

Abstract
Background: Temozolomide-resistant (TMZ-R) glioblastoma is very difficult to treat, and a novel approach to overcome resistance is needed. Materials and Methods: The efficacy of a combination treatment of STAT3 inhibitor, STX-0119, with rapamycin was investigated against our established TMZ-resistant U87 cell line. Results: The growth-inhibitory effect of the combination treatment was significant against the TMZ-R U87 cell line (IC50: 78 μM for STX-0119, 30.5 μM for rapamycin and 11.3 μM for combination of the two). Western blotting analysis demonstrated that the inhibitory effect of STX-0119 on S6 and 4E-BP1 activation through regulation of YKL-40 expression occurred in addition to the inhibitory effect of rapamycin against the mTOR pathway. Conclusion: These results suggest that the STAT3 pathway is associated with the mTOR downstream pathway mediated by YKL-40 protein, and the combination therapy of the STAT3 inhibitor and rapamycin could be worth developing as a novel therapeutic approach against TMZ-resistant relapsed gliomas.
Abbreviations: GB: Glioblastoma, TMZ: temozolomide, MGMT: O6-methylguanine-O6-methylguanine-DNAmethyltransferase, STAT: signal transducer and activator of transcription, mTOR: mammalian target of rapamycin, shRNA: small hairpin RNA.
Keywords: STAT3 inhibitor, mTOR inhibitor, temozolomide resistance, glioblastoma
Glioblastoma (GB) is one of the most malignant and aggressive tumors and has a very poor prognosis, with a mean survival time of less than 2 years even with the recent development of temozolomide (TMZ)-based intensive treatment (1,2). Once recurrence develops, there are few therapeutic approaches to control the growth of glioblastoma. Therefore, TMZ-resistant GB is very difficult to treat, and a novel approach to overcome resistance is needed.
With regard to the mechanism of TMZ resistance, O6-methylguanine-DNA -methyltransferase (MGMT) removes methylation from the O6 position of guanine and contributes to TMZ resistance induction (3). It is generally accepted that high MGMT expression through the methylation of the MGMT promoter is one of the mechanisms responsible for TMZ resistance. Alternatively, several novel biomarkers linked to MGMT expression and the methylation status such as the HOX signature and epidermal growth factor receptor (EGFR) expression (4), somatic mutation of mismatch repair gene mutS homolog (MSH)6 (5), prolyl 4-hydroxylase, beta polypeptide (P4HB), EGFR mutation (EGFRvIII) (6), CD74 and signal transducer and activator of transcription (STAT)3 signaling have been reported. Kohsaka et al. reported the association of STAT3 expression with MGMT expression level using small interfering (si)RNA-mediated STAT3 gene inhibition (7).
Additionally, mammalian target of rapamycin (mTOR) signaling is activated in TMZ-resistant glioma cells as a result of EGFR, phosphoinositide 3 kinase (PI3K) and Akt signaling activation. mTOR is a Ser/Thr kinase that belongs to the phosphoinositide kinase-related family of protein kinases (PIKKs). mTOR acts as an essential integrator of growth-factor-activated and nutrient-sensing pathways to control and coordinate various cellular functions, including survival, proliferation, differentiation, autophagy and metabolism (8-11). More interestingly, Johnson et al. identified a novel mTOR-specific single nucleotide variant (SNV) in a clinical tumor specimen from TMZ-resistant glioma patients (12), which can contribute to mTOR signal activation in TMZ resistance.
Previously, we identified a novel inhibitor of STAT3 dimerization, STX-0119, that exhibited a potent anti-tumor effect on a TMZ-resistant U87 glioma cell line, and demonstrated that anti-tumor activity was partly mediated by a down-regulation of YKL-40 (13-15). In the present study, we focused on cancer signaling in the TMZ-resistant glioma cell line, in which the STAT3 and PI3K/Akt/mTOR pathways were highly activated, and investigated the effect of a combination therapy of the STAT3 inhibitor (STX-0119) and mTOR inhibitor (rapamycin) on a TMZ-resistant glioblastoma cell line in vitro and in vivo. We demonstrated that combination therapy effectively inhibited the proliferation of even a TMZ-resistant cell line through the possible association of the STAT3 and mTOR signaling pathways.
