

Abstract
Background
To examine methylation of the peroxisome proliferator-activated receptor γ (PPARγ) gene and its relationship with child weight status, at birth and 9 years.
Methods
We measured PPARγ methylation across 23 CpG sites using the Infinium Illumina 450 k array for children from the Center for the Health Assessment of Mothers and Children of Salinas (CHAMACOS) cohort at birth (N = 373) and 9 years (N = 245).
Results
Methylation level correlation patterns across the 23 PPARγ CpG sites were conserved between birth and 9-year ages. We found high inter-CpG correlations between sites 1–3 (methylation block 1) and also between sites 18–23 (methylation block 2) for both time points, although these patterns were less pronounced at 9 years. Additionally, sites 1–3 (north shore) had the highest intra-CpG correlations over time (r = 0.24, 0.42, and 0.3; P = 0.002, P < 0.001, P < 0.001, respectively). PPARγ methylation levels tended to increase with age, and the largest differences were observed for north shore sites (7.4%). Adjusting for sex, both site 1 and site 20 (gene body) methylation at birth was significantly and inversely associated with birth weight (β = −0.13, P = 0.033; β = −0.09, P = 0.025, respectively). Similarly, we found that site 1 and site 20 methylation at 9 years was significantly and inversely associated with 9-year BMI z-score (β = −0.41, P = 0.015; β = −0.23, P = 0.045, respectively).
Conclusion
Our results indicate that PPARγ methylation is highly organized and conserved over time, and highlight the potential functional importance of north shore sites, adding to a better understanding of regional human methylome patterns. Overall, our results suggest that PPARγ methylation may be associated with child body size.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-016-0365-4) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, Birth cohort study, PPARγ, Obesity, Newborns, Children, Whole blood, DNA methylation
Background
It has been hypothesized that prenatal environmental exposures may cause lasting epigenetic changes during child development, leading to adverse health outcomes in later life [1, 2]. Epigenetics refers to heritable changes regulating gene expression that do not affect the DNA base pair sequence. DNA methylation is the most commonly studied epigenetic mark [3–6]. This modification involves attachment of a methyl group to the cytosine base within cytosine-guanine dinucleotides, also known as ‘CpG methylation’. Higher CpG methylation (hypermethylation) within the promoter region of a gene can reduce gene expression [7].
Our understanding of how the human methylome is organized is rapidly evolving and current literature outlines a classification system for CpG sites, highlighting that their function may be intimately related to their location in the gene [8, 9]. For example, approximately 70% of gene promoter regions are thought to contain a CpG island – a region with densely concentrated CpGs that typically have low levels of methylation [10]. CpG sites flanking the island are located in regions termed north shore (upstream, 5′ end) and south shore (downstream, 3′ end) and are thought to be particularly important in regulating gene expression [6, 9].
There is a growing interest in examining interactions between environmental and genetic factors on differential CpG methylation. A classic example is from animal studies on the Agouti mouse, where hypomethylation of the intracisternal A particle (IAP) increases expression of the Agouti gene, resulting in yellow coat color and obese phenotype. Using this model, Waterland et al. [11] showed that inheritance of the Agouti gene was associated with trans-generational amplification of obesity and that maternal methyl-donor supplementation could prevent this effect.
In humans, DNA methylation has been proposed to mediate direct intra-uterine associations between maternal and offspring phenotypes. Differential DNA methylation has been reported when assessing offspring exposed in utero to extreme maternal undernutrition [12, 13], maternal morbid obesity [14] and less extreme maternal underweight and maternal obesity [15]. However, several important challenges remain. There is an ongoing effort to determine the causal direction between DNA methylation and an offspring phenotype. In their 2016 study, Richmond et al. apply a causal framework to parse out whether HIF3A methylation has a causal effect on BMI or vice versa [16]. Their results argue for the potential of a phenotype to affect methylation status and highlight the potential for inter-generational influence of maternal BMI on offspring methylation, possibly confounding the offspring HIF3A methylation and obesity association.
Another important challenge has been replication of results from epigenome-wide association studies (EWAS) studies. For example, the EWAS study by Sharp et al. identified 28 CpGs in newborns that were associated with maternal pre-pregnancy BMI. Four of these hits had previously been reported in literature, but their results did not replicate the direction and magnitude of the earlier analyses [17]. Additionally, in their EWAS, Aslibekyan et al. found only 8 CpGs in 3 genes (CPT1A, PHGDH, CD38) associated with body mass index (BMI) in adults that withstood replication and multiple testing adjustment [18].
To avoid the limitations of multiple testing, candidate genes can be selected a priori. With respect to obesity development, the peroxisome proliferator-activated receptor γ (PPARγ) gene may play a critical role, functioning as the only gene that is both necessary and sufficient for fat cell production [19, 20]. PPARγ upregulation has been linked to improvement of critical metabolism-related hormones (increased adiponectin and decreased leptin) and increased insulin sensitivity at the expense of greater body weight in adults [21] and animals [22–24]. Importantly, while methylation affects PPARγ expression in animal and in vitro studies, only limited human data on PPARγ methylation, its relationship with obesity and/or with perinatal factors are available [25, 26].
In the Center for the Health Assessment of Mothers and Children of Salinas (CHAMACOS) cohort, we have previously examined a subset of PPARγ CpG sites and their relationship with gene expression in a cohort of children with a high prevalence of obesity [27]. We reported that hypomethylation of the PPARγ CpG site cg10499651 was associated with increased PPARγ expression as measured by both real-time polymerase chain reaction (RT-PCR) and nCounter assays. In the current investigation, we build on this finding and add to current data gaps on PPARγ methylation and its relationship with obesity. We use the Illumina 450 k assay to examine methylation of 23 CpG sites spanning the PPARγ promoter and gene body regions in children at birth (N = 373) and at 9 years (N = 245) and 1) analyze the correlation structure between the 23 PPARγ CpG sites, 2) characterize associations between perinatal factors, including maternal pre-pregnancy BMI, and PPARγ methylation at birth and at 9 years, and 3) examine associations between PPARγ methylation, child birthweight and BMI at 9 years.
