

SUMMARY
Host-to-host transmission is a critical step for infection. Here we studied transmission of the opportunistic pathogen Streptococcus pneumoniae in an infant mouse model. Transmission from nasally colonized pups required high levels of bacterial shedding in nasal secretions and was temporally correlated with, and dependent upon, the acute inflammatory response. Pneumolysin, a pore-forming cytotoxin and major virulence determinant, was both necessary and sufficient to promote inflammation, which increased shedding and allowed for intralitter transmission. Direct contact between pups was not required for transmission indicating the importance of an environmental reservoir. An additional in vivo effect of pneumolysin was to enhance bacterial survival outside of the host. Our findings provide experimental evidence of a microbial strategy for transit to new hosts and explain why an organism expresses a toxin that damages the host upon which it depends.
In Brief
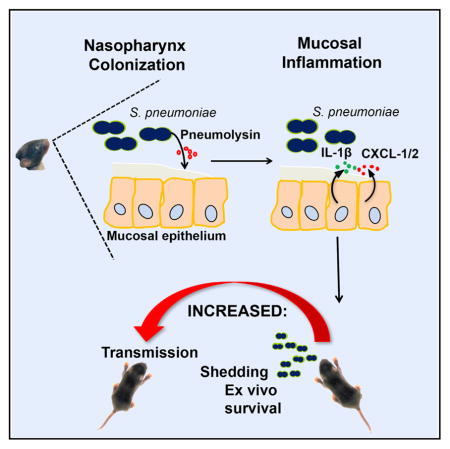
The requirements for host-to-host transmission of the leading respiratory pathogen Streptococcus pneumoniae were examined in an infant mouse model. Shedding of colonizing bacteria by the donor animal is increased by inflammation. The bacterial toxin pneumolysin promotes inflammation, increases bacterial survival after exit from the host, and is required for transmission.
INTRODUCTION
The mucosal surfaces of the airways are colonized by a complex and extensive flora (de Steenhuijsen Piters et al., 2015). Many of these species are highly adapted to and reliant on their mammalian host. These microbes must overcome two fundamental challenges not encountered by organisms inhabiting the gut. Because their niche is not regularly exposed to food consumed by their host, they must depend on and compete with their host, as well as other resident microbes, for limited and required nutrients (Siegel and Weiser, 2015). Extensive investigation into the physiology and metabolism of respiratory tract bacteria has defined many of the ways in which respiratory tract colonizers obtain critical nutrients, such as iron, from within this environment (Bachman and Weiser, 2015). A further challenge is that the healthy host does not regularly expel contents of the airways to allow for the transit of microbes from one host to another. Therefore, the process of transmission for inhabitants of the respiratory tract is likely to be inherently different and less efficient compared to inhabitants of the gut. Although a critical step for organisms that reside within the respiratory tract of their obligate host, there is currently minimal understanding of either microbial or host factors involved in host-to-host transmission.
A paradigm for our understanding of transmission is the opportunistic pathogen Vibrio cholerae. Local production of cholera toxin within the gut lumen stimulates secretions causing profuse diarrhea that promotes transmission through direct fecal-oral contact and by spread of organisms into the environmental reservoir as another source of contagion (Nelson et al., 2009). In contrast, for the leading opportunistic pathogen of the human respiratory tract, Streptococcus pneumoniae (the pneumococcus), person-to-person transmission from its niche in the upper respiratory tract (URT) occurs largely from asymptomatic individuals (carriers), particularly young children, and is thought to require intimate contact with nasal secretions or contaminated surfaces (Hodges et al., 1946; Musher, 2003). Pneumococcal transmission is more common in the setting of concurrent respiratory viral infection, especially during seasonal or pandemic influenza (Gwaltney et al., 1975; Vu et al., 2011). The contribution of influenza to pneumococcal transmission has been modeled in an infant mouse model of co-infection, whereby intranasally (IN) inoculated pups become colonized and transmit the bacterium to their littermates (Diavatopoulos et al., 2010; Richard et al., 2014). There are several effects of influenza that contribute to bacterial transmission in this model. Viral infection induces inflammation that may render the ‘‘recipient pups’’ more susceptible to acquiring the bacteria (Short et al., 2012). Viral inflammation also increases the availability of nutrients, such as sialic acid, resulting in greater proliferation of colonizing pneumococci (Siegel et al., 2014). The density of colonizing pneumococci is also elevated during episodes of viral rhinitis in children (Rodrigues et al., 2013). In the infant mouse model, the higher density of colonizing bacteria, together with increased nasal secretions triggered by influenza, leads to greater numbers of shed organisms exiting from the co-infected ‘‘donor pups’’ (Richard et al., 2014). Analysis of this model suggests that very few organisms succeed in transiting from one host to another, and increased shedding in the setting of influenza A virus (IAV) allows the pneumococcus to overcome the constraints of this tight population bottleneck (Kono et al., 2016).
Pneumococcal colonization of the URT also induces an acute inflammatory response in the infected nasal spaces, albeit more mild than during IAV infection, when modeled in mice (Kadioglu et al., 2008; Weiser, 2010). Several factors are known to contribute to the host response to the pneumococcus. These include the expression of a pore-forming, cholesterol-dependent cytotoxin (pneumolysin [Ply]) and recognition by Toll-like receptors (TLRs), chiefly TLR2 (Matthias et al., 2008; Mitchell and Dalziel, 2014; Zhang et al., 2009). Pneumococci, like other successful opportunistic bacterial pathogens of the URT, stimulate the expression of and modulate the activity of platelet-activating factor (PAF), a potent phospholipid mediator of neutrophil recruitment and function (Hergott et al., 2015). Other proposed inflammatory pathways—such as signaling through TLR3, TLR4, and TLR9 and the cytotoxity of hydrogen peroxide produced during aerobic growth—are of unknown significance during carriage (Albiger et al., 2007; Malley et al., 2003; Rai et al., 2015; Spelmink et al., 2016).
We recently described modifications of the infant mouse model that allows for the study of pneumococcal transmission during mono-infection (Zafar et al., 2016). Since it remains unclear how pneumococci are transmitted in the absence of viral co-infection, in this report, we addressed whether pneumo-coccal-induced inflammation is sufficient to promote its shedding and transmission. Here we show that inflammation induced by pneumococcal colonization, particularly in response to pneumolysin, promotes bacterial shedding that allows for transmission between hosts.
