

Abstract
Oligosaccharide natural products target a wide spectrum of biological processes including disruption of cell wall biosynthesis, interference of bacterial translation, and inhibition of human α-amylase. Correspondingly, oligosaccharides possess potential for development as treatments of such diverse diseases as bacterial infections and type II diabetes. Despite their potent and selective activities and potential clinical relevance, isolated bioactive secondary metabolic oligosaccharides are less prevalent than other classes of natural products and their biosynthesis has received comparatively less attention. This review highlights the unique modes of action and biosynthesis of four classes of bioactive oligosaccharides: the orthosomycins, moenomycins, saccharomicins, and acarviostatins.
1. Introduction
A large fraction of secondary metabolites may be considered oligomeric in nature, derived via sequences of condensation and modification of monomeric precursors that originate from various branches of primary and secondary metabolism. For instance, amino acids, isopentyl pyrophosphate, and malonates condense into polypeptide, polyisoprenoid, and polyketide secondary metabolites, respectively, or mixtures thereof. In many cases these classes provide core scaffolds that are further elaborated by oxidation, cyclization, and frequently glycosylation. Derivatization via glycosylation, which is typically effected via glycosyltransferase-mediated condensation of nucleoside diphosphate (NDP)-sugars onto aglycone scaffolds, is an essential determinant of bioactivity for many secondary metabolites.1-4 Multiple glycosylation is not uncommon and indeed is a recurrent theme in many bioactive natural products.5 Given that poly-glycosylation is a common property of bioactive secondary metabolites, one would expect that oligomeric natural products derived predominantly from monomeric sugar precursors may also be common. Indeed, while oligosaccharide natural products possessing a wide range of discerned biological activities and molecular targets are reported, they are not as well represented in nature’s isolated pharmacopeia as other classes. This is perhaps surprising given the importance of oligosaccharide structural relatives found in primary metabolism which are important mediators of molecular recognition, particularly in cellular recognition, cancer, and the immunology of microbial pathogenesis.6-9 The reasons for the oligosaccharide natural products’ relative underrepresentation in secondary metabolic databases are unknown. Possibly these large, highly functionalized, polar metabolites may be expressed at lower levels, be more difficult to detect, isolate and identify, and be less stable than other classes of compounds.
The biosynthesis of oligomeric secondary metabolites has been primarily studied in the context of their appendage to polyketides, polypeptides, and polyisoprenoid scaffolds. However, the biochemistry of assembly of oligosaccharide secondary metabolites, either assembled on aglycones or into oligosaccharides, is only marginally understood at this time. Moreover, the inferred existence of secondary metabolic polysaccharides in genomically sequenced organisms is also less common. This may be a result of the scarcity of annotated and biochemically rationalized oligosaccharide secondary metabolite gene clusters.
Herein we collect and discuss the subset of oligosaccharide natural products (see figure 1) reported to possess biological activity, with a focus on oligosaccharides larger than tetrasaccharides that have been biosynthetically characterized. Often these compounds comprise moderate and high molecular weight oligosaccharides that compete with binding sites of very large substrates, such as in the case of the moenomycins, or target large surface area macromolecular interfaces, as with the orthosomycin antibiotics. Understanding the biosynthesis of oligosaccharide natural products will aid in the identification of new members of this relatively underrepresented class of secondary metabolite via genomic analysis and potentially enable opportunities for rational reengineering of this compound class for improved pharmacological properties.
Figure 1.