Methods
Subject and study design
The CHAMACOS study is a longitudinal birth cohort designed to assess the health effects of pesticides and other environmental exposures on growth and development of primarily Mexican-American children living in Salinas Valley, an agricultural region of California [28, 29]. Mothers were enrolled during pregnancy between October 1999 and October 2000, with 537 mother-child pairs in the study at delivery and 327 pairs participating at the 9-year visit. Eligible women were ≥18 years of age, <20 weeks gestation at enrollment, English or Spanish speaking, eligible for low-income health insurance (Medi-Cal) and planning to deliver at the county hospital. Women were interviewed twice during pregnancy, shortly after delivery, and when their children were 6 months, and 1, 2, 3½, 5, 7, and 9 years of age. This study used a subset of the CHAMACOS cohort (373 children at birth and 245 children at 9 years) who had blood samples available for methylation analyses. Study protocols (2010-01-620 & 2010-03-949) ethics were approved by the University of California, Berkeley Committee for Protection of Human Subjects. Written informed consent was obtained from all mothers and assent was provided by the children at the 9-year assessment.
Questionnaire data
Interviews were conducted in Spanish or English by bilingual, bicultural trained interviewers. Maternal age was assessed during the first prenatal interview at 14 ± 5 weeks gestation. Maternal pre-pregnancy BMI was calculated using the mother’s self-reported pre-pregnancy weight and measured height. Data on infant birth weight and gestational age were obtained from delivery medical records abstracted by a registered nurse.
Anthropometric measurements
An electronic scale (Tanita Mother-Baby Scale Model 1582, Tanita Corp.) was used to measure child weight at the 9-year visit. Child 9-year height was measured in triplicate using a stadiometer (Seca 222) and the average of measurements was used. Child height and weight were converted to age- and sex-specific BMI z-scores using the 2000 Centers for Disease Control and Prevention (CDC) child growth data and children were categorized as normal weight, overweight, or obese using the sex and age-specific cutoffs (85th and 95th percentile, respectively) [30]. Monthly rate of weight gain during the first 6 months of life was calculated as weight at the 6-month visit minus birth weight divided by exact age in months at the 6-month visit and reported in 100 grams/month. This approach to examining infancy weight gain has been previously validated [31].
Child CpG methylation measurement
DNA was isolated from blood clots previously collected and stored at −80 °C using the QIAamp DNA blood maxi kit (Qiagen, CA). To measure CpG methylation, we used the Infinium Illumina 450 k array, which is based on multiplexed genotyping of bisulfite converted genomic DNA. This technology is currently considered the leading method to measure genome-wide methylation, providing both broad and dense coverage, in total interrogating 485,577 CpG sites over 99% of RefSeq genes. The workflow involves bisulfite conversion of DNA, performed using Zymo Bisulfite Conversion Kits (Zymo Research, Orange, CA). Subsequently, each sample is whole-genome amplified, enzymatically fragmented, purified and applied to the BeadChips according to the Illumina methylation protocol. BeadChips were processed with robotics and analyzed using the Illumina Hi-Scan system at the Genomics Core. Samples included in the analysis had detection P values below 0.01 for 95% of CpG sites and poor performing CpG sites with P value > 0.01 were excluded. Raw signal intensities were background corrected and then normalized for color-channel bias using the all sample mean normalization method as described by Yousefi et al. (2013) [32]. Beta mixture quantile normalization was also applied to make interpretation between type I and type II probes comparable [33]. One of the CpG sites in our analysis (CpG site 13, cg04632671) was found to have a common single nucleotide polymorphism (SNP), minor allele frequency > 5%, within 50 base pairs in the Mexican ancestry in Los Angeles, California, (MXL) HapMap population. In our sensitivity analysis, excluding this site had no impact on our results and a decision was made to retain this site. Additionally, although we did not have data on the rs1801282 Pro12Ala PPARγ SNP, adjusting our analyses for potential effects of admixture did not affect the relationships between PPARγ DNA methylation and birthweight or BMI in our study.
Cell composition
To examine the relationship of blood cell composition with CpG methylation in PPARγ CpG sites, we performed differential cell counts in a subset of cord samples (N = 111) as described previously [34]. To prepare heparinized whole blood smears, we used the “gold standard” Wright-Push blood smearing technique followed by staining utilizing a DiffQuikVR staining kit [35]. At least 100 cells were scored for each slide, and a percentage of each cell type (lymphocytes, monocytes, neutrophils, eosinophils, and basophils) was used for data analysis. The coefficient of variation (CV) for the repeat scoring in this subset was less than 10%.
Whole blood smears were not available for differential cell count in 9-year-old CHAMACOS children. For these children we used the Bioconductor R package minfi (v1.10.2) to estimate the distribution of six different white blood cell types (CD8+ T and CD4+ T lymphocytes, CD56+ natural killer cells, CD19+ B cells, CD14+ monocytes, and granulocytes) based on their methylation signatures in 450 k data [36]. We did not use minfi to estimate cell composition at birth as we have previously shown that proportions of white blood cells in newborns are significantly different from the adult reference samples on which minfi estimates are based [34, 37, 38].
For comparison of cell type composition in cord bloods to those estimated by minfi in 9-year-olds, we used proportions of lymphocytes, granulocytes, and monocytes. For minfi estimates, this required summation of the frequencies for CD8+ T, CD4+ T, natural killer cells, and B cells to calculate the proportion of lymphocytes. For differential cell count, proportions of neutrophils, eosinophils, and basophils were summed to give an estimate of granulocytes.
Additionally, we used data from four separate cohorts, including Bakulski et al. 2016 [39], characterizing cell composition in cord blood in relation to DNA methylation [40–42]. Re-running our analysis using data from these four cohorts as a reference did not change our findings.
Statistical analyses
Our statistical analyses used methylation levels expressed as M-values, which are calculated as the log2 ratio of the intensities of methylated probe to unmethylated probe, M-value = log2(beta/(1-beta)) [43]. In addition, we also presented relative methylation betas (% methylation) in some of the tables for ease of interpretation. Importantly, these untransformed values were not used in analyses given the reported heteroscedasticity for highly methylated or unmethylated CpG sites [43]. To analyze PPARγ methylation structure across the 23 sites, we plotted methylation levels by site and examined inter-site correlations at birth and 9 years using Pearson’s correlation coefficients (r). Linkage disequilibrium (LD) methylation blocks were established based on several criteria slightly modified from Shoemaker et al. (2010) and Liu et al. (2014): (i) they had to contain at least 3 contiguous CpG sites and (ii) at least 50% of the CpG site pairs had to have methylation levels that were highly correlated with each other (r2 > 0.4) [44, 45]. Additionally, we calculated correlation coefficients for each CpG site comparing values at birth to 9 years and tested whether PPARγ methylation levels changed from birth to 9 years using generalized estimating equations (GEE) and whether methylation levels differed at each site and at each age by sex using Student’s t-Test. We examined associations between PPARγ methylation at birth and child birthweight and PPARγ methylation at 9 years and 9-year BMI z-score for each of the 23 CpG sites. We used directed acyclic graphs (DAGs) to select our covariates for multiple regression models. We examined whether methylation levels varied by gestational age, maternal age, maternal BMI, parity, and weight gain in the first six months of life [46–50]. Bonferroni correction was used to account for multiple testing. Testing for associations between 23 CpG sites with 4 variables at birth and 5 variables at the 9-year time point comprises 207 tests. Thus for this analysis, we used an adjusted alpha of 0.05/207 = 2.4E-4. Statistical analyses were conducted using STATA 12 (College Station, TX) for Windows and R statistical software (R Foundation for Statistical Computing, Vienna, Austria).