RESULTS
Role of URT Inflammation in Pneumococcal Shedding
Initially, we examined the relationship between pneumococcal shedding and mucosal inflammation in the URT. After colonization was established with streptomycin-resistant mutant of strain TIGR4 (T4S) in 4-day-old C57/BL6J (wild-type [WT]) mice, daily shedding was measured by gently tapping the nares onto an agar surface for quantitative culture (Figure 1A). Shedding peaked between days 5 and 7 of age and was followed until it began to decline at 9 days of age (Figure 1B). Events greater than ~300 colony-forming units (CFUs)/pup were considered a threshold, since in a prior study, intralitter transmission from colonized pups was observed only under conditions in which shedding exceeded this level (Richard et al., 2014). The gradual decrease in shedding over the 5 days post-challenge occurred despite continued robust colonization as assessed by quantitative culture of nasal lavages (Figure 1C). Thus, the pattern of shedding following inoculation could not be attributed to differences in the density of colonizing organisms. Maximal shedding was temporally correlated with acute inflammation as assessed by numbers of neutrophils in nasal lavages, which peaked at age 7 days (Figure 1D). The peak inflammatory response was also characterized by increased URT expression of chemokines (CXCL1 and CXCL2) and the proinflammatory cytokine Interleukin-1β (IL-1β ) that contribute to neutrophil recruitment and activity (Figure 1E). Elevated expression of CXCL2 and IL-1β persisted through age 9 days. Although heavily diluted in the process of collecting URT lavages, increased levels of IL-1β were detectable in colonized pups as measured by a sensitive ELISA (Figure 1F).
Figure 1. Pneumococcal Shedding Correlates with Upper Respiratory Tract Inflammation.

(A) 4-day-old pups were intranasally (IN) inoculated with ~2,000 CFUs S. pneumoniae strain T4S.
(B) Daily shedding was collected and quantified from nasal secretions on the days shown with median values indicated and each symbol representing the CFU observed from a single pup. Statistical analysis compares shedding on ages 6 and 9 days. Dashed line represents the 300 CFU threshold level described in the Results.
(C) Colonization density for T4S in cultures of upper respiratory tract (URT) lavages obtained from pups at the age indicated with the median value shown.
(D) Numbers of neutrophils as determined by flow cytometry (CD45+, CD11b+, and Ly6G+ events) in URT lavages obtained at the age indicated in colonized pups. Values are ±SEM (n = 5–11).
(E) The URT was lavaged with RLT RNA lysis buffer to isolate RNA from the epithelium to make cDNA from T4S colonized pups. Gene expression relative to PBS (mock)-inoculated mice was measured by qRT-PCR for the chemokine/cytokine shown. Values are ±SEM (n = 5–8).
(F) IL-1β measured by ELISA in URT lavages obtained at age 7 days for T4S or PBS (mock)-colonized pups. Values are ±SEM (n = 6–11).
*p < 0.05, **p < 0.01.
Having established a correlation between acute inflammation and above-threshold shedding events, we examined whether modulation of URT inflammation impacts shedding. Pups were treated twice daily with an IN dose of dexamethasone or PBS as vehicle control. Overall mean shedding on days of age 5–9, and in particular events >300 CFUs/pup, was significantly reduced by dexamethasone treatment (Figure 2A) and was not attributable to an effect on colonization density (Figure 2B). The anti-inflammatory effect of dexamethasone treatment was confirmed by lack of upregulation of URT expression of CXCL1, CXCL2, and IL-1β (Figure 2C). Similarly, in the absence of PAF-mediated inflammation, which was tested by comparison of pafr−/−to WT mice, there was also diminished mean shedding without a concomitant effect on colonization (Figures 2D and 2E). The diminished inflammatory response in colonized pafr−/− mice was confirmed by the lack of increased URT expression of CXCL1, CXCL2, or IL-1β compared to mock-infected controls (Figure 2F).
Figure 2. Pneumococcal Shedding Requires URT Inflammation.

(A–C) Pups colonized at age 4 days with T4S were treated IN twice daily with dexamethasone (Dex) or PBS-vehicle control. Daily shedding quantified in secretions from ages 5 to 9 days from nasal secretions with median values indicated and each symbol representing the CFU observed from a single pup on a single day (A). Colonization density in URT lavages obtained from Dex- or PBS-treated pups at age 9 days with the median value shown (B). Gene expression for the Dex-treated relative to PBS-treated pups at age 9 days measured by qRT-PCR for the chemokine/cytokine shown (C). Values are ±SEM (n = 5–7).
(D–F) WT or congenic pafr−/− pups were colonized at age 4 days with T4S. Daily shedding quantified in secretions from ages 5 to 9 days from nasal secretions with median values indicated and each symbol representing the CFU observed from a single pup (D). Colonization density in URT lavages obtained at age 9 days with the median value shown (E). Gene expression in colonized pafr−/− pups at age 9 days relative to PBS (mock)-inoculated pups measured by qRT-PCR for the chemokine/cytokine shown (F). Values are ±SEM (n = 7–9).
Dashed line represents the 300 CFU threshold level described in the Results. **p < 0.01. ****p < 0.0001.
How Pneumococci Stimulate Inflammation and Shedding
We then examined whether pneumococcal products known to stimulate inflammation are involved in increased shedding. Inflammation and the recruitment of neutrophils are impaired in tlr2−/− mice that are unable to sense and respond to pneumococcal lipoproteins (Tomlinson et al., 2014; Zhang et al., 2009). In tlr2−/− mice, mean shedding and the number of high-shedding events were reduced without an effect on colonization density compared to WT controls (Figures 3A and 3B). Without TLR2 signaling, there was no increase in neutrophil numbers in nasal lavages or elevated URT expression of CXCL1, CXCL2, and IL-1β during early colonization (Figures 3C and 3D).
Figure 3. Recognition of Pneumococci and Pneumococcal Products Increases URT Inflammation and Promotes Shedding.

(A–D) WT or congenic tlr2−/− pups were colonized at age 4 days with T4S. Daily shedding quantified in secretions from ages 5 to 9 days from nasal secretions with median values indicated and each symbol representing the CFU observed from a single pup on a single day (A). Dashed line represents the 300 CFU threshold level described in the Results. Colonization density in URT lavages obtained at age 9 days with the median value shown (B). Numbers of neutrophils as determined by flow cytometry (CD45+, CD11b+, and Ly6G+ events) in URT lavages obtained from tlr2−/− or PBS (mock)-inoculated pups at age 7 days (C). Values are ±SEM (n = 8–11). Gene expression in colonized tlr2−/− pups relative to PBS (mock)-inoculated pups at age 7 days measured by qRT-PCR for the chemokine/cytokine shown (D). Values are ±SEM (n = 8).