Materials and Methods
TMZ-resistant U87 cell line and antibodies. TMZ-resistant (R) U87 cells were established using a TMZ-dose-escalation method up to 150 μM, were maintained at a dose of 100 μM TMZ and were used for in vitro and in vivo experiments (14). Antibodies against STAT3, phospho-STAT3 (Y705), cleaved caspase-3, EGFR, phospho-EGFR (Y845, Y1173), Ras, PI3 kinase p85, phospho-PI3 kinase p85 (Y458), Akt, phospho-Akt1 (S473), mTOR, phospho-mTOR (S2448), S6, phospho-S6 (S235, S236), 4E-BP1, phospho-4E-BP1 (T37, T46), B-Raf, phospho-B-Raf (S445), extracellular signal-regulated kinase (ERK)1/2, mitogen-activated protein kinase (MAPK), phospho-ERK1/2 pMAPK (T202, Y204), p38 MAPK and phospho-p38 MAPK (T180, Y182) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) and Becton-Dickinson (BD) Biosciences (Franklin Lakes, NJ, USA) for Western blotting (WB). Mouse anti-human YKL-40 antibody was purchased from Abcam (Cambridge, MA, USA).
Chemicals. STX-0119 was supplied by the Center for Drug Discovery, University of Shizuoka (Shizuoka, Japan). These compounds were suspended and diluted in a sterile 0.5%w/v methyl cellulose 400cp solution (Wako, Tokyo, Japan) for use in animal experiments. TMZ and rapamycin were purchased from SIGMA-ALDRICH (St. Louis, MO, USA) and Selleck chemicals (Houston, TX, USA).
Cell proliferation assay. Cell proliferation was examined using the WST-1 assay (Dojin Kagaku Corp., Kumamoto, Japan) described previously (14). Briefly, 1×104 parental U87 or TMZ-R U87 cells were seeded into each well of a 96-well micro-culture plate (Corning, NY, USA) and compounds ranging from 0-200 μM were added alone or combination. After 4 days, the WST-1 substrate was added to the culture and optical density (OD) was measured at 450 and 620 nm using an immunoreader (Immuno Mini NJ-2300; Nalge Nunc International, Roskilde, Denmark). The IC50 value was defined as the dose needed for a 50% reduction in OD calculated from the survival curve. Percent survival was calculated as follows: (mean O.D. of test wells – mean O.D. of background wells)/(mean O.D. of control wells – mean O.D. of background wells) ×100.
Western blotting. TMZ-R U87 cells were treated with STX-0119 or rapamycin at the determined doses for 24 h. The cells were lysed using RIPA buffer (Thermo Fisher Scientific Inc., Rockford, IL, USA) containing protease inhibitors and phosphatase inhibitors and used for western blotting as described previously (13). Briefly, cell lysate was subjected to SDS-PAGE with a 7.5% polyacrylamide separating gel, and then transferred to PVDF membranes. After blocking, the membranes were incubated at 4 °C overnight with various primary antibodies in blocking solution. After washing, the membranes were incubated for 1 h with horseradish peroxidase (HRP)-conjugated anti-mouse IgG. Membranes were treated with ECL plus reagent (GE Healthcare, Piscataway, NJ, USA) and analyzed on a chemiluminescence scanner (LAS-4000; GE Healthcare). Apoptosis induction in TMZ-R U87 cells treated with chemical reagents for 24 h was investigated using a Caspase-3 Western detection kit including the primary antibody against cleaved caspase-3 (Cell Signaling).
Inhibition of YKL-40 gene expression using shRNA transfection into TMZ-resistant U87 cells. The shRNA gene transfection into the TMZ-R U87 cell line was performed using a lipofection FreeStyle MAX reagent (Life Technology, Carlsbad, CA, USA) as reported previously (15). Briefly, YKL-40 shRNA-containing plasmid (SureSilencing shRNA vector; Qiagen GmbH, Hilden, Germany) and FreeStyle MAX reagent was suspended in opti-MEM I reduced-serum medium (Life Technologies), that was then mixed and incubated for 15 min at room temperature (RT). The solution was added to 2×106 TMZ-resistant U87 cells, which were incubated in DMEM plus 10% FBS and utilized for in vitro experiments on day3. Similarly, plasmid pcDNA3.1 (Life Technologies) containing YKL-40 cDNA was transfected using lipofection into the parental U87 cell line, and transiently obtained U87 cells producing a high amount of YKL-40 protein were used for western blot analysis.
Animal experiments. Male nude mice (BALB/cA-nu/nu, 5-6 weeks old) were obtained from Nippon Clea (Tokyo, Japan). All of the animals were cared for and used humanely according to the guidelines for the welfare and use of animals in cancer research published in Br J Cancer in 2010, and the procedures were approved by the Animal Care and Use Committee of Shizuoka Cancer Center Research Institute.
TMZ-R U87 cells (1×106) were inoculated into the flank of BALB/cA-nu/nu mice. To evaluate the anti-tumor activity against subcutaneous (s.c.) inoculated tumors, tumor volume was calculated based on the National Cancer Institute formula as follows: tumor volume (mm3)=length (mm) × [width (mm)]2 × 1/2.