Results
Maternal and child characteristics
Our study sample included a total of 444 children who had DNA samples available for methylation analysis at birth and/or age 9 in addition to birthweight and BMI at 9 years. Of these children, 174 had samples available at both time points, 199 had samples only at birth, and 71 had samples at age 9 only. Overall, there were 373 children available for analyses at birth and 245 children at 9 years.
At pregnancy, mothers tended to be young (25.6 years, 95% CI 25.1, 26.1 years) and overweight or obese (61%), with an average BMI of 27.0 kg/m2 (95% CI of 26.5, 27.5 kg/m2). Of the 444 children in this study, there were similar numbers of boys (N = 221) and girls (N = 223) (Table 1). Mean gestational duration was 38.9 weeks (95% CI 38.7, 39.0 weeks) and mean birthweight was 3.46 kg (95% CI 3.41, 3.5 kg). Their average weight gain in the first 6 months of life was 0.73 kg (95% CI 0.71, 0.75 kg). At 9 years, the majority of children were overweight or obese (56%), with mean BMI of 20.7 kg/m2 (95% CI of 20.1, 21.3 kg/m2) and mean BMI-z-score of 1.11 (95% CI of 0.98, 1.24). We did not find any statistically significant differences between the maternal and child characteristics, including anthropometric measures, in this subset compared to the overall CHAMACOS dataset.
Table 1.
CHAMACOS maternal and child characteristics
| Cohort Characteristics | Number | Mean | Range | 95% CI |
|---|---|---|---|---|
| Sex (male/female) | 221/223 | --- | --- | --- |
| Gestational age (weeks) | 444 | 38.9 | 33–42 | 38.7, 39.0 |
| Parity | 444 | 1.3 | 0–9 | 1.1, 1.4 |
| Birth weight (kg) | 444 | 3.46 | 1.93–4.89 | 3.41, 3.50 |
| Weight gain in 1st 6 months (kg) | 229 | 0.73 | 0.34–1.22 | 0.71, 0.75 |
| BMI at 9 years (kg/m2) | 240 | 20.7 | 13.9–33.8 | 20.1, 21.3 |
| BMI z-score | 240 | 1.11 | −1.57–2.70 | 0.98, 1.24 |
| Maternal age at pregnancy (years) | 444 | 25.6 | 18–43 | 25.1, 26.1 |
| Maternal pre-pregnancy BMI (kg/m2) | 444 | 27.0 | 17.7–57.3 | 26.5, 27.5 |
BMI body mass index
PPARγ methylation
Additional file 1: Figure S1 shows the distribution of 23 CpG sites (red squares) measured by the Illumina Methylation 450 k array across the PPARγ gene promoter and body. Blue squares indicate all other CpG sites (N = 183). This figure was generated using the DNA sequence provided in the Genome Reference Consortium Human Build 38 patch release 2 (GRCh38.p2) and serves as a visual reference showing the relative locations of the 23 CpG sites. Illumina annotation classifies sites 1–3 as north shore (cg01412654, cg25929976, and cg18063278), 4–15 as island (cg26364899, cg27095527, cg06573644, cg21946299, cg23514324, cg04748988, cg15722404, cg13518792, cg09405169, cg04632671, cg07556134, and cg18887186), 16–17 as south shore (cg21859053 and cg04908300), 18 as south shelf (cg16827534), 19 as 5′ untranslated region (5′ UTR) (cg16197186), and 20–23 as PPARγ gene body (cg18537222, cg07895576, cg07676920, and cg10499651) [8, 9].
Figure 1 shows methylation M-Values across the 23 PPARγ CpG sites at birth and 9-years. As expected, CpG sites in the north shore (average beta 61.4% at birth, 68.8% at 9 years), south shore (average beta 12.5% at birth, 18.8% at 9 years) and shelf (average beta 88.2% at birth, 89.5% at 9 years), 5′UTR (average beta 91.2% at birth, 91.9% at 9 years) and gene body (average beta 76.5% at birth, 78.0% at 9 years) tended to be highly methylated while island sites had significantly lower methylation (average beta 4.5% at birth, 4.6% at 9 years). Interestingly, we observed a highly conserved pattern of methylation across the 23 sites comparing both time points. For example, sites 21, 22, 23 follow the same pattern of decreasing methylation at birth (90.2%, 85.1%, and 78.9%, respectively) and at 9 years (90.7%, 85.7%, and 77.7%, respectively).
Fig. 1.
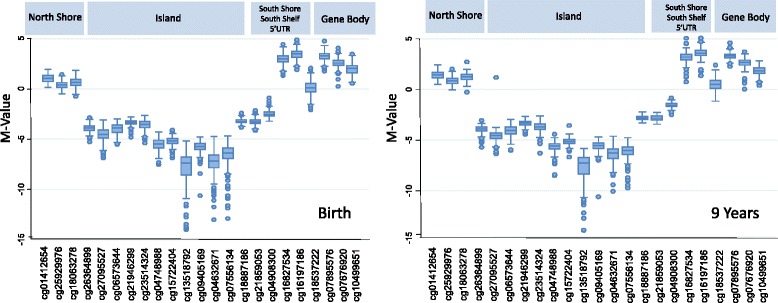
Methylation M-values across 23 PPARγ CpG Sites at birth and 9-years. Sites are labeled according to their Illumina 450K annotation. Methylation levels are highly conserved comparing the two time points
PPARγ methylation blocks
There is a growing body of literature showing that neighboring CpG sites are often highly correlated with each other [45, 51]. We examined inter-CpG correlations for the 23 PPARγ sites and our results are presented in Fig. 2. Both at birth and 9-years, we identified two methylation blocks. Block 1 was comprised of north shore sites 1–3 spanning approximately 1 kilobase (kb) pairs. Block 2 contained sites 18–23 located in the south shelf, 5′UTR, and gene body, and spanned approximately 130 kb. Additionally, we noted that correlations within the blocks were consistently stronger at birth compared to 9 years. At birth, correlations ranged from 0.66 to 0.79 and from 0.21 to 0.79 for blocks 1 and 2, respectively, while at 9 years, block 1 ranged from 0.55 to 0.69 and block 2 ranged 0.12 to 0.57. Interestingly, we also found that CpG site 1 was highly correlated with methylation at gene body sites 20–23 at both birth (r = 0.74, 0.54, 0.31, and 0.59, all P < 0.001) and 9-years (r = 0.66, 0.50, 0.30, and 0.53, all P < 0.001).