(E–H) WT pups were colonized with the ply− strain at age 4 days. From ages 4 to 8 days, pups were given a daily IN dose (100 ng/pup) of purified pneumolysin (Ply), a non-pore-forming toxoid with the W433F point mutation (PdB), or PBS-vehicle control. Daily shedding quantified in secretions from ages 5 to 9 days from nasal secretions with median values indicated and each symbol representing the CFU observed from a single pup (E). Colonization density in URT lavages obtained at age 9 days with the median value shown (F). Numbers of neutrophils as determined by flow cytometry (CD45+, CD11b+, and Ly6G+ events) in URT lavages at age 9 days (G). Values are ±SEM (n = 8). Gene expression relative to PBS (mock)-inoculated pups measured by qRT-PCR for the chemokine/cytokine shown at age 9 days (H). Values are ±SEM (n = 10–18).
*p < 0.05, **p < 0.01, ***p < 0.001.
The effect of the pneumolysin toxin was initially tested by daily IN dosing and compared to PBS (vehicle control). In pilot studies, we tested a range of recombinant Ply doses from 20 to 200 ng/day (Figures S1A and S1B, related to Figure 3). Once daily administration of 100 ng/pup was sufficient to increase mean shedding above PBS controls (Figure 3E). Ply treatment increased numbers of neutrophils in nasal lavages and the expression of CXCL2 and IL-1β but had no impact on colonization density (Figures 3F–3H). The requirement for pore formation by Ply was assessed in experiments using a recombinant toxoid containing a W433F mutation affecting conformational changes following membrane insertion (Rossjohn et al., 1998). In contrast to the toxin, the same dose of the toxoid (PdB) did not elevate shedding, neutrophil recruitment, or expression of chemokines/cytokines. Studies with Ply, PdB, and PBS were carried out using a strain carrying an unmarked, in-frame deletion of the pneumolysin gene (ply−) to avoid any confounding effects of endogenous toxin expression.
Experiments described above suggest that Ply expression during colonization could contribute to inflammation and shedding. During the period of peak shedding (days of age 5–7), there was a significantly lower proportion of shedding events of >300 CFUs/pup for the ply− compared to parent strain (Figure 4A). A strain containing the W433F modification in ply leading to expression of a pore-formation-deficient toxin showed reduced shedding. Parental levels of shedding were restored when this mutation was corrected with the native ply gene (ply+). Reduced shedding of the ply− and plyW433F mutants and its restoration in the ply+ strain was not associated with altered colonization density (Figure 4B). Both the ply− and plyW433F mutants failed to stimulate neutrophil recruitment or increased expression of IL-1β unlike the parental strain and corrected mutant (Figures 4C and 4D).
Figure 4. Pneumolysin Is Required for Robust Inflammation and Shedding.

WT pups were colonized at age 4 days with T4S, ply−, a point mutant deficient in pore formation (plyW433F), or a corrected mutant (ply+) and monitored over the period of peak inflammation until age 7 days.
(A) Daily shedding quantified in secretions from nasal secretions with median values indicated and each symbol representing the CFU observed from a single pup on a single day. Horizontal dashed line represents the 300 CFU threshold level described in the Results. T4S and ply− were tested among littermates and the proportion of shedding events >300 CFUs compared for significance using the Fisher’s exact test.
(B) Colonization density in URT lavages obtained with the median value shown.
(C) Numbers of neutrophils as determined by flow cytometry (CD45+, CD11b+, and Ly6G+ events) in URT lavages. (Data for T4S is also shown in Figure 1C and provided here for comparison.) Values are ±SEM (n = 12–19).
(D) Gene expression of IL-1β relative to PBS (mock)-inoculated pups measured by qRT-PCR. Values are ±SEM (n = 10–19).
*p< 0.05, **p < 0.01, ***p < 0.001.
Contribution of Pneumolysin to Pneumococcal Transmission
Next, we determined whether the acute inflammatory response to the products of colonizing pneumococci that promote shedding also allowed for pup-to-pup transmission. We had previously reported that transmission of strain T4S during mono-infection requires a high ratio (~1:1) of ‘‘index’’ pups colonized at day 4 of age to uncolonized ‘‘contact’’ littermates (Zafar et al., 2016). Under these experimental conditions, T4S transmission from index to contact pups was observed in 28% of pups (Figure 5A). In tlr2−/− pups with diminished inflammation and lower average shedding, no transmission events were detected (Figure 5C). Likewise, in the absence of endogenous expression of Ply (ply− mutant) or in a strain expressing a non-pore-forming toxin (plyW433F mutant), there was no detectable transmission. Restoration of the deletion with the full ply gene (ply+) allowed for parental levels of transmission, confirming the requirement for toxin expression (Figure 5A). The increased inflammation and shedding from IN treatment with exogenous Ply to index pups was sufficient to allow for transmission of the ply− mutant, whereas no transmission was seen from the PBS-treated index pup controls (Figure 5B). These findings demonstrated that inflammation induced by its toxin (Ply) is necessary and sufficient to drive pneumococcal transmission.
Figure 5. Pneumolysin Increases Host-to-Host Transmission.

(A) At age 4 days, half the pups in a litter were colonized with the strain indicated (index pups, I) and the other half left uncolonized (contact pups, C). At age 14 days, nasal lavages were obtained to quantify colonization density with each pup, and median values are shown. Intralitter transmission is shown by acquisition of the strain by contact pups.
(B) Index pups colonized with the ply− strain were treated daily from age 4 days with either purified Ply (100 ng/dose) or PBS-vehicle control. For IAV co-infection, both index and contact pups were infected with strain x31 at age 8 days.
(C) Summary of transmission data for the strain indicated. Unless otherwise indicated, the pups were C57/Bl6 (WT). The p value was calculated using the Fisher’s exact test. * in comparison to ply− without IAV. ns, non-significant.
Our results suggested that conditions in which there is elevated URT inflammation could enable transmission in the absence of Ply. This was tested in pups co-infected with IAV at age 8 days (Figure 6A). Following viral inoculation, shedding increased and, although significantly higher for the T4S compared to the ply− mutant, generally exceeded the threshold level for both strains despite similar levels of colonization (Figures 6B and 6C). Accordingly, there was no longer a requirement for Ply expression for pneumococcal transmission in IAV co-infected pups (Figure 5B).
Figure 6. Experiments with IAV Co-infection Showing Increased Shedding with Pneumolysin Expression and the Route of Pup-to-Pup Transmission.

(A) WT pups were colonized at age 4 days with the T4S or ply− strain and at age 8 days co-infected IN with IAV virus strain x31.
