carboxyethyl)phosphine (TCEP), was reacted with 5 mM BS3 (using a 50:50 ratio of d0 and d4) for 30 min at 37°C. The reaction was quenched by adding 50 mM ammonium bicarbonate for 20 min at 37°C and then dried to completion using vacuum centrifugation. The sample was resuspended at a concentration of 1 mg/mL using 8 M urea, reduced with 2.5 mM TCEP, and alkylated with 5 mM iodoacetamide. The sample was diluted with 50 mM ammonium bicarbonate to reduce the urea concentration to 1 M, and trypsin (Promega) was added at a 1:50 enzyme:substrate ratio and left to digest overnight at 37°C. The solution was then acidified, and peptides were purified using C18 in house solid-phase elution tips (Empore, 3M).
For size exclusion fractionation, the peptides were resuspended in SEC buffer (70% H2O, 30% acetonitrile, and 0.1% trifluoroacetic acid), and chromatographically separated using an ÄKTAmicro (GE Healthcare) with a Superdex Peptide PC 3.2/30 column (GE Healthcare) and detected using 215 nm absorbance. The flow rate was set to 50 μL/min, and 100 μL fractions were collected and dried to completion using vacuum centrifugation. For nano-LC-MS acquisition, the SEC fractions were resuspended into 30 μL of 0.1% trifluoroacetic acid in H2O, and 1 μL was injected and chromatographically resolved using an Ultimate 3000 RSLCnano (Dionex) with an EASY-Spray column (2-μm particles, PepMap C18, 100-Å pore size, 50 cm × 75 μm ID) (Thermo Scientific). A flow rate of 0.25 μL/min was used, starting at 98% mobile A (0.1% formic acid, 5% DMSO, 95% H2O) and 2% mobile B (0.1% formic acid, 5% DMSO, 80% acetonitrile, 10% H2O). Over 80 min, Mobile B was then increased to 40% followed by a further increase to 90% over 10 min. Spectra were acquired in real time using an LTQ Orbitrap Velos Mass Spectrometer (Thermo Scientific) with a 400 to 2,000 m/z (mass-to-charge ratio) acquisition window. The top ten most intense ions with a charge state of +3 or greater were then selected for tandem MS (MS/MS) using data-dependent acquisition and CID (collision-induced dissociation) fragmentation (at 35% normalized collision energy). A dynamic exclusion list, with a repeat count of 1, repeat duration of 20 s, exclusion list size of 100, and exclusion duration of 30 s, was used to prevent repeat sampling. Data analysis was carried out using the xQuest/xProphet (v2.1.1) workflow. The data were searched for BS3 mono-links (+156.079 Da for water quench, +155.096 Da for ammonium quench), intra-links, and intra- and inter-protein crosslinks (+138.068 Da) on lysine residues, using isotopic pairing with a mass shift of +4.025 Da for BS3 d4 ions, in addition to a fixed carbamidomethylation modification (+57.021 Da) and variable methionine oxidation (+ 15.994 Da). The data were searched using a 10-ppm mass tolerance for charge states +3 or above. xProphet was then used, and peptides with a false discovery rate (FDR) below 5% were then manually assessed for adequate signal intensity and peak assignment.
Author Contributions
P.S., A.J.D., and A.C. designed the study. A.J.D., V.J.M., S.v.T., and F.S. constructed all baculoviruses and performed preliminary protein preparations. P.S. optimized purification protocols for structural studies, prepared all proteins and EM grids, collected EM data (with L.R.), and performed all image analysis under the supervision of A.C. A.B. performed the XL/MS analysis supervised by B.S. P.S. and A.C. wrote the paper, with input from A.D. and the other authors.
Acknowledgments
We would like to thank J. Gannon, A. Davies, and Cell Services at The Francis Crick Institute for help with the baculovirus work; and R. Carzaniga, L. Collinson, and K. MacLellan-Gibson for technical support with EM. We acknowledge Diamond for access and support of the cryo-EM facilities at eBIC, proposal EM14266, funded by Wellcome Trust/MRC/BBSRC. We thank A. Siebert, D. Clare, and S. Welsch for help with the Titan Krios. A.J.D. is funded by the National Breast Cancer Foundation (Australia), the Victorian Cancer Agency, the Fanconi Anemia Research Fund, the Victorian Government’s OIS Program, and the Cancer Council of Victoria. Research in the A.C. lab is supported by The Francis Crick Institute (which receives its core funding from Cancer Research UK, the UK Medical Research Council, and the Wellcome Trust).
Published: December 13, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, one table, and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.11.013.