Fig. 2.
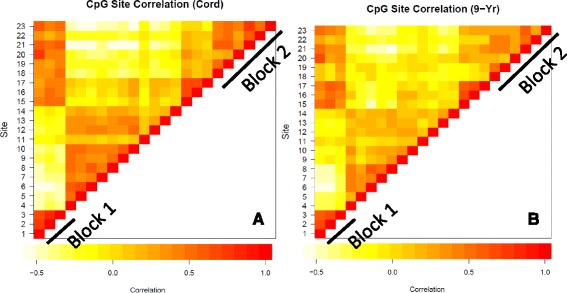
Correlations for the 23 PPARγ CpG sites spanning the promoter and gene body region at (a) birth and (b) 9 years. White/yellow squares represent none/poor correlations, while orange/red squares represent moderate/high correlations. Two methylation blocks were identified for both time points, Block 1 consists of north shore sites 1-3 and Block 2 consists of south shelf, 5’ untranslated region (5’UTR), and gene body sites 18-23
PPARγ methylation by age
Figure 3 shows correlations for PPARγ CpG sites between the birth and 9-year time points. We found significant correlations for all three of the north shore sites (r = 0.24, 0.42, and 0.31; P = 0.002, <0.001, and < 0.001, respectively). Island CpG sites 5 and 8 (r = 0.18 and 0.28; P = 0.02 and <0.001, respectively), south shore site 16 (r = 0.18; P = 0.02), and gene body site 23 (r = 0.3; P < 0.001) were also significantly correlated over time. We used GEE to account for repeated measures over time and found that for all regions (north shore, island, south shore, south shelf, 5′UTR and gene body) methylation values had statistically significant increases with age. The greatest increases were observed for north and south shore sites, 7.4% and 6.3%, respectively. Averaging over all 23 sites, methylation levels had a statistically significant increase of 2% over the 9-year span. Increases in methylation remained statistically significant for all regions adjusting for cell composition (data not shown).
Fig. 3.
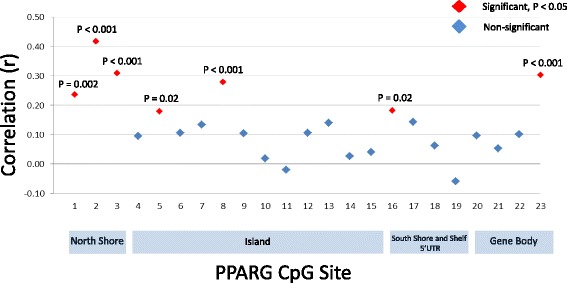
Correlations between birth and 9-year methylation values for the 23 PPARγ CpG Sites. All three north shore sites were significantly correlated over time
PPARγ methylation by sex
Table 2 shows methylation betas for all 23 PPARγ CpG sites stratified by sex at both the birth and 9-year time points. For all three north shore sites, females had significantly higher methylation betas compared to males at both birth (site 1: 67.8 vs. 65.8, P = 0.002, site 2: 58.1 vs. 55.6, P < 0.001, site 3: 62.1 vs. 59.1, P < 0.001) and 9 years (site 1: 73.1 vs. 71.5, P = 0.018, site 2: 65.4 vs. 62.5, P < 0.001, site 3: 71.1 vs. 68.3, P < 0.001). Females also had significantly higher methylation at 9 years at site 17 (25.7 vs. 24.8, P = 0.03) and at birth at site 18 (88.6 vs. 87.8, P = 0.021).
Table 2.
PPPPARγ methylation by sex
| PPARγ Site | CpG Illumina ID | Classificationa | Methylation at birth (%) | Methylation at 9 years (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Male (N = 188) | Female (N = 185) | P valueb | Male (N = 113) | Female (N = 132) | P valueb | |||
| 1 | cg01412654 | North Shore | 65.8 | 67.8 | 0.002 | 71.5 | 73.1 | 0.018 |
| 2 | cg25929976 | North Shore | 55.6 | 58.1 | <0.001 | 62.5 | 65.4 | <0.001 |
| 3 | cg18063278 | North Shore | 59.1 | 62.1 | <0.001 | 68.3 | 71.1 | <0.001 |
| 4 | cg26364899 | Island | 6.3 | 6.3 | 0.931 | 6.0 | 6.2 | 0.156 |
| 5 | cg27095527 | Island | 4.3 | 4.5 | 0.289 | 4.0 | 4.6 | 0.380 |
| 6 | cg06573644 | Island | 6.2 | 6.4 | 0.434 | 5.8 | 5.8 | 0.887 |
| 7 | cg21946299 | Island | 9.0 | 8.9 | 0.445 | 9.1 | 9.0 | 0.724 |
| 8 | cg23514324 | Island | 8.0 | 8.0 | 0.943 | 7.4 | 7.3 | 0.626 |
| 9 | cg04748988 | Island | 2.3 | 2.3 | 0.636 | 2.0 | 2.0 | 0.570 |
| 10 | cg15722404 | Island | 2.8 | 2.7 | 0.430 | 2.8 | 2.7 | 0.119 |
| 11 | cg13518792 | Island | 1.0 | 1.0 | 0.297 | 1.0 | 1.0 | 0.564 |
| 12 | cg09405169 | Island | 1.9 | 1.8 | 0.550 | 2.1 | 2.1 | 0.707 |
| 13 | cg04632671 | Island | 1.0 | 1.1 | 0.115 | 1.4 | 1.2 | 0.054 |
| 14 | cg07556134 | Island | 1.3 | 1.3 | 0.797 | 1.5 | 1.5 | 0.457 |
| 15 | cg18887186 | Island | 9.8 | 9.9 | 0.372 | 12.5 | 12.5 | 0.709 |
| 16 | cg21859053 | South Shore | 9.4 | 9.5 | 0.459 | 12.3 | 12.4 | 0.657 |
| 17 | cg04908300 | South Shore | 15.4 | 15.9 | 0.107 | 24.8 | 25.7 | 0.030 |
| 18 | cgl6827534 | South Shelf | 87.8 | 88.6 | 0.021 | 89.7 | 89.4 | 0.506 |
| 19 | cg16197186 | 5′ UTR | 91.1 | 91.3 | 0.398 | 92.0 | 91.9 | 0.996 |
| 20 | cg18537222 | Gene Body | 51.5 | 52.1 | 0.612 | 57.1 | 58.4 | 0.345 |
| 21 | cg07895576 | Gene Body | 90.1 | 90.3 | 0.244 | 90.7 | 90.6 | 0.707 |
| 22 | cg07676920 | Gene Body | 85.1 | 85.0 | 0.930 | 85.9 | 85.4 | 0.513 |
| 23 | cg10499651 | Gene Body | 79.0 | 78.8 | 0.632 | 77.1 | 78.2 | 0.084 |
PPARγ peroxisome proliferator-activated receptor gamma
UTR untranslated region
aBased on Illumina 450 K annotation
bStudent’s t-Test
Associations between perinatal factors and PPARγ methylation
Additional file 2: Table S1 and Additional file 3: Table S2 show regression results for testing associations between the selected perinatal characteristics and PPARγ methylation at birth and 9 years, respectively. Of note, at both birth and 9 years, methylation at PPARγ site 22 was inversely associated with maternal pre-pregnancy BMI. However, this relationship was not significant after adjustment for multiple testing. All other perinatal variables were not associated with methylation at any of the sites.