STX-0119 was administered orally daily from day 0 to day 4 followed by 2 day of rest (15 administrations at 40 mg/kg for 18 day). Rapamycin was administered intraperitoneally every other day followed by 2 days of rest (9 administrations at 4 mg/kg for 18 days). The efficacy of the treatment was expressed as the mean V/V0 value or tumor/control ratio, where V is the tumor volume on the day of evaluation and V0 is the tumor volume on the day of treatment.
Whole-exome sequencing (WES) analysis of the TMZ-R U87 cell line using next-generation sequencing. WES was performed using an Ion Proton system with the AmpliSeq Exome kit as reported previously (16). Briefly, raw data were processed, filtered and converted to sequence reads by Torrent Suite Software and sequenced reads were mapped to the reference human genome (UCSC hg19) by tmap. Variant call was performed for each cell line separately by using Torrent Variant Caller. Quality <30, depth of coverage <20 and variant allele frequency <10% were applied to filter out low quality mutations. The IonReporter tumor-normal workflow was applied to identify those mutations that were observed in the TMZ-R cell line only.
Statistical analysis. Statistical differences were analyzed using Student’s t-test. Values of p<0.05 were considered statistically significant. For the in vivo experiment, statistical analysis was performed with corrected p-values to compare with the untreated control using Mann-Whitney’s rank-sum test.
Results
Cancer signal pathway profiling of the TMZ-R U87 cell line. Phospho-EGFR, phospho-PIK3, phospho-Akt, phospho-mTOR (S6 and 4E-BP1), and phospho-STAT3 signaling molecule protein levels were found up-regulated in TMZ-R U78 cells compared to the parental U87 cells (Figure 1). In contrast, the levels of the RAS downstream molecules phospho-raf, phospho-ERK, and phospho-MAPK were down-regulated.
Figure 1. Cancer cell signaling profile in the TMZ-R U87 cell line. Cancer cell-associated signal molecules, such as EGFR, PI3K, Akt, mTOR, S6, 4E-BP1, Ras, Raf, ERK, MAPK and STAT3, were investigated using western blotting analysis. For each blot: Left, U87 parental cell line; right, TMZ-R U87 cell line; upper, whole protein; lower, phosphorylated protein.

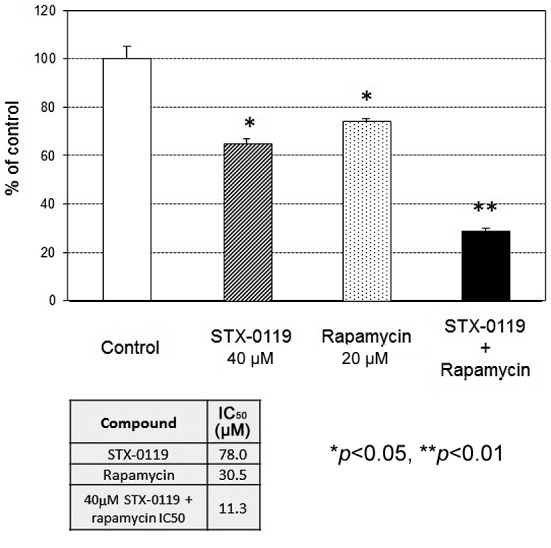
Cell proliferation assay. STX-0119 and rapamycin displayed a moderate inhibitory effect on TMZ-R U87 cells (STX-0119 IC50=87 μM, rapamycin IC50=30.5 μM) (Figure 2). Remarkably, a combination of a suboptimal dose of STX-0119 (40 μM) and rapamycin (20 μM) significantly suppressed the proliferation of TMZ-R U87 cells by more than 70% compared to a single reagent (IC50=11.3 μM).
Figure 2. Effect of STX-0119 and/or rapamycin on the proliferation of the TMZ-R U87 cell line. The proliferation of the TMZ-R U87 cell line without treatment was designed 100 as a control, and the growthinhibitory effect of the drug was expressed as % control. Each column shows the mean±SD of triplicate samples. Open column: Control, shaded column: STX-0119, hatched column: rapamycin, closed column: STX-0119 and rapamycin. *p<0.05, **p<0.01, statistically significant.
Effect of a combination treatment on signaling pathways of the TMZ-R U87 cell line. STX-0119 alone moderately inhibited the expression of both STAT3 and mTOR signaling molecules; however, rapamycin alone inhibited only mTOR. Remarkably, a combination of STX-0119 and rapamycin at 40 μM and 20 μM, respectively, significantly suppressed STAT3 and PI3K/Akt/mTOR signaling molecule levels (Figure 3).