(B) Daily shedding was quantified in secretions from ages 9 to 14 days from nasal secretions with median values indicated and each symbol representing the CFU observed from a single pup on a single day. Dashed line represents the 300 CFU threshold level described in the Results. *p < 0.05.
(C) Colonization density in URT lavages obtained with the median value shown.
(D–F) WT index pups in one cage were colonized at age 4 days with T4S. At age 8 days, both index pups and contact pups (housed in a separate cage) were inoculated with IAV. Colonization was measured in cultures of nasal lavages at age 14 days after index and contact pups were switched between cages 2–3 times/day (D), cages containing the bedding material were switched 3 times/day (E), or the dams only were switched between cages (F). Horizontal line indicates limit of detection.
Pneumolysin toxoids have been evaluated as components of protein-based pneumococcal vaccines (Chen et al., 2015; Odutola et al., 2016). Since ~90% of estimated overall efficacy of currently licensed pneumococcal vaccine is due to reduced transmission within the population (Odutola et al., 2016), we tested whether immunization with PdB could block transmission among infant mice. We induced high serum and detectable mucosal IgG titers to PdB inpups by systemic immunization of dams prior to delivery (Figure S1C, related to Figure 5). There was no reduction, however, in transmission among immune pups (Figure 5C).
Mechanisms of Pneumolysin-Mediated Transmission
Secretions containing shed pneumococci from an index pup could directly access the nasal mucosa of a littermate to establish colonization in a contact pup. Alternatively, organisms might be acquired by contact pups from the surface of the dam or via a fomite, such as the cage bedding. To test these later possibilities, we kept colonized index pups with different dams in separate cages from uncolonized contact pups. These experiments were carried out with IAV co-infection because the transmission rate without co-infection was limiting. Three times a day, only the pups were switched between cages so that the index and contact pups were never in direct contact. The transmission rate exceeded 50%, demonstrating the importance of an environmental reservoir for transmission in this model (Figure 6D). Switching of the dams, but not the bedding, was sufficient to observe transmission in the absence of direct contact with colonized index pups (Figures 6E and 6F).
Since transmission via the surface of the dam requires some period that the organism must survive after exiting the nasal mucosa, we tested whether Ply expression in vivo affects viability of the pneumococcus when tested ex vivo. Colonizing pneumococci were collected from nasal lavages and incubated in PBS at 30° C with viable counts followed over time. Viable counts dropped more rapidly over the first hours in lavages obtained from ply− colonized, compared to T4S colonized, pups (Figure 7A). Restoration of the native ply gene (ply+ strain) increased ex vivo survivability to a level equivalent to T4S, confirming the role of Ply expression. In contrast, for pneumococci grown in vitro in nutrient broth, collected, and then followed for their viability in PBS, survival was more prolonged, and there was no difference between the T4S and ply− strains (Figure 7B). Similarly, daily IN administration of Ply (100 ng/dose) increased ex vivo viability significantly more compared to the PBS- or PdB-treated controls (Figure 7C).
Figure 7. Pneumolysin Expression Promotes Bacterial Survival and Infectivity outside of the Host.

(A–C) To compare ex vivo survival, we incubated pneumococci in PBS and viable counts determined at the times indicated. Prior to incubation in PBS, bacteria were obtained from nasal lavages obtained at age 9 days from mice colonized at age 4 days with the strain indicated (A), broth culture in nutrient medium (B), or nasal lavages at age 9 days from mice colonized at age 4 days with ply− and then treated daily with an IN dose of Ply or PdB (100 ng/day) or PBS control (C). Statistical comparisons are to the group(s) without pneumolysin. *p < 0.05, **p < 0.01.
(D) Nasal lavages obtained from T4S or ply− colonized mice were diluted to the desired density in PBS and incubated for 4 hr at 30° C. Aliquots then used to compare the infectious dose by IN inoculation of 4-day-old pups. Colonization density at age 6 days is shown relative to the inoculum size.
Finally, we tested whether these differences in ex vivo survival affected the ability of pneumococci to establish in the naso-pharynx of a new host. Lavages from T4S or ply− colonized pups were diluted in PBS, incubated for 4 hr, and then used to compare the infectious dose (ID) needed to colonize 4-day-old pups (Figure 7D). The infectious dose (ID50) for ex vivo organisms obtained from T4S or ply− colonized mice was <30 CFUs versus >200 CFUs, respectively. Thus, the expression of Ply improved infectivity for pneumococci surviving outside the host.
DISCUSSION
The success of the pneumococcus depends on its commensal relationship with its obligate human host, since none of the multiple invasive diseases it causes facilitate its transmission between individuals (Musher, 2003). Colonization studies in mice have shown that the increased inflammation induced by its sole toxin, pneumolysin, accelerates the eventual clearance of the organism from its niche on the mucosal surface of the URT (Das et al., 2014; Matthias et al., 2008; van Rossum et al., 2005). This raises the question of why an organism dependent on a commensal lifestyle expresses a toxin that is both damaging to its obligate host and promotes its clearance (Weiser, 2010). This report demonstrates the importance of Ply, long recognized as one of the major virulence factors of the pneumococcus, to its ability to transit between hosts (Kadioglu et al., 2008; Mitchell and Dalziel, 2014). We propose that the benefit to the organism of a higher rate of transmission compensates for the non-beneficial within-host effects of the toxin.
Our findings suggest that it is the inflammatory response to Ply, through its potent lytic, pore-forming function, that mediates its effect on transmission. Although we could not quantify secretions directly, our results showed that shedding of the organism to levels permissive for transmission is increased by inflammation and that Ply promotes inflammation and is both necessary and sufficient for levels of shedding that allow for transmission. The effects of Ply were apparent despite the variability of the shedding assay, as during pneumococcal mono-infection, non-pore-forming mutants showed fewer high-shedding events, and during IAV co-infection, mean shedding was lower in the absence of Ply. For transmission, Ply expression was a requirement unless complemented by exogenous Ply or another source of URT inflammation, such as concurrent IAV infection.
Several aspects of Ply-dependent inflammation merit further comment. Ply is unusual for a bacterial toxin because it lacks an N-terminal signal sequence or known secretion mechanism, leaving it unclear how it is released from the bacterial cytosol to access its mammalian target (Price et al., 2012). We have suggested that the release of Ply occurs primarily within professional phagocytes, following bacterial uptake through the degradative activity of lysozyme and other components of the phagolysosomes (Davis et al., 2011; Lemon and Weiser, 2015). In this manner, toxin release and pore formation from within the phagolysosome causes cytosolic access of bacterial products that trigger inflammasome activation and eventual pro-inflammatory death of the phagocyte. Thus, the organism may utilize the cellular host immune response that is capable of eliminating it as a signal of a hostile environment and means of inducing secretions that facilitate its exit and establishment in a new more hospitable host.