Relationship between PPARγ methylation and birth weight and 9-year BMI z-score
Table 3 summarizes our analysis of the relationships between PPARγ methylation and child birthweight and BMI at age 9. Given evidence in literature highlighting importance of island-flanking CpG sites, our analyses were focused on sites 1–3 and 16–23. Adjusting for sex, we found that methylation at birth for both site 1 and site 20 was significantly and inversely associated with birth weight (β = −0.13, P = 0.033; β = −0.09, P = 0.025, respectively). Similarly, we found methylation at 9 years at site 1 and site 20 was significantly and inversely associated with 9-year BMI z-score (β = −0.41, P = 0.015; β = −0.23, P = 0.045, respectively). We subsequently tested whether methylation at birth at either of these sites was associated with 9-year BMI z-score but did not find any significant associations. Adjusting for cell composition did not appreciably change results of these analyses.
Table 3.
Associations between PPARγ methylation and child size at birth and 9 years of age
| CpG Site Birth | Birth Weight (kg) (N = 373) |
9-Year BMI i-Score (N = 240) |
||
|---|---|---|---|---|
| Betaa | P value | Beta | P value | |
| 1 | −0.13 | 0.033 | −0.41 | 0.015 |
| 2 | −0.06 | 0.367 | 0.11 | 0.514 |
| 3 | 0.01 | 0.825 | −0.08 | 0.625 |
| 16 | −0.11 | 0.191 | 0.43 | 0.099 |
| 17 | −0.04 | 0.537 | 0.40 | 0.151 |
| 18 | −0.02 | 0.739 | −0.05 | 0.689 |
| 19 | −0.05 | 0.400 | −0.16 | 0.260 |
| 20 | −0.09 | 0.025 | −0.23 | 0.045 |
| 21 | −0.01 | 0.917 | −0.05 | 0.825 |
| 22 | 0.04 | 0.517 | −0.17 | 0.293 |
| 23 | −0.01 | 0.987 | −0.14 | 0.380 |
PPARγ peroxisome proliferator-activated receptor gamma
BMI body mass index
aAdjusted for sex
Discussion
In this study, we aimed to address several knowledge gaps on the 1) correlation structure of PPARγ methylation, 2) relationships between perinatal factors and PPARγ methylation, and 3) associations between PPARγ methylation, birth weight and child BMI. We found that PPARγ methylation displays a highly conserved pattern and report on two methylation blocks comprised of sites 1–3 (block 1) and 18–23 (block 2) present at both birth and 9-year time points. Additionally, we observed high intra-CpG correlations comparing the birth to 9-year time points for all three north shore CpG sites. With respect to aim 2, we found that none of the perinatal variables examined, including gestational age, parity, maternal age and pre-pregnancy BMI and in addition, for 9 years, weight gain in the first 6 months, were significantly associated with PPARγ methylation at either birth or 9 years. Further, we observed that girls had significantly greater methylation at north shore sites 1–3 compared to boys at both time points. Adjusting for sex, we found that methylation at birth for sites 1 and 20 was significantly and inversely associated with birth weight. Similarly, we found that methylation at these sites at 9 years was also significantly and inversely associated with 9-year BMI z-score. Taken together, these results indicate that PPARγ methylation may be involved in regulating child body size and highlight the potential functional importance of north shore sites.
PPARγ CpG organization is typical of many other genes, with its promoter region containing a CpG island flanked by north and south shore sites [52]. Additionally, in agreement with studies showing complex inter-CpG correlations over both short and long regions, PPARγ contained two methylation blocks spanning 1 kb over the north shore (block 1) and 130 kb over the south shore, 5′ UTR, and gene body (block 2) [45, 53]. Interestingly, we also found that north shore CpG site 1 from block 1 was correlated with methylation at sites 20–23 from block 2. There is a growing understanding that the location of a particular CpG site may be functionally important and several studies have highlighted the role of shore sites in gene expression, tissue differentiation, and overall phenotype [9, 54, 55]. For example, Doi et al. (2009) showed that CpG shore methylation distinguished between several cell lines, including brain, liver, spleen cells, their pluripotent stem cells and parental fibroblasts [54]. Similarly, Irizarry et al. (2009) showed that most methylation changes associated with colon cancer occurred in CpG shores [9]. Our observations of methylation blocks surrounding the PPARγ CpG island and high correlations between the north shore and gene body sites add evidence that shore sites may be of particular relevance in regulating biological pathways.
With respect to changes in CpG methylation over time, although some reports indicate stable methylation patterns [53, 56] others do not [57–59]. In their analyses of blood samples from the Netherlands Twin Register, Talens et al. (2010) showed that of 8 regions examined, 5 displayed stable methylation patterns for up to 20 years [56]. Additionally, using Illumina 450 k data, we have previously shown that methylation across 16 paraoxonase 1 gene (PON1) shore, shelf, and island sites was highly conserved comparing birth and 9-year time points [53]. On the other hand, Fraga et al. (2004) showed that while 3-year-old monozygotic (MZ) twins showed relatively few epigenetic differences, there was considerably larger variability in older twin pairs [57]. Our results indicate that PPARγ methylation is stable over the birth to 9-year period and that even minute differences between CpG sites are conserved.