Figure 3. Effect of a combination treatment of STX-0119 and rapamycin on cancer cell signaling in the TMZ-R U87 cell line. The cells were treated with STX-0119 and/or rapamycin for 24 h and then used for WB analysis of cancer cell signaling molecules, such as EGFR, PI3K, Akt, mTOR, S6, 4E-BP1, HIF-1, Ras, Raf, ERK, MAPK and STAT3. (A) Whole proteins, (B) phosphorylated proteins.

Apoptosis induction by a combination of STX-0119 with rapamycin in TMZ-R U87 cell line. Cleaved caspase-3 expression increased in TMZ-R U87 cells treated with rapamycin. Additionally, a combination of STX-0119 and rapamycin demonstrated the highest increase of cleaved caspase-3 expression in TMZ-R U87 cells (Figure 3).
Impact of the regulation of YKL-40 gene expression on PIK3/Akt/mTOR signaling. YKL-40 mRNA inhibition by shRNA inhibited mTOR and 4E-BP1, but not S6. Alternatively, YKL-40 overexpression in the parental U87 cells using YKL-40 cDNA transduction induced a significant up-regulation of the S6 signaling molecule. In contrast, mTOR phosphorylation was inhibited (Figure 4).
Figure 4. Impact of YKL-40 expression level on mTOR pathway signals. The expression levels of mTOR, S6 and 4E-BP1 phosphoproteins were investigated in the case of (A) YKL-40-downregulation (TMZ-R U87 cells) or (B) YKL-40-upregulation (U87 parental cells). At the bottom, each phosphoprotein level is shown as % of control (C) Open column: Control, closed column: YKL-40 down-regulated by shRNA, (D) Open column: control, closed column:YKL-40 overexpressed by gene transfection. (E) A speculated pathways between STAT3-YKL-40 and mTOR signaling. Solid line: Stimulating signal, dashed line: inhibiting signal.

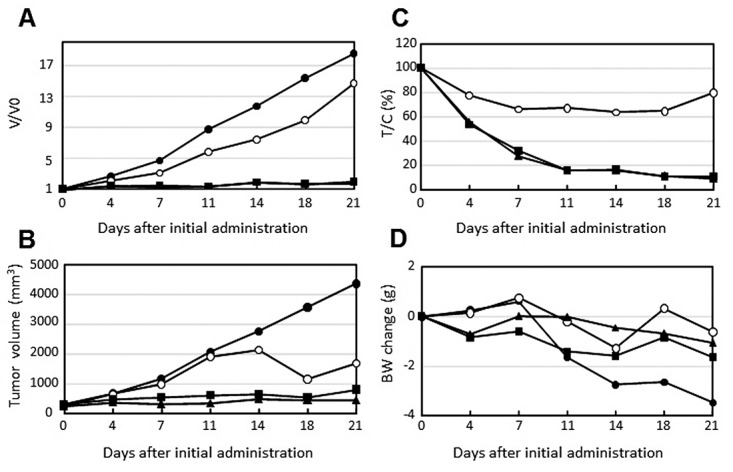
Effect of the combination of STX-0119 with rapamycin on TMZR U87 tumor growth in vivo. TMZ-R U87 cell-transplanted mice showed significant resistance to TMZ and a shorter survival time in vivo compared to the parental U87 cell line, as shown in our previous study (14). STX-0119 alone showed a moderate inhibitory effect on TMZR U87 tumor growth in nude mice. In contrast, rapamycin alone exhibited improved growth inhibition of TMZR U87 tumors. Therefore, a combination of STX-0119 with rapamycin did not show a significant additive effect on TMZR U87 tumor growth (Figure 5).
Figure 5. Inhibitory effect of STX-0119 and/or rapamycin on in vivo tumor growth of TMZ-R U87 cells. Nude mice transplanted with TMZ-R U87 cells were used. (●) Control, (●) STX-0119, (■) rapamycin, (▲) STX-0119 and rapamycin. The tumor growth was indicated as V/V0 value (A), actual tumor volume (B) or tumor/control ratio (%) (C). To evaluate adverse effects, the change in body weight is shown in (D). Each point shows the mean value of 5 mice.