Supplemental Information
{kind=link}
{kind=link}
{kind=link}
References
- Alpi A.F., Pace P.E., Babu M.M., Patel K.J. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Araki H. Elucidating the DDK-dependent step in replication initiation. EMBO J. 2016;35:907–908. doi: 10.15252/embj.201694227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzovic P.S., Rajagopal P., Hoyt D.W., King M.C., Klevit R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- Bui K.H., von Appen A., DiGuilio A.L., Ori A., Sparks L., Mackmull M.T., Bock T., Hagen W., Andrés-Pons A., Glavy J.S., Beck M. Integrated structural analysis of the human nuclear pore complex scaffold. Cell. 2013;155:1233–1243. doi: 10.1016/j.cell.2013.10.055. [DOI] [PubMed] [Google Scholar]
- Ceccaldi R., Sarangi P., D’Andrea A.D. The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa S.C., Lach F.P., Kimble D.C., Kamat A., Teer J.K., Donovan F.X., Flynn E., Sen S.K., Thongthip S., Sanborn E., NISC Comparative Sequencing Program Massively parallel sequencing, aCGH, and RNA-seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood. 2013;121:e138–e148. doi: 10.1182/blood-2012-12-474585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Speck C., Wendel P., Tang C., Stillman B., Li H. The architecture of the DNA replication origin recognition complex in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 2008;105:10326–10331. doi: 10.1073/pnas.0803829105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Ling C., Coulthard R., Yan Z., Xue Y., Meetei A.R., Laghmani H., Joenje H., McDonald N., de Winter J.P. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Cole A.R., Lewis L.P., Walden H. The structure of the catalytic subunit FANCL of the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2010;17:294–298. doi: 10.1038/nsmb.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A., Renault L., Swuec P., Petojevic T., Pesavento J.J., Ilves I., MacLellan-Gibson K., Fleck R.A., Botchan M.R., Berger J.M. DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. eLife. 2014;3:e03273. doi: 10.7554/eLife.03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulthard R., Deans A.J., Swuec P., Bowles M., Costa A., West S.C., McDonald N.Q. Architecture and DNA recognition elements of the Fanconi anemia FANCM-FAAP24 complex. Structure. 2013;21:1648–1658. doi: 10.1016/j.str.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans A.J., West S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan T.D., Yeeles J.T., Diffley J.F. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016;35:961–973. doi: 10.15252/embj.201593552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A., Cherepanov P. Retroviral integrase structure and DNA recombination mechanism. Microbiol. Spectr. 2014;2 doi: 10.1128/microbiolspec.MDNA3-0024-2014. MDNA3-0024-2014. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I., Taniguchi T., Ganesan S., Meyn M.S., Timmers C., Hejna J., Grompe M., D’Andrea A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gordon S.M., Alon N., Buchwald M. FANCC, FANCE, and FANCD2 form a ternary complex essential to the integrity of the Fanconi anemia DNA damage response pathway. J. Biol. Chem. 2005;280:36118–36125. doi: 10.1074/jbc.M507758200. [DOI] [PubMed] [Google Scholar]
- Hodson C., Cole A.R., Lewis L.P., Miles J.A., Purkiss A., Walden H. Structural analysis of human FANCL, the E3 ligase in the Fanconi anemia pathway. J. Biol. Chem. 2011;286:32628–32637. doi: 10.1074/jbc.M111.244632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson C., Purkiss A., Miles J.A., Walden H. Structure of the human FANCL RING-Ube2T complex reveals determinants of cognate E3-E2 selection. Structure. 2014;22:337–344. doi: 10.1016/j.str.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Leung J.W., Lowery M., Matsushita N., Wang Y., Shen X., Huong D., Takata M., Chen J., Li L. Modularized functions of the Fanconi anemia core complex. Cell Rep. 2014;7:1849–1857. doi: 10.1016/j.celrep.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itou H., Shirakihara Y., Araki H. The quaternary structure of the eukaryotic DNA replication proteins Sld7 and Sld3. Acta Crystallogr. D Biol. Crystallogr. 2015;71:1649–1656. doi: 10.1107/S1399004715010457. [DOI] [PubMed] [Google Scholar]
- Joo W., Xu G., Persky N.S., Smogorzewska A., Rudge D.G., Buzovetsky O., Elledge S.J., Pavletich N.P. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–316. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.M., Kee Y., Gurtan A., D’Andrea A.D. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P., Räschle M., Smogorzewska A., Enoiu M., Ho T.V., Schärer O.D., Elledge S.J., Walter J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann M.C., Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A., Walzthoeni T., Aebersold R. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 2014;9:120–137. doi: 10.1038/nprot.2013.168. [DOI] [PubMed] [Google Scholar]
- Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Zhai Y., Zhang Y., Li W., Yang M., Lei J., Tye B.K., Gao N. Structure of the eukaryotic MCM complex at 3.8 A. Nature. 2015;524:186–191. doi: 10.1038/nature14685. [DOI] [PubMed] [Google Scholar]
- Liang C.C., Li Z., Lopez-Martinez D., Nicholson W.V., Vénien-Bryan C., Cohn M.A. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat. Commun. 2016;7:12124. doi: 10.1038/ncomms12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew C.W., Sun H., Hunter T., Day C.L. RING domain dimerization is essential for RNF4 function. Biochem. J. 2010;431:23–29. doi: 10.1042/BJ20100957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorick K.L., Jensen J.P., Fang S., Ong A.M., Hatakeyama S., Weissman A.M. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA. 1999;96:11364–11369. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida Y.J., Machida Y., Chen Y., Gurtan A.M., Kupfer G.M., D’Andrea A.D., Dutta A. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol. Cell. 2006;23:589–596. doi: 10.1016/j.molcel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Meetei A.R., de Winter J.P., Medhurst A.L., Wallisch M., Waisfisz Q., van de Vrugt H.J., Oostra A.B., Yan Z., Ling C., Bishop C.E. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- Mindell J.A., Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Nookala R.K., Hussain S., Pellegrini L. Insights into Fanconi anaemia from the structure of human FANCE. Nucleic Acids Res. 2007;35:1638–1648. doi: 10.1093/nar/gkm033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace P., Johnson M., Tan W.M., Mosedale G., Sng C., Hoatlin M., de Winter J., Joenje H., Gergely F., Patel K.J. FANCE: the link between Fanconi anaemia complex assembly and activity. EMBO J. 2002;21:3414–3423. doi: 10.1093/emboj/cdf355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Polito D., Cukras S., Wang X., Spence P., Moreau L., D’Andrea A.D., Kee Y. The carboxyl terminus of FANCE recruits FANCD2 to the Fanconi Anemia (FA) E3 ligase complex to promote the FA DNA repair pathway. J. Biol. Chem. 2014;289:7003–7010. doi: 10.1074/jbc.M113.533976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda J.N., Stoll K.E., Bolton L.J., Brzovic P.S., Klevit R.E. Ubiquitin in motion: structural studies of the ubiquitin-conjugating enzyme∼ubiquitin conjugate. Biochemistry. 2011;50:1624–1633. doi: 10.1021/bi101913m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra E., Oestergaard V.H., Langevin F., Wang M., Dornan G.L., Patel K.J., Passmore L.A. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol. Cell. 2014;54:858–869. doi: 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond T.J., Finch J.T., Rushton B., Rhodes D., Klug A. Structure of the nucleosome core particle at 7 A resolution. Nature. 1984;311:532–537. doi: 10.1038/311532a0. [DOI] [PubMed] [Google Scholar]
- Rohou A., Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Ishiai M., Toda K., Furukoshi S., Osakabe A., Tachiwana H., Takizawa Y., Kagawa W., Kitao H., Dohmae N. Histone chaperone activity of Fanconi anemia proteins, FANCD2 and FANCI, is required for DNA crosslink repair. EMBO J. 2012;31:3524–3536. doi: 10.1038/emboj.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims A.E., Spiteri E., Sims R.J., 3rd, Arita A.G., Lach F.P., Landers T., Wurm M., Freund M., Neveling K., Hanenberg H. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R., 3rd, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D’Andrea A.D., Elledge S.J. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Timmers C., Taniguchi T., Hejna J., Reifsteck C., Lucas L., Bruun D., Thayer M., Cox B., Olson S., D’Andrea A.D. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell. 2001;7:241–248. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- van Twest S., Murphy V.J., Hodson C., Tan W., Swuec P., O’Rourke J.J., Heierhorst J., Crismani W., Deans A.J. Mechanism of ubiquitination and deubiquitination in the Fanconi anemia pathway. Mol. Cell. 2017;65 doi: 10.1016/j.molcel.2016.11.005. Published online December 13, 2016. [DOI] [PubMed] [Google Scholar]
- Walden H., Deans A.J. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- Wang A.T., Smogorzewska A. SnapShot: Fanconi anemia and associated proteins. Cell. 2015;160:354–354.e1. doi: 10.1016/j.cell.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Watson J.D., Crick F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- Yan Z., Delannoy M., Ling C., Daee D., Osman F., Muniandy P.A., Shen X., Oostra A.B., Du H., Steltenpool J. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell. 2010;37:865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.