Although the pattern of CpG sites remained similar over time (Fig. 1), we found that north shore sites had slightly but significantly higher beta values (7.4%) at 9 years compared to birth. Previous literature has identified both hypo and hyper-methylation changes with age and taken together, these findings suggest that different genomic regions may have varying stability over time [5, 28, 60, 61]. Additionally, we observed small differences by sex, with girls having slightly higher methylation compared to boys, at both birth and 9-year time points. However, these differences were limited to north shore sites 1–3. Although the significance of this remains unclear, our previous work using 450 k data identified that overall ~ 3% of CpG sites are differentially methylated by sex and are enriched for genes related to nervous system development and behavior [53]. Interestingly, Hall et al. (2014) showed that genome-wide CpG methylation in pancreatic islets differentially clustered between males and females, suggesting that methylation may be involved in sex-specific metabolic differences [62]. Our results are in line with this and overall show that PPARγ CpG methylation is carefully maintained, emphasizing its potentially important role in regulating PPARγ function.
In addition to data gaps on methylation structure and organization, very little is known about the epigenetic changes that accompany obesity development. Our report of an inverse relationship between PPARγ methylation and body size is consistent with the idea that higher methylation downregulates PPARγ, suppressing adipogenesis. To date, few studies have examined these relationships in PPARγ, providing mixed results [63, 64]. Yan et al. (2014) examined PPARγ gene expression and methylation in offspring of dams exposed to polycyclic aromatic hydrocarbons (PAHs), reporting that increased PAH exposure was associated with increased weight, fat mass, higher gene expression of PPARγ and lower PPARγ CpG methylation [64]. In contrast to this inverse relationship between PPARγ methylation and weight, Drogan et al. (2015) analyzed subcutaneous adipose tissue (SAT) samples, showing that tissues from individuals with higher visceral fat mass had increased PPARγ CpG methylation [63]. Additionally, Nilsson et al. (2014) found differential PPARγ methylation in adipose tissues from subjects with type 2 diabetes compared to controls but did not report on this relationship’s direction [65]. We did not find that PPARγ methylation at birth could predict 9-year BMI z-score and more work is needed to further elucidate its role in PPARγ function and adipogenesis over time. Of note, site 1 was located in the north shore, further emphasizing the potentially critical role of north shore sites in regulating gene expression.
Lastly, there are several important points to consider with respect to our analyses. We measured methylation in blood samples, which can introduce bias if cell heterogeneity affects both methylation and obesity. However, our sensitivity analyses accounting for differences in cell composition did not substantially alter associations between PPARγ methylation and child size. Furthermore, our data displayed a consistent pattern of CpG methylation in blood samples over both birth and 9-year time points suggesting that heterogeneity of blood cell types may not significantly affect PPARγ methylation.
Nevertheless, whether PPARγ methylation in blood is a suitable marker for its activity in adipocytes remains unknown. Several studies have indicated that molecular changes in blood do reflect pathological changes in the body and gene expression in blood is highly concordant (>80%) with expression in other tissues [66, 67, 68–70]. With respect to body size, Ghosh et al. (2010) used principal components analysis to show that blood-based gene expression signals could distinguish between obese and lean subjects [71]. Interestingly, Charriere et al. (2003) found that based on transcriptome profiling, pre-adipocytes were more closely related to macrophages than adipocytes [72]. Further, a large genome-wide association study found that BMI was associated with methylation of HIF3A in both blood and adipose tissue [70]. Taken together, these data suggest that assessing PPARγ function in blood may be biological relevant however more work is needed to determine this in the context of methylation.
Additionally, although we had previously shown that methylation at PPARγ site 23 (gene body) was associated with PPARγ gene expression [27], this site was not significantly associated with child birth weight or BMI. Reasons for this inconsistency remain unclear and further research is warranted to examine relationships between CpG location and potential effects on gene expression. Overall, while our research argues that PPARγ methylation has a relationship with child body weight and that north shore sites may be of particular functional importance, key questions remain on factors that influence site-specific methylation and whether it can be used to predict metabolic outcomes over time.
Conclusion
In summary, PPARγ CpG methylation is highly organized and conserved over time. We found high inter-CpG correlations between sites 1–3 (methylation block 1) and also between sites 18–23 (methylation block 2) for both birth and 9-year time points. Additionally, we report that methylation at birth for sites 1 (north shore) and 20 (gene body) was significantly and inversely associated with birth weight. Similarly, we found that methylation at these sites at 9 years was also significantly and inversely related to 9-year BMI z-score. Taken together, these results indicate that PPARγ methylation may be associated with child body size.
Acknowledgements
We are grateful to the laboratory and field staff and participants of the CHAMACOS study for their contributions. We are thankful to Dr. Lisa Barcellos and Ms. Hong Quach for their help with the Illumina 450 k methylation assay. We are also thankful to Kelly Street for his help generating heat maps in R.
Funding
This work was supported by grants from the National Institute of Environmental Health Science (NIEHS) [PO1 ES009605, R01ES023067, R01 ES021369] and from the US Environmental Protection Agency (EPA) [RD83451301, R82670901].
Availability of data and material
The CHAMACOS data used in this analysis cannot be shared as they are collected from a vulnerable, minority population of Mexican-American children and mothers and require strict protection.
Authors’ contributions
VV, NH, KH, BE conceived the study and were involved in its design and data gathering. VV, PY, KHuen carried out analysis of Illumina 450 k and obesity-related data. VV, NH drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not Applicable.
Ethics approval and consent to participate
Study protocols (2010-01-620 & 2010-03-949) ethics were approved by the University of California, Berkeley Committee for Protection of Human Subjects. Written informed consent was obtained from all mothers and assent was provided by the children at the 9-year assessment.
Abbreviations
- BMI
Body mass index
- CDC
Centers for disease control and prevention
- CHAMACOS
Center for the Health Assessment of Mothers and Children of Salinas
- CpG
Cytosine-guanine dinucleotides
- CV
Coefficient of variation
- DAGs
Directed acyclic graphs
- DNA
Deoxyribonucleic acid
- EWAS
Epigenome-wide association study
- GEE
Generalized estimating equations
- HIF3A
Hypoxia-inducible factor 3-alpha
- IAP
Intracisternal A particle
- MXL
Mexican ancestry in Los Angeles, California
- MZ
Monozygotic
- PAHs
Polycyclic aromatic hydrocarbons
- PON1
Paraoxonase 1 gene
- PPARγ
Peroxisome proliferator-activated receptor γ
- SAT
Subcutaneous adipose tissue
- SNP
Single nucleotide polymorphism
- UTR
Untranslated region
Additional files
PPARγ Promoter and Gene Body CpG Sites. Figure S1 shows the distribution of 23 CpG sites (red squares) measured by the Illumina Methylation 450 K Array. Blue squares indicate all other CpG sites (N = 183). Numbers in the DNA sequence, such as “2100” are shorthand for number of base pairs. (PPTX 295 kb)
{kind=link}
Associations between perinatal characteristics and PPARγ methylation at birth. (PNG 42 kb)
{kind=link}
Associations between perinatal characteristics and PPARγ methylation at 9 years. (PNG 50 kb)
Contributor Information
Vitaly Volberg, Email: vvolberg@gmail.com.