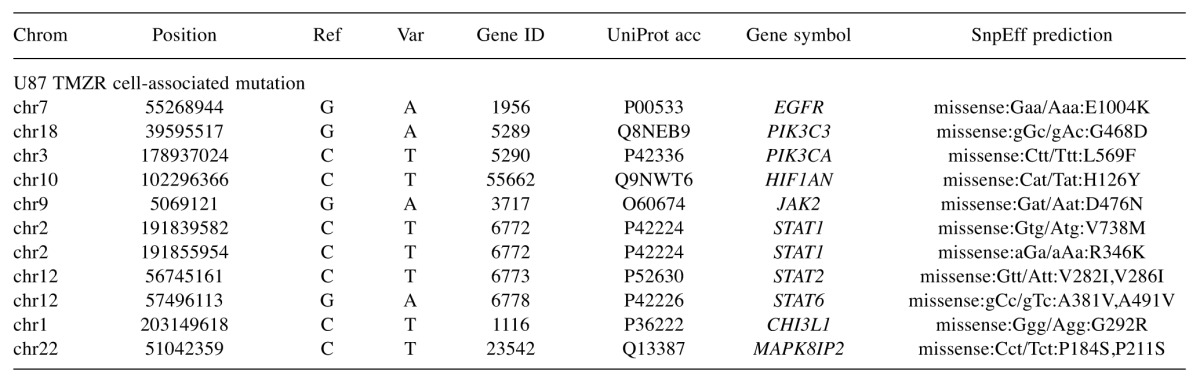
Whole-exome sequencing (WES) analysis of the TMZ-R U87 cell line. The number of mutated genes and SNVs per cell line was 9,533 and 22,824 in the U87 parental cell line and 11,837 and 30,872 in the U87 TMZ-R cell line, respectively. U87 TMZ-R cell-specific SNVs related to cancer signaling pathway-associated genes are shown in Table I. The number of non-synonymous SNVs detected per cell line was 389 in the U87 parental and 686 in the U87 resistant cell line. U87 TMZ-R cell-specific SNVs were found in the EGFR, PIK3C3, STAT1, STAT2, STAT6 and YKL-40 (CHI3L1) genes. Interestingly, a somatic mutation of the EGFR C-terminal domain (E1004K) was verified. Additionaly, an adjacent mutation (ER1005-1006KD) was reported to induce spontaneous activation of EGFR phosphorylation (17). Alternatively, a somatic mutation of the YKL-40 gene (G292R) was also found, but it was unlikely to be functional in signal transduction.
Table I. U87-TMZR signaling-associated SNV gene list.
Discussion
Glioblastomas are the most malignant and aggressive of tumors and have a very poor prognosis and a high recurrence rate, with a less than 5% survival rate at 2 years.
Frequent recurrence even after chemo-radiation treatment is a crucial problem in the clinical field, which must be overcome to extend the overall survival of GBM patients. Recently, genomic studies using exome-based clinical sequencing revealed tumor-specific genetic alterations according to the tumor development stage from primary to relapse as follows; loss of heterozygosity (LOH) 10q (69%), EGFR amplification (34%), TP53 mutation (31%), p16 (INK4a) deletion (31%) and phosphatase and tensin homolog deleted on chromosome ten (PTEN) mutations (24%) in primary glioblastomas which account for 95% of all glioblastomas (18,19).
PTEN is a phosphatase that was, originally identified as a tumor suppressor gene and is frequently altered in a variety of human cancers, such as brain, breast and prostate (20,21); these alterations lead to deregulation of protein synthesis, the cell cycle, migration, growth, DNA repair and survival signaling. PTEN inactivation by deletion and mutations occurs in approximately 40% of glioblastoma patients. PTEN inactivation is known to induce PI3K/Akt/mTOR signal activation in various tumors. Targeting these pathways is often complex and can result in pathway activation depending on the presence of upstream mutations (22,23). In glioblastomas, PTEN inactivation-based mTOR signal activation is frequently seen together with PI3K mutation-based amplification; therefore, those tumors are more sensitive to mTOR inhibitors (24,25). Additionally, as for mTOR activation in glioblastomas, Pelloski et al. showed that higher expression of phospho (p)-mTOR, p-70S6K and p-MAPK was associated with worse outcome, as shown by immunohistochemical staining in 268 cases of newly diagnosed glioblastomas. Importantly, Cloughesy et al. reported anti-tumor activity of the mTOR inhibitor rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastomas; however, the result of the trial was disappointing (25).
Cell signaling profiling of TMZ-R U87 cells using WB analysis demonstrated that p-EGFR, p-PIK3, p-Akt, p-mTOR (S6 and 4E-BP1), and phosho-STAT3 signaling protein levels were up-regulated in TMZ-R U87 cells compared to parental U87 cells. These results seemed to be similar to the observation verified in TMZ-resistant glioma cells or tissues (11,23). The inhibitory effect of rapamycin seemed to be efficient but was restricted to p-mTOR and p-70S6, and did not affect the p-PIK3 or p-Akt proteins. STX-0119 was specific and potent for p-STAT3, and the inhibitory effect on p-mTOR and p-Akt protein was positive but moderate. Remarkably, a combination of rapamycin with STX-0119 diminished both PI3K/Akt/mTOR and STAT3 signaling and significantly suppressed the growth of TMZ-R U87 cells.