The nature of specific host pathways that increase URT secretions consisting mostly of fluid and mucus in response to pro-inflammatory stimuli remain incompletely understood. In this study, we followed the influx of neutrophils and chemokines involved in their recruitment since shedding was temporally correlated with the acute inflammatory response. It is technically challenging to completely eliminate neutrophils at mucosal surfaces using standard depletion protocols and so the requirement for these cells on the effects described in this study have not been established. Ply has been shown to be a potent inducer of CXC-motif chemokines involved in neutrophil recruitment in human dendritic cells (Bernatoniene et al., 2008). IL-1β , another proinflammatory mediator detected in our study, is secreted through activation of the inflammasome, in conjunction with a TLR2-dependent increase in its transcription, during colonization in a Ply-dependent manner (Lemon et al., 2015).
Rare pneumococcal isolates express non-hemolytic forms of Ply, while other strains display reduced pore-forming activity because of minor sequence variations (Jefferies et al., 2007; Lock et al., 1996). Our observations with strain T4S suggest a requirement for Ply and its pore-forming activity in transmission. Strains deficient in Ply hemolytic activity may escape innate immune surveillance at the expense of a lower transmission rate. Our study depended on a model where high levels of shedding are needed to detect transmission from pup to pup (Zafar et al., 2016). Transmission rates during natural carriage could be higher or lower, and this could reflect the volume or type of secretions the organism induces. Strains that are most common because of high rates of transmission might be more likely to cause disease since the expression of Ply provides a molecular link between the ability of the organism to damage its host and its transmission. The association of microbial virulence and transmissibility has been considered previously (Lipsitch and Moxon, 1997).
Our data suggest the possibility of a second role for Ply apart from its enhancement of bacterial shedding. Transmission among infant mice did not require direct contact between donor and recipient. Switching of the dam between cages was sufficient for cage-to-cage spread among pups. Since the dams do not become nasally colonized from the pups, this result suggests that the surface of the dam is the source of contagion in this model as previously proposed (Diavatopoulos et al., 2010). Pneumococci are known to survive desiccation in the environment for substantial periods (Walsh and Camilli, 2011), and it is proposed that the organism may be acquired from fomites and that bio-film growth characteristics affect how long it remains infectious outside of the host (Marks et al., 2014). Although the absence of Ply has previously been linked to inferior in vitro formation of biofilm structures, this effect did not involve its hemolytic activity, a characteristic required in our report for inflammation and transmission (Shak et al., 2013). Evidence present herein suggests that growth on the nutritionally poor mucosal surface of the URT limits the survival of secreted organisms. Endogenous or exogenous Ply provided during in vivo growth partially rescued this ex vivo survival deficiency. We speculate that the damaging effects of host cell lysis from pore formation releases critical nutrients in vivo that reprogram the organism to protect it when metabolically stressed in the environment. Minor differences in viability outside of the host could have a major impact on the transmission rate, since experimental evidence suggests there is a tight population bottleneck for pneumococci in passage between hosts (Kono et al., 2016). Findings from our study demonstrate a relationship between pneumolysin-dependent ex vivo survival and infectivity.
Despite its critical role in the life cycle of infectious agents, there is little understanding of microbial factors involved in transmission, especially for organisms residing in the respiratory tract. This gap in knowledge is largely a consequence of a lack of tractable models to study transmission. Our study took advantage of a model that recapitulates many of the key features of pneumococcal transmission, including higher rates among infants, increase by concurrent IAV, and requirement for close contact (Zafar et al., 2016). Our report details the contributions of a specific bacterial factor in respiratory transmission. Considering the importance of transmission to the infectious life cycle, it is likely there will be other mediators of transmission expressed by the pneumococcus as well as by other respiratory pathogens. These could be attractive targets of prevention since interfering with this key step would greatly amplify the effectiveness of vaccination. We were, however, unable to block transmission by generating antibody to pneumolysoid in pups via maternal immunization in this study. This could be because the important toxin-mediated events occur inside host cells where they are shielded from the immune response. Alternatively, maternal immunization may have failed because of insufficient levels of mucosal immunity in their offspring. In fact, there may already be a precedent for blocking transmission through immunization with a toxoid. A retrospective epidemiologic analysis of diphtheria toxoid concluded that the dramatic decline in disease incidence from immunization must be due to herd immunity through a reduction in host-to-host transmission (Chen et al., 1985).
In summary, Ply has two effects that each could impact transmission success. Ply contributed to the numbers of bacteria shed in secretions—an effect that correlated with the potent local inflammatory effect of the toxin. Additionally, the activity of Ply in vivo enhanced survival of the pneumococcus outside of the host. Both the effects on dissemination and viability of the organism could increase its chances of accessing and establishing in a new host.
EXPERIMENTAL PROCEDURES
Ethics Statement
This study was conducted according to the guidelines outlined by National Science Foundation Animal Welfare Requirements and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. The New York University Medical Center IACUC oversees the welfare, well-being, and proper care and use of all vertebrate animals.
Growth Conditions and Strain Construction
Pneumococcal strains were grown statically in Tryptic Soy (TS) broth (Becton Dickinson) to mid-exponential phase at 37° C. Upon reaching the desired optical density at 620 nm, cells were washed and diluted in sterile PBS for inoculation. For quantitative culture, serial dilutions were plated on TS agar supplemented with either 5% sheep blood or catalase (6,300 U/plate) (Worthington Biochemical Corporation) and incubated overnight at 37° C with 5% CO2.
A streptomycin-resistant (200 μg/mL) derivative of strain TIGR4 (T4S, type 4) was used throughout the study (Zafar et al., 2016). An inframe, unmarked deletion of ply was constructed by generating a PCR product on genomic DNA from strain P2408C (ply::Janus) and P1726 (ply−) and primers Ply-1000-US 5′-CGCCCTTGCTCTGGTTAAAAAAAGA-3′ and Ply-1000-DS 5′-ATCTGGAT CACCTTTTTTAGCTGC-3. To construct the point mutant (plyW433F), we obtained the PCR product using genomic DNA from strain P1727 (plyW433F; type 23F) (Davis et al., 2011; Matthias et al., 2008). To construct the corrected mutant (ply+), we obtained the PCR product using genomic DNA from T4S as template. The genotype of each construct was confirmed via PCR/sequencing. The pore-forming phenotype of each construct was confirmed with a horse erythrocyte lysis assay (Lemon and Weiser, 2015). Hemolysis of the ply minus; and plyW433F mutants was <1% of strain T4S.