Paul Yousefi, Email: yousefi@berkeley.edu.
Karen Huen, Email: khuen@berkeley.edu.
Kim Harley, Email: kharley@berkeley.edu.
Brenda Eskenazi, Email: eskenazi@berkeley.edu.
Nina Holland, Phone: 510-455-0561, Email: ninah@berkeley.edu.
References
- 1.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 2.Lustig RH. Obesity Before Birth: Maternal and Prenatal Influences on the Offspring. Springer Sceince. 2011;30:1–25. [Google Scholar]
- 3.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 4.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1(1):11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 5.Bell JT, Spector TD. A twin approach to unraveling epigenetics. Trends Genet. 2011;27(3):116–125. doi: 10.1016/j.tig.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell JT, Spector TD. DNA methylation studies using twins: what are they telling us? Genome Biol. 2012;13(10):172. doi: 10.1186/gb-2012-13-10-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 8.Baccarelli A, Rienstra M, Benjamin EJ. Cardiovascular epigenetics: basic concepts and results from animal and human studies. Circ Cardiovasc Genet. 2010;3(6):567–573. doi: 10.1161/CIRCGENETICS.110.958744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103(5):1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes (Lond) 2008;32(9):1373–1379. doi: 10.1038/ijo.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guénard F, Tchernof A, Deshaies Y, Cianflone K, Kral JG, Marceau P, et al. Methylation and expression of immune and inflammatory genes in the offspring of bariatric bypass surgery patients. J Obes. 2013;2013:492170. doi: 10.1155/2013/492170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharp GC, Lawlor DA, Richmond RC, Fraser A, Simpkin A, Suderman M, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2015;44(4):1288–304. doi: 10.1093/ije/dyv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richmond RC, Sharp GC, Ward ME, Fraser A, Lyttleton, McArdle WL, Ring SM, Gaunt TR, Lawlor DA, Davey Smith G, Relton CL. DNA Methylation and BMI: Investigating Identified Methylation Sites at HIF3A in a Causal Framework. Diabetes. 2016;65(5):1231–44. doi: 10.2337/db15-0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharp GC, Lawlor DA, Richmond RC, Fraser A, Simpkin A, Suderman M, Shihab HA, Lyttleton O, McArdle W, Ring SM, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2015;44(4):1288–1304. doi: 10.1093/ije/dyv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L, Sha J, Pankow JS, Liu C, Irvin MR, et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring, Md) 2015;23(7):1493–1501. doi: 10.1002/oby.21111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 20.Vidal-Puig A, Jimenez-Linan M, Lowell BB, Hamann A, Hu E, Spiegelman B, Flier JS, Moller DE. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest. 1996;97(11):2553–2561. doi: 10.1172/JCI118703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50(9):2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 22.Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4(4):597–609. doi: 10.1016/S1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- 23.Larsen TM, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int J Obes Relat Metab Disord. 2003;27(2):147–161. doi: 10.1038/sj.ijo.802223. [DOI] [PubMed] [Google Scholar]
- 24.Toruner F, Akbay E, Cakir N, Sancak B, Elbeg S, Taneri F, Akturk M, Karakoc A, Ayvaz G, Arslan M. Effects of PPARgamma and PPARalpha agonists on serum leptin levels in diet-induced obese rats. Horm Metab Res. 2004;36(4):226–230. doi: 10.1055/s-2004-814452. [DOI] [PubMed] [Google Scholar]
- 25.Noer A, Sorensen AL, Boquest AC, Collas P. Stable CpG hypomethylation of adipogenic promoters in freshly isolated, cultured, and differentiated mesenchymal stem cells from adipose tissue. Mol Biol Cell. 2006;17(8):3543–3556. doi: 10.1091/mbc.E06-04-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujiki K, Kano F, Shiota K, Murata M. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009;7:38. doi: 10.1186/1741-7007-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dave V, Yousefi P, Huen K, Volberg V, Holland N. Relationship between expression and methylation of obesity-related genes in children. Mutagenesis. 2015;30(3):411–420. doi: 10.1093/mutage/geu089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eskenazi B, Gladstone EA, Berkowitz GS, Drew CH, Faustman EM, Holland NT, Lanphear B, Meisel SJ, Perera FP, Rauh VA, et al. Methodologic and logistic issues in conducting longitudinal birth cohort studies: lessons learned from the Centers for Children's Environmental Health and Disease Prevention Research. Environ Health Perspect. 2005;113(10):1419–1429. doi: 10.1289/ehp.7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eskenazi B, Harley K, Bradman A, Weltzien E, Jewell NP, Barr DB, Furlong CE, Holland NT. Association of in utero organophosphate pesticide exposure and fetal growth and length of gestation in an agricultural population. Environ Health Perspect. 2004;112(10):1116–1124. doi: 10.1289/ehp.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, Wei R, Curtin LR, Roche AF, Johnson CL. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11. 2002;(246):1–190. Data from the national health survey. [PubMed]
- 31.Stettler N, Zemel BS, Kumanyika S, Stallings VA. Infant weight gain and childhood overweight status in a multicenter, cohort study. Pediatrics. 2002;109(2):194–199. doi: 10.1542/peds.109.2.194. [DOI] [PubMed] [Google Scholar]
- 32.Yousefi P, Huen K, Aguilar Schall R, Decker A, Elboudwarej E, Quach H, Barcellos L, Holland N. Considerations for normalization of DNA methylation data by Illumina 450 k BeadChip assay in population studies. Epigenetics. 2013;8(11):1141–1152. doi: 10.4161/epi.26037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yousefi P, Huen K, Quach H, Motwani G, Hubbard A, Eskenazi B, Holland N. Estimation of blood cellular heterogeneity in newborns and children for epigenome-wide association studies. Environ Mol Mutagen. 2015;56(9):751–758. doi: 10.1002/em.21966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houwen B. The differential cell count. Lab Hematol. 2001;7:89–100. [Google Scholar]
- 36.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxford, England) 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dallman P. Blood and blood-forming tissues. In: Rudolph Q, editor. Paediatrics. New York: Appleton; 1977. pp. 1109–1112. [Google Scholar]
- 38.Nathan D, Oski F, Orkin S. Nathan and Oski’s Hematology of Infancy and Childhood. 6. Philadelphia: Saunders; 2003. [Google Scholar]
- 39.Bakulski KM, Feinberg JI, Andrews SV, et al. DNA methylation of cord blood cell types: Applications for mixed cell birth studies. Epigenetics. 2016;11:354–362. doi: 10.1080/15592294.2016.1161875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andrews S, Bakulski K. FlowSorted.CordBlood.450k: Illumina 450k data on sorted cord blood cells. 2016. Available at: https://www.bioconductor.org/packages/devel/data/experiment/html/FlowSorted.CordBlood.450k.html. Accessed 25 May 2016.