The signaling relationship between mTOR and STAT3 has been suggested in a few studies regarding mTOR inhibitor development (26-28). Specifically, Hu et al. (27) identified brain-expressed X-linked 2 (BEX2), a novel downstream molecule of the mTOR pathway; they found that mTOR signal activation can be mediated by BEX2 and transmitted to the STAT3 and NF-ĸB pathways. Oroxylin, a natural mono-flavonoid, reported by Zou et al., inhibited Akt and ERK activation and the downstream phosphorylation of mTOR and STAT3, which was mediated by Beclin 1, a key autophagy-related protein (28). Our shRNA-mediated gene inhibition study showed that both STAT3 and YKL-40 gene inhibition suppressed mTOR signaling; however, inhibition of the mTOR signaling pathway was greater when YKL-40 was inhibited (data not shown); this result suggests that mTOR is located downstream of the STAT3/YKL-40 pathway, because STAT3 signal inhibition by STX-0119 resulted in a significant reduction of the YKL-40 protein level in our previous study (14).
Another important issue regarding mTOR signal activation in TMZ-R U87 cells is genetic alteration of the mTOR gene (29-31). Briefly, three studies regarding mTOR mutation research have been cited. Sato et al. identified two mTOR point mutations (S2215Y and R2505P) out of 750 cancer samples from the COSMIC library; however, these mutations were not functional (29). Gerlinger et al. identified a tumor-specific functional mTOR mutation (L2431P) from four cases of metastatic renal cancer that showed mTOR kinase activation with S6 and 4EBP phosphorylation (30). Finally, Johnson et al. reported that a novel mTOR mutation (S2215F) was identified in relapsed secondary glioblastoma tissue, which induced p-RPS6 and p-4EBP1 activation (12).
Our whole-exome sequencing of TMZ-R U87 cells revealed that tumor-specific SNVs (non-synonymous mutations) were identified, and some SNVs belonged to the Vogelstein cancer driver gene group. The list of genes that were specifically mutated in TMZ-resistant U87 cells contained EGFR, PIK3CA, MSH2, ARID1A, PRDM1, and SMARCA4, some of which had already been reported in relapsed glioblastoma tissue (data not shown). However, specific mTOR mutations were not recognized, as demonstrated by Johnson et al. (12). Interestingly, a novel mutation (E1004K) located in the C-terminal tail domain of EGFR was found in Table I. Considering that the N-terminal half of the EGFR C-terminal tail domain has an inhibitory function in EGFR signal activation (31) and that a neighboring mutation (ER1005-1006KD) was reported to induce spontaneous activation of EGFR phosphorylation (17), this mutation (E1004K) might be involved in EGFR signaling. Alternatively, a novel somatic mutation of the YKL-40 gene (G292R) was also found, but it likely dose not function in signal transduction.
In contrast to the in vitro study, the in vivo study of a combination treatment of STX-0119 and rapamycin against TMZ-R U87 tumors did not show a significant additive inhibitory effect. There is a concern regarding the dose of rapamycin used in the in vivo study. Mukhopadhyay et al. (32) demonstrated the enigma of rapamycin dosage: a low dose of rapamycin inhibits mainly S6 phosphorylation, and a high dose inhibits both S6 and 4E-BP1 phosphorylation. Therefore, different doses of rapamycin can be investigated to identify the optimal combination for in vivo study.
The present study is the first report of a successful combination therapy of an mTOR inhibitor and a STAT3 inhibitor against TMZ-R glioblastoma cells in vitro. Additionally, we made the novel observation of an association between mTOR and STAT3 signaling that is mediated by the YKL-40 protein (Figure 4E). Considering that novel dual or triple inhibitor development against PI3K/Akt/mTOR signal activation has been in progress since the development of rapalogs, everolimus and temsirolimus (33-38), the novel effective combination of an mTOR inhibitor and a STAT3 inhibitor shown in the present study deserves attention as a novel therapeutic approach against relapsed TMZ-resistant glioblastomas.
Conflicts of Interest
The Authors have no competing interests to declare.