Shedding and Colonization in Infant Mice
C57BL/6J mice or congenic knockout mice were obtained from The Jackson Laboratory or were previously described and bred and maintained in a conventional animal facility (Hergott et al., 2015; Zhang et al., 2009). The pups were housed with a dam (mother) for the duration of the experiment and gained weight similar to uninfected animals.
4-day-old pups were given an IN inoculation without anesthesia containing ~2,000 CFUs of S. pneumoniae (Sp) suspended in 3 μL of PBS as described previously (Richard et al., 2014). Shedding was quantified by gently tapping the nares (20 taps/pup) on TS agar plate supplemented with streptomycin (200 μg/mL) to prevent the growth of contaminants and spreading the secretions over the agar surface with a sterile cotton-tipped swab. To measure colonization density, we euthanized pups at the age indicated by CO2 asphyxiation followed by cardiac puncture. The upper respiratory tract was lavaged with 200 μL of sterile PBS from a needle inserted into the trachea, and fluid was collected from the nares. The limit of detection in lavages was 33 CFUs/mL unless otherwise noted.
Where indicated, pups within a litter were treated IN with 20 ng of dexamethasone 21-phosphate disodium salt (Sigma) or PBS (vehicle control) twice daily in a 3 μL volume beginning at age 3 days. Pups were colonized with T4S at age 4 days and shedding measured each day beginning at age 5 days immediately prior to the first daily dose.
In other experiments, strain ply minus; colonized pups were treated with purified Ply or its toxoid (PdB) administered IN (20–200 ng/3 μL) once daily to half the pups in a litter with the remaining pups receiving PBS (3 μL). Shedding was collected from days 5 to 9 of life prior to the daily treatment.
To determine the effect of IAV, we inoculated colonized pups at age 8 days IN with IAV/HKx31 strain (2 × 104 TCID50) suspended in 3 μL of PBS as previously described (Richard et al., 2014).
Transmission in Infant Mice
The transmission model for mono- and IAV co-infection was described in previous studies (Richard et al., 2014; Zafar et al., 2016). Briefly, half the pups in the litter were randomly selected and, at age day 4, infected with the strain indicated. These index mice were then returned to the dam and the other uninfected pups (contact mice). To detect bacterial transmission from the index to contact pups, we euthanized all pups at age 14 days and cultured nasal lavages. For transmission experiment with IAV co-infection, at 8 days of age, all pups in the litter were inoculated IN with IAV virus/HKx31 strain as described above. All the pups were euthanized at age 14 days to determine the rate of transmission. Where indicated, index pups infected with strain ply minus; were given a daily IN dose of Ply (100 ng/3 μL) or PBS (vehicle control) beginning at age 4 days until sacrifice at age 14 days. Pilot experiments established no transmission from colonized pups to the URT of dams.
For cage-switch transmission experiments, pups were randomly divided into index and contact groups and kept in two separate cages with their dam. At age 4 days, index pups were infected with T4S as described above. At age 8 days, pups of both index and contact groups were then infected with IAV. From days 9 to 13, both the pups and dam, the dam only, or the pups only were switched between the two cages 2–3 times per day so that index and contact mice never had direct contact but shared only the cage bedding, only dam, or both the cage bedding and the dam. Transmission rate was determined by quantifying colonization in pups sacrificed at age 14 days as described above.
qRT-PCR
The URT of pups was lavaged using RLT lysis buffer (QIAGEN) to obtain RNA from the epithelium. Total RNA and cDNA generation was carried out as previously described (Lemon et al., 2015). qRT-PCR reactions were performed with Power SYBR Green Master Mix (Applied Biosystems) using 10 ng cDNA and 0.5 μM primers per reaction. Samples were run in duplicate, and each experiment run was repeated. Samples were run on a StepOnePlus real-time PCR system (Applied Biosystems). Primers directed toward gapDH were used as an internal control. RNA expression was quantified using the ΔΔCT method. Primers used in this study were previously described (Lemon et al., 2015; Siegel et al., 2015).
Flow Cytometry
To quantify neutrophils, we pelleted nasal lavage samples from individual pups at 500 × g for 5 min and resuspended them in PBS containing 1% bovine serum albumin (BSA). Samples were blocked with a 1:200 dilution of a rat anti-mouse CD16/32 (clone 93; BioLegend). Cells were stained for 30 min at 4° C with fluorophore-conjugated antibodies (diluted 1:150) against the following surface markers: CD11b-V450 (BD), Ly6G-peridinin chlorophyll protein (PerCP)-Cy5.5 (BD), and CD45-allophycocyanin (APC)-Cy7 (BD). Samples were run on BD LSR II flow cytometer and analyzed with BD FACSDiva software.
Protein Purification and Immunization
Purification and activity of recombinant pneumolysin (Ply) and its toxoid (PdB) containing the W433F point mutation were carried out as previously described (Gelber et al., 2008; Lemon et al., 2015). Proteins were stored at −20° C with 50% (w/v) glycerol to maintain biological activity.
4-week-old female C57BL6J mice were immunized via subcutaneous injection of purified PdB protein (10 μg) using Freud’s adjuvant. Mice in control group received PBS and adjuvant. Mice were immunized three times at an interval of 14 days. 1 week after third immunization, serum samples were collected from tail vein to measure anti-PdB IgG titers by ELISA. Immunity in pups born to immunized dams was determined by ELISA on serum obtained at time of sacrifice by cardiac puncture.
ELISA
Purified PdB protein was diluted at a final concentration of 1 μg/mL in PBS and used to coat Immulon 1B 96-well plates (Thermo Fisher Scientific). Following overnight incubation at 4° C and washing with PBS-T (PBS with 0.05% Tween 20), the plates were blocked for 1 hr at 37° C with 1% BSA in PBS. Serially diluted serum samples in PBS containing 1% BSA were applied to each well, and the plate was incubated at 37° C for 2 hr. Subsequently, the wells were washed, 100 μL of AP-conjugated secondary antibody goat-anti-mouse IgG (1:4,000 diluted in PBS containing 1% BSA) was applied to each well, and the plate was incubated at 37° C for 1.5 hr. The wells were then washed and secondary antibody quantified using pNPP substrate (Sigma).
To quantify Interleukin-1β (IL-1β ) levels in nasal lavages, we used a mouse ELISA kit (Abcam) in accordance with manufacturer’s directions. The limit of detection was 2.7 pg/mL.