- 41.de Goede OM, Razzaghian HR, Price EM, et al. Nucleated red blood cells impact DNA methylation and expression analyses of cord blood hematopoietic cells. Clin Epigenetics. 2015;7(1):95. doi: 10.1186/s13148-015-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gervin K, Hansen KD. Bioconductor - FlowSorted.CordBloodNorway.450k. 2016. Available at: https://bioconductor.org/packages/release/data/annotation/html/FlowSorted.CordBloodNorway.450k.html. Accessed 25 May 2016.
- 43.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20(7):883–889. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Li X, Aryee MJ, Ekstrom TJ, Padyukov L, Klareskog L, Vandiver A, Moore AZ, Tanaka T, Ferrucci L, et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet. 2014;94(4):485–495. doi: 10.1016/j.ajhg.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volberg V, Harley KG, Aguilar RS, Rosas LG, Huen K, Yousefi P, Dave V, Phan N, Lustig RH, Eskenazi B, et al. Associations between perinatal factors and adiponectin and leptin in 9-year-old Mexican-American children. Pediatr. Obes. 2013;8(6):454–463. doi: 10.1111/j.2047-6310.2012.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dulloo AG, Jacquet J, Seydoux J, Montani JP. The thrifty 'catch-up fat' phenotype: its impact on insulin sensitivity during growth trajectories to obesity and metabolic syndrome. Int J Obes (Lond) 2006;30(Suppl 4):S23–35. doi: 10.1038/sj.ijo.0803516. [DOI] [PubMed] [Google Scholar]
- 48.Ong KK, Preece MA, Emmett PM, Ahmed ML, Dunger DB. Size at birth and early childhood growth in relation to maternal smoking, parity and infant breast-feeding: longitudinal birth cohort study and analysis. Pediatr Res. 2002;52(6):863–867. doi: 10.1203/00006450-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 49.Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004;114(1):e29–36. doi: 10.1542/peds.114.1.e29. [DOI] [PubMed] [Google Scholar]
- 50.Hediger ML, Overpeck MD, McGlynn A, Kuczmarski RJ, Maurer KR, Davis WW. Growth and fatness at three to six years of age of children born small- or large-for-gestational age. Pediatrics. 1999;104(3):e33. doi: 10.1542/peds.104.3.e33. [DOI] [PubMed] [Google Scholar]
- 51.Sofer T, Schifano ED, Hoppin JA, Hou L, Baccarelli AA. A-clustering: a novel method for the detection of co-regulated methylation regions, and regions associated with exposure. Bioinformatics (Oxford, England) 2013;29(22):2884–2891. doi: 10.1093/bioinformatics/btt498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huen K, Yousefi P, Street K, Eskenazi B, Holland N. PON1 as a model for integration of genetic, epigenetic, and expression data on candidate susceptibility genes. Envrionmental Epigenetics. 2015;1(1):1–11. doi: 10.1093/eep/dvv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41(12):1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar R, Tan PP, Portales-Casamar E, Pavlidis P. Meta-analysis of human methylomes reveals stably methylated sequences surrounding CpG islands associated with high gene expression. Epigenetics Chromatin. 2014;7(1):28. doi: 10.1186/1756-8935-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Talens RP, Boomsma DI, Tobi EW, Kremer D, Jukema JW, Willemsen G, Putter H, Slagboom PE, Heijmans BT. Variation, patterns, and temporal stability of DNA methylation: considerations for epigenetic epidemiology. FASEB J. 2010;24(9):3135–3144. doi: 10.1096/fj.09-150490. [DOI] [PubMed] [Google Scholar]
- 57.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, Moffitt TE, Mill J. A longitudinal study of epigenetic variation in twins. Epigenetics. 2010;5(6):516–526. doi: 10.4161/epi.5.6.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299(24):2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130(4):234–239. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20(3):332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hall E, Volkov P, Dayeh T, Esguerra JL, Salo S, Eliasson L, Ronn T, Bacos K, Ling C. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 2014;15(12):522. doi: 10.1186/s13059-014-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drogan D, Boeing H, Janke J, Schmitt B, Zhou Y, Walter J, Pischon T, Tierling S. Regional distribution of body fat in relation to DNA methylation within the LPL, ADIPOQ and PPARgamma promoters in subcutaneous adipose tissue. Nutr Diabetes. 2015;5:e168. doi: 10.1038/nutd.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan Z, Zhang H, Maher C, Arteaga-Solis E, Champagne FA, Wu L, McDonald JD, Yan B, Schwartz GJ, Miller RL. Prenatal polycyclic aromatic hydrocarbon, adiposity, peroxisome proliferator-activated receptor (PPAR) gamma methylation in offspring, grand-offspring mice. PLoS One. 2014;9(10):e110706. doi: 10.1371/journal.pone.0110706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, Poulsen P, Ribel-Madsen R, Pedersen NL, Almgren P, et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes. 2014;63(9):2962–2976. doi: 10.2337/db13-1459. [DOI] [PubMed] [Google Scholar]
- 66.Mohr S, Liew CC. The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol Med. 2007;13(10):422–432. doi: 10.1016/j.molmed.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147(3):126–132. doi: 10.1016/j.lab.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Zhu H, Snieder H, et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010;8:87. doi: 10.1186/1741-7015-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chadwick LH, Sawa A, Yang IV, et al. New insights and updated guidelines for epigenome-wide association studies. Neuroepigenetics. 2015;1:14–19. doi: 10.1016/j.nepig.2014.10.004. [DOI] [Google Scholar]
- 70.Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383:1990–8. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 71.Ghosh S, Dent R, Harper ME, Gorman SA, Stuart JS, McPherson R. Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Med Genomics. 2010;3:56. doi: 10.1186/1755-8794-3-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Charriere G, Cousin B, Arnaud E, Andre M, Bacou F, Penicaud L, Casteilla L. Preadipocyte conversion to macrophage. Evidence of plasticity. J Biol Chem. 2003;278(11):9850–9855. doi: 10.1074/jbc.M210811200. [DOI] [PubMed] [Google Scholar]