Acknowledgements
The Authors thank Drs Nagashima and Shimoda, Cancer Diagnostics Division, Shizuoka Cancer Center Research Institute for excellent technical support for whole-exome sequencing of glioma cell lines.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Mirimanoff RO, Gorlia T, Mason W, Van den Bent MJ, Kortmann RD, Fisher B, Reni M, Brandes AA, Curschmann J, Villa S, Cairncross G, Allgeier A, Lacombe D, Stupp R. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol. 2006;24(16):2563–2569. doi: 10.1200/JCO.2005.04.5963. [DOI] [PubMed] [Google Scholar]
- 3.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozo-lomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 4.Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26(18):3015–3024. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 5.Xie C, Sheng H, Zhang N, Li S, Wei X, Zheng X. Association of MSH6 mutation with glioma susceptibility, drug resistance and progression. Mol Clin Oncol. 2016;5(2):236–240. doi: 10.3892/mco.2016.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camorani S, Crescenzi E, Colecchia D, Carpentieri A, Amoresano A, Fedele M, Chiariello M, Cerchia L. Aptamer targeting EGFRvIII mutant hampers its constitutive autophosphorylation and affects migration, invasion and proliferation of glioblastoma cells. Oncotarget. 2015;6(35):37570–37587. doi: 10.18632/oncotarget.6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, Tanino M, Kimura T, Nishihara H, Tanaka S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by donregulating MGMT expression. Mol Cancer Ther. 2012;11(6):1289–1299. doi: 10.1158/1535-7163.MCT-11-0801. [DOI] [PubMed] [Google Scholar]
- 8.Majewska E, Szeliga M. AKT/GSK3β signaling in glioblastoma. Neurochem Res. 2016 doi: 10.1007/s11064-016-2044-4. 10.1007/s11064-016-2044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin F, de Gooijer MC, Hanekamp D, Chandrasekaran G, Buil L, Thota N, Sparidans RW, Beijnen JH, Wurdinger T, van Tellingen O. PI3K-mmTOR pathway inhibition exhibits efficacy against high-grade glioma in clinically relevant mouse models. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1276. 10.1158/1078-0432.CCR-16-1276. [DOI] [PubMed] [Google Scholar]
- 10.Neshat MS, Mellinghoff IK, Trans C, Stiles B, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001;98(18):10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Basecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, Tafuri A, Cocco L, Evangelisti C, Chiarini F, Martelli AM. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3(9):954–987. doi: 10.18632/oncotarget.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, Weiss WA, Smirnov IV, Song JS, Olshen AB, Cha S, Zhao Y, Moore RA, Mungall AJ, Jones SJ, Hirst M, Marra MA, Saito N, Aburatani H, Mukasa A, Berger MS, Chang SM, Taylor BS, Costello JF. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashizawa T, Miyata H, Ishii H, Oshita C, Matsuno K, Masuda Y, Furuya T, Okawara T, Otsuka M, Ogo N, Asai A, Akiyama Y. Antitumor activity of a novel small molecules STAT3 inhibitor against a human lymphoma cell line with high STAT3 activation. Int J Oncol. 2011;38(5):1245–1252. doi: 10.3892/ijo.2011.957. [DOI] [PubMed] [Google Scholar]
- 14.Ashizawa T, Akiyama Y, Miyata H, Iizuka A, Komiyama M, Kume A, Omiya M, Sugino T, Asai A, Hayashi N, Mitsuya K, Nakasu Y, Yamaguchi K. Effect of the STAT3 inhibitor STX-0119 on the proliferation of a temozolomide-resistant glioblastoma cell line. Int J Oncol. 2014;45(1):411–418. doi: 10.3892/ijo.2014.2439. [DOI] [PubMed] [Google Scholar]
- 15.Akiyama Y, Ashizawa T, Komiyama M, Miyata H, Oshita C, Omiya M, Iizuka A, Kume A, Sugino T, Hayashi N, Mitsuya K, Nakasu Y, Yamaguchi K. YKL-40 down-regulation is a key factor to overcome temozolomide resistance in a glioblastoma cell line. Oncol Rep. 2014;32(1):159–166. doi: 10.3892/or.2014.3195. [DOI] [PubMed] [Google Scholar]
- 16.Urakami K, Shimoda Y, Ohshima K, Nagashima T, Serizawa M, Tanabe T, Saito J, Usui T, Watanabe Y, Naruoka A, Ohnami S, Ohnami S, Mochizuki T, Kusuhara M, Yamaguchi K. Next generation sequencing approach for detecting 491 fusion genes from human cancer. Biomed Res. 2016;37(1):51–62. doi: 10.2220/biomedres.37.51. [DOI] [PubMed] [Google Scholar]