Ex Vivo Viability and Infectivity Assays
Nasal lavages (PBS, 200 μL/pup), obtained at age 9 days from pups colonized with the strain indicated at age 4 days, were serially diluted 10-fold in PBS and plated to determine the starting CFU. The time course of viability was assayed using microtiter plates with serial dilutions with PBS of the nasal lavage incubated at 30° C, and every 30 min aliquots were plated on selective media to determine survival relative to T = 0. For other ex vivo viability assays, lavages were obtained from pups treated IN with Ply or PdB (100 ng/day) or PBS vehicle control. To compare viability in TS broth, we grew the strain indicated until reaching an OD620 = 1.0. Pneumococci were then pelleted, serially diluted in PBS, and incubated at 30° C to follow viability over time as described above. To compare infectivity of ex vivo pneumococci, we diluted lavages to the desired density in PBS and incubated for 4 hr at 30° C. 3 μL aliquots were used to inoculate IN 4-day-old pups, and colonization density was assessed in lavages at age 6 days.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad). Unless otherwise specified, differences were determined using the Mann-Whitney U test (comparing two groups) or the Kruskal-Wallis test with Dunn’s post-analysis (comparing multiple groups).
Supplementary Material
Highlights.
Shedding of colonizing S. pneumoniae from mice correlates with mucosal inflammation
The pneumococcal toxin pneumolysin promotes mucosal inflammation
Pneumolysin expression is required for pup-to-pup transmission of S. pneumoniae
Ex vivo survival of pneumococci is affected by pneumolysin expression in the donor
Acknowledgments
This project was supported by grants from the U.S. Public Health Service to J.N.W. (AI038446 and AI105168).
Footnotes
Supplemental Information includes one figure and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2016.12.005.
AUTHOR CONTRIBUTIONS
M.A.Z., Y.W., and J.N.W. conceived the experiments; M.A.Z., Y.W., and S.H. performed the experiments; J.N.W. secured funding; M.A.Z. and J.N.W. wrote the manuscript; and M.A.Z., Y.W., and S.H. analyzed data.
References
- Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol. 2007;9:633–644. doi: 10.1111/j.1462-5822.2006.00814.x. [DOI] [PubMed] [Google Scholar]
- Bachman M, Weiser J. The competition for metals among microbes and their host. In: Nriagu J, Skaar E, editors. Trace Metals and Infectious Disease. MIT Press; 2015. pp. 29–38. [PubMed] [Google Scholar]
- Bernatoniene J, Zhang Q, Dogan S, Mitchell TJ, Paton JC, Finn A. Induction of CC and CXC chemokines in human antigen-presenting dendritic cells by the pneumococcal proteins pneumolysin and CbpA, and the role played by toll-like receptor 4, NF-kappaB, and mitogen-activated protein kinases. J Infect Dis. 2008;198:1823–1833. doi: 10.1086/593177. [DOI] [PubMed] [Google Scholar]
- Chen RT, Broome CV, Weinstein RA, Weaver R, Tsai TF. Diphtheria in the United States, 1971–81. Am J Public Health. 1985;75:1393–1397. doi: 10.2105/ajph.75.12.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Mann B, Gao G, Heath R, King J, Maissoneuve J, Alderson M, Tate A, Hollingshead SK, Tweten RK, et al. Multivalent pneumococcal protein vaccines comprising pneumolysoid with epitopes/fragments of CbpA and/or PspA elicit strong and broad protection. Clin Vaccine Immunol. 2015;22:1079–1089. doi: 10.1128/CVI.00293-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, LaRose MI, Hergott CB, Leng L, Bucala R, Weiser JN. Macrophage migration inhibitory factor promotes clearance of pneumococcal colonization. J Immunol. 2014;193:764–772. doi: 10.4049/jimmunol.1400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest. 2011;121:3666–3676. doi: 10.1172/JCI57761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Steenhuijsen Piters WA, Sanders EA, Bogaert D. The role of the local microbial ecosystem in respiratory health and disease. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140294. doi: 10.1098/rstb.2014.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diavatopoulos DA, Short KR, Price JT, Wilksch JJ, Brown LE, Briles DE, Strugnell RA, Wijburg OL. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J. 2010;24:1789–1798. doi: 10.1096/fj.09-146779. [DOI] [PubMed] [Google Scholar]
- Gelber SE, Aguilar JL, Lewis KL, Ratner AJ. Functional and phylogenetic characterization of Vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J Bacteriol. 2008;190:3896–3903. doi: 10.1128/JB.01965-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwaltney JM, Jr, Sande MA, Austrian R, Hendley JO. Spread of Streptococcus pneumoniae in families. II Relation of transfer of S pneumoniae to incidence of colds and serum antibody. J Infect Dis. 1975;132:62–68. doi: 10.1093/infdis/132.1.62. [DOI] [PubMed] [Google Scholar]
- Hergott CB, Roche AM, Naidu NA, Mesaros C, Blair IA, Weiser JN. Bacterial exploitation of phosphorylcholine mimicry suppresses inflammation to promote airway infection. J Clin Invest. 2015;125:3878–3890. doi: 10.1172/JCI81888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges RG, MacLEOD CM, Bernhard WG. Epidemic pneumococcal pneumonia; pneumococcal carrier studies. Am J Hyg. 1946;44:207–230. doi: 10.1093/oxfordjournals.aje.a119090. [DOI] [PubMed] [Google Scholar]
- Jefferies JM, Johnston CH, Kirkham LA, Cowan GJ, Ross KS, Smith A, Clarke SC, Brueggemann AB, George RC, Pichon B, et al. Presence of nonhemolytic pneumolysin in serotypes of Streptococcus pneumoniae associated with disease outbreaks. J Infect Dis. 2007;196:936–944. doi: 10.1086/520091. [DOI] [PubMed] [Google Scholar]
- Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- Kono M, Zafar MA, Zuniga M, Roche AM, Hamaguchi S, Weiser JN. Single cell bottlenecks in the pathogenesis of Streptococcus pneumoniae. PLoS Pathog. 2016;12:e1005887. doi: 10.1371/journal.ppat.1005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon JK, Weiser JN. Degradation products of the extracellular pathogen Streptococcus pneumoniae access the cytosol via its pore-forming toxin. MBio. 2015;6:e02110–e02114. doi: 10.1128/mBio.02110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon JK, Miller MR, Weiser JN. Sensing of interleukin-1 cytokines during Streptococcus pneumoniae colonization contributes to macrophage recruitment and bacterial clearance. Infect Immun. 2015;83:3204–3212. doi: 10.1128/IAI.00224-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsitch M, Moxon ER. Virulence and transmissibility of pathogens: what is the relationship? Trends Microbiol. 1997;5:31–37. doi: 10.1016/S0966-842X(97)81772-6. [DOI] [PubMed] [Google Scholar]