- 17.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtramembrane segment. Cell. 2009;137(7):1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lütolf UM, Kleihues P. Genetic pathways to glioblastoma:a population-based study. Cancer Res. 2004;64(19):6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 19.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Lavarone A. The transcriptional network for mesenchymal transformation of brain tumors. Nature. 2010;463(7279):318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function:the long and the short of it. Trends Biochem Sci. 2014;39(4):183–190. doi: 10.1016/j.tibs.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27(41):5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 22.Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, Neshat M, Wang H, Yang L, Gibbons J, Frost P, Dreisbach V, Blenis J, Gaciong Z, Fisher P, Sawyers C, Hedrick-Ellenson L, Parsons R. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in PTEN+/– mice. Proc Natl Acad Sci USA. 2001;98(18):10320–10325. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nocoletti F, Bäsecke J, Mijatovic M, Maksimovic-Ivanic D, Milella M, Tafuri A, Chiarini F, Evangelisti C, Cocco L, Martelli AM. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors:how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3(10):1068–1111. doi: 10.18632/oncotarget.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL, Lee JS. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011;18(4):666–677. doi: 10.1038/cdd.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng W, Zhang C, Ren X, Jiang Y, Han S, Liu Y, Cai J, Li M, Wang K, Liu Y, Hu H, Li Q, Yang P, Bao Z, Wu A. Bioinformatic analyses reveal a distinct Notch activation induced by STAT3 phosphorylation in the mesenchymal subtype of glioblastoma. J Neurosurg. 2016 doi: 10.3171/2015.11.JNS15432. 10.3171/2015.11.jns15432. [DOI] [PubMed] [Google Scholar]
- 27.Hu Z, Wang Y, Huang F, Chen R, Li C, Wang F, Goto J, Kwiatkowski DJ, Wdzieczak-Bakala J, Tu P, Liu J, Zha X, Zhang H. Brain-expressed X-linked 2 is pivotal for heperactive mechanistic target of rapamycin (mTOR)-mediated tumorigenesis. J Biol Chem. 2015;290(42):25756–25765. doi: 10.1074/jbc.M115.665208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou M, Hu C, You Q, Zhang A, Wang X, Guo Q. Oroxylin A induces autophagy in human malignant glioma cells via the mTOR-STAT3-Notch signaling pathway. Mol Carcinog. 2015;54(11):1363–1375. doi: 10.1002/mc.22212. [DOI] [PubMed] [Google Scholar]
- 29.Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;29(18):2746–2752. doi: 10.1038/onc.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovacs E, Das R, Wang Q, Collier TS, Cantor A, Huang Y, Wong K, Mirza A, Barros T, Grob P, Jura N, Bose R, Kuriyan J. Analysis of the role of the C-terminal tail in the regulation of the epidermal growth factor receptor. Mol Cell Biol. 2015;35(17):3083–3102. doi: 10.1128/MCB.00248-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukhopadhyay S, Frias MA, Chatterjee A, Yellen P, Faoter DA. The enigma of rapamycin dosage. Mol Cancer Ther. 2016;15(3):347–353. doi: 10.1158/1535-7163.MCT-15-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grabiner BC, Nordi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4(5):554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Z, Xie G, Zhou G, Cheng Y, Zhang G, Yao G, Chen Y, Li Y, Zhao G. NVP-BEZ235, a novel dual PISK-mTOR inhibitor displays anti-glioma activity and reduce chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015;367(1):58–68. doi: 10.1016/j.canlet.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Ströbele S, Schneider M, Schneele L, Siegelin MD, Nonnenmacher L, Zhou S, Karpel-Massier G, Westhoff MA, Halatsch ME, Debatin KM. A potential role for the inhibition of PIK3 signaling in glioblastoma therapy. PLoS One. 2015;10(6):e0131670. doi: 10.1371/journal.pone.0131670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K, Tofilon PJ. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Nature Oncol. 2014;16(1):29–37. doi: 10.1093/neuonc/not139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blumenthal DT, Dvir A, Lossos A, Tzuk-Shina T, Lior T, Limon D, Yust-Katz S, Lokiec A, Ram Z, Ross JS, Ali SM, Yair R, Soussan-Gutman L, Bokstein F. Clinical utility and treatment outcome of comprehensive genomic profiling in high grade glioma patients. J Neurooncol. 2016;130(1):211–219. doi: 10.1007/s11060-016-2237-3. [DOI] [PubMed] [Google Scholar]
- 38.Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, Robins HI, Lieberman FS, Gilbert MR, Mehta MP, Drappatz J, Groves MD, Santagata S, Ligon AH, Yung WK, Wright JJ, Dancey J, Aldape KD, Prados KD, Ligon KL. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014;16(4):567–578. doi: 10.1093/neuonc/not247. [DOI] [PMC free article] [PubMed] [Google Scholar]