- Lock RA, Zhang QY, Berry AM, Paton JC. Sequence variation in the Streptococcus pneumoniae pneumolysin gene affecting haemolytic activity and electrophoretic mobility of the toxin. Microb Pathog. 1996;21:71–83. doi: 10.1006/mpat.1996.0044. [DOI] [PubMed] [Google Scholar]
- Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks LR, Reddinger RM, Hakansson AP. Biofilm formation enhances fomite survival of Streptococcus pneumoniae and Streptococcus pyogenes. Infect Immun. 2014;82:1141–1146. doi: 10.1128/IAI.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias KA, Roche AM, Standish AJ, Shchepetov M, Weiser JN. Neutrophiltoxin interactions promote antigen delivery and mucosal clearance of Streptococcus pneumoniae. J Immunol. 2008;180:6246–6254. doi: 10.4049/jimmunol.180.9.6246. [DOI] [PubMed] [Google Scholar]
- Mitchell TJ, Dalziel CE. The biology of pneumolysin. Subcell Biochem. 2014;80:145–160. doi: 10.1007/978-94-017-8881-6_8. [DOI] [PubMed] [Google Scholar]
- Musher DM. How contagious are common respiratory tract infections? N Engl J Med. 2003;348:1256–1266. doi: 10.1056/NEJMra021771. [DOI] [PubMed] [Google Scholar]
- Nelson EJ, Harris JB, Morris JG, Jr, Calderwood SB, Camilli A. Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat Rev Microbiol. 2009;7:693–702. doi: 10.1038/nrmicro2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odutola A, Ota MO, Ogundare EO, Antonio M, Owiafe P, Worwui A, Greenwood B, Alderson M, Traskine M, Verlant V, et al. Reactogenicity, safety and immunogenicity of a protein-based pneumococcal vaccine in Gambian children aged 2–4 years: A phase II randomized study. Hum Vaccin Immunother. 2016;12:393–402. doi: 10.1080/21645515.2015.1111496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price KE, Greene NG, Camilli A. Export requirements of pneumolysin in Streptococcus pneumoniae. J Bacteriol. 2012;194:3651–3660. doi: 10.1128/JB.00114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P, Parrish M, Tay IJ, Li N, Ackerman S, He F, Kwang J, Chow VT, Engelward BP. Streptococcus pneumoniae secretes hydrogen peroxide leading to DNA damage and apoptosis in lung cells. Proc Natl Acad Sci USA. 2015;112:E3421–E3430. doi: 10.1073/pnas.1424144112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AL, Siegel SJ, Erikson J, Weiser JN. TLR2 signaling decreases transmission of Streptococcus pneumoniae by limiting bacterial shedding in an infant mouse Influenza A co-infection model. PLoS Pathog. 2014;10:e1004339. doi: 10.1371/journal.ppat.1004339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F, Foster D, Nicoli E, Trotter C, Vipond B, Muir P, Gonçalves G, Januário L, Finn A. Relationships between rhinitis symptoms, respiratory viral infections and nasopharyngeal colonization with Streptococcus pneumoniae,Haemophilus influenzaeandStaphylococcusaureus in children attending daycare. Pediatr Infect Dis J. 2013;32:227–232. doi: 10.1097/INF.0b013e31827687fc. [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Gilbert RJ, Crane D, Morgan PJ, Mitchell TJ, Rowe AJ, Andrew PW, Paton JC, Tweten RK, Parker MW. The molecular mechanism of pneumolysin, a virulence factor from Streptococcus pneumoniae. J Mol Biol. 1998;284:449–461. doi: 10.1006/jmbi.1998.2167. [DOI] [PubMed] [Google Scholar]
- Shak JR, Ludewick HP, Howery KE, Sakai F, Yi H, Harvey RM, Paton JC, Klugman KP, Vidal JE. Novel role for the Streptococcus pneumoniae toxin pneumolysin in the assembly of biofilms. MBio. 2013;4:e00655–e13. doi: 10.1128/mBio.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Reading PC, Wang N, Diavatopoulos DA, Wijburg OL. Increased nasopharyngeal bacterial titers and local inflammation facilitate transmission of Streptococcus pneumoniae. MBio. 2012;3:e00255–e12. doi: 10.1128/mBio.00255-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Weiser JN. Mechanisms of bacterial colonization of the respiratory tract. Annu Rev Microbiol. 2015;69:425–444. doi: 10.1146/annurev-micro-091014-104209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Roche AM, Weiser JN. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe. 2014;16:55–67. doi: 10.1016/j.chom.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Tamashiro E, Weiser JN. Clearance of pneumococcal colonization in infants is delayed through altered macrophage trafficking. PLoS Pathog. 2015;11:e1005004. doi: 10.1371/journal.ppat.1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelmink L, Sender V, Hentrich K, Kuri T, Plant L, Henriques-Normark B. Toll-like receptor 3/TRIF-dependent IL-12p70 secretion mediated by Streptococcus pneumoniae RNA and its priming by influenza A virus coinfection in human dendritic cells. MBio. 2016;7:e00168–e16. doi: 10.1128/mBio.00168-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson G, Chimalapati S, Pollard T, Lapp T, Cohen J, Camberlein E, Stafford S, Periselneris J, Aldridge C, Vollmer W, et al. TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J Immunol. 2014;193:3736–3745. doi: 10.4049/jimmunol.1401413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum AM, Lysenko ES, Weiser JN. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect Immun. 2005;73:7718–7726. doi: 10.1128/IAI.73.11.7718-7726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu HT, Yoshida LM, Suzuki M, Nguyen HA, Nguyen CD, Nguyen AT, Oishi K, Yamamoto T, Watanabe K, Vu TD. Association between nasopharyngeal load of Streptococcus pneumoniae, viral coinfection, and radiologically confirmed pneumonia in Vietnamese children. Pediatr Infect Dis J. 2011;30:11–18. doi: 10.1097/INF.0b013e3181f111a2. [DOI] [PubMed] [Google Scholar]
- Walsh RL, Camilli A. Streptococcus pneumoniae is desiccation tolerant and infectious upon rehydration. MBio. 2011;2:e00092–e11. doi: 10.1128/mBio.00092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser JN. The pneumococcus: why a commensal misbehaves. J Mol Med. 2010;88:97–102. doi: 10.1007/s00109-009-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafar MA, Kono M, Wang Y, Zangari T, Weiser JN. Infant mouse model for the study of shedding and transmission during Streptococcus pneumoniae monoinfection. Infect Immun. 2016;84:2714–2722. doi: 10.1128/IAI.00416-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. 2009;119:1899–1909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.