Representative members of the families of bioactive oligosaccharide natural products discussed in this review.
2. Orthosomycins
The orthosomycins were first coined as a class of antibiotics in 1979 although hygromycin B, produced by S. hygroscopicus, was isolated two decades earlier.10,11 All orthosomycins contain a non-canonical orthoester linkage between sugar residues rather than the typical glycosidic linkage. The orthosomycins can be divided into two classes based on the number of orthoester linkages and sugar residues. Class I orthosomycins may be defined as hepta- and octasaccharides with two orthoester linkages and a dichloroisoeverninic acid ester. Avilamycins, everninomicins, flambamycin, and curamycin are all members of this class. Class II orthosomycins are pseudotrisaccharides with one orthoester linkage and an aminocyclitol. Hygromycin B and the destomycins are examples of this class. Class II orthosomycins will not be discussed in this review as they are more aptly classified with the aminoglycosides.12
Because little is known about the activity and biosynthesis of flambamycin and curamycin, this review will focus on the avilamycins and everninomicins. A complex of everninomicins, produced by Micromonospora carbonacea, was first described in 1964,13 and in 1975 the structure of everninomicin D was reported.14 To date, twelve everninomicins have been reported.10,14-22 Figure 2 shows the variety of everninomicins isolated from Micromonospora carbonacea. All everninomicins, with the exception of Ever-2 which lacks the A ring nitro sugar, are octasaccharides containing dichloroisoeverninic acid. The majority of everninomicins also contain orsellinic acid at the opposite end of the saccharide chain. Everninomicins possess three unique oxidative features. The first is a methylenedioxy bridge attached to ring F. The second is its namesake orthoester linkages located between rings C and D and rings G and H. Finally, L-evernitrose (ring A) is a nitrosugar unique to everninomicins. In contrast with the other polysaccharides discussed in this review, the everninomicins contain a large proportion of deoxy sugars. Rings A, B (D-olivose), and C (D-olivose), and sometimes ring D (D-evalose) are all 2,6-dideoxy sugars while ring E (4-O-methyl-D-fucose) is 6-deoxygenated. Ring F is 2,6-di-O-methyl-D-mannose, ring G is L-lyxose, and ring H is eurekanate.
Figure 2.

Naturally occurring everninomicins and avilamycins.
Avilamycins, produced by Streptomyces viridochromogenes Tü57, are heptasaccharides similar to everninomicin but lacking the nitrosugar. At least sixteen avilamycins have been characterized to date (Figure 2).10,23 Avilamycins have the same seven-sugar core as the everninomicins. All avilamycins contain dichloroisoeverninic acid but lack orsellinic acid at the eastern side of the molecule. The main points of differentiation among the avilamycins are the decorations of rings G and H. As in the everninomicins, the avilamycins also contain a methylenedioxy bridge and two orthesters located between rings C and D and rings G and H.
Avilamycin antibiotics have found wide spread application as growth promoters in animal feed.24 Interest in the everninomicin series of orthosomycins peaked in the early 2000s when Schering-Plough Corporation was developing everninomicin A (Ziracin) as an antimicrobial agent. Development of this product reached phase III clinical trials prior to withdrawal for unstated pharmacological complications.25 However, investigation of the orthosomycins are still of interest as members of this class possess potent activity against clinically important strains such as methicillin-resistant staphylococci, vancomycin-resistant enterococci, and penicillin-resistant streptococci and may be especially useful for treating infective endocarditis.26-28
2.1 Mode of Action
The orthosomycins act as bacterial translation inhibitors although by a different mechanism than antibiotics currently in clinical use. Early studies revealed that everninomicin is a potent inhibitor of prokaryote but not eukaryote protein synthesis, specifically targeting the 50S ribosomal subunit.29, 30,14C-labeled everninomicin bound specifically to the E. coli and S. aureus 50S subunit. In competitive binding assays, known inhibitors of protein synthesis which also target the 50S subunit such as chloramphenicol, clindamycin, erythromycin, linezolid, and thiostrepton were unable to block binding of 14C-labeled everninomicin. Only avilamycin and unlabelled everninomicin were able to block its binding suggesting that the orthosomycins target a unique site of the ribosome.29
Specifically, everninomicin appears to interact with ribosomal protein L16 and r23S RNA helices 89 and 91 (Figure 3). Streptococcus pneumonia strains with low levels of resistance to everninomicin were found to contain an isoleucine 52 to serine mutation in ribosomal protein L16.31 Later studies confirmed this mutation as well as identifying two other L16 protein mutations which conferred everninomicin resistance. Mutation of arginines 56 and 51 to histidines resulted in everninomicin resistance in E. faecium, E. faecalis, and S. aureus.32, 33
Figure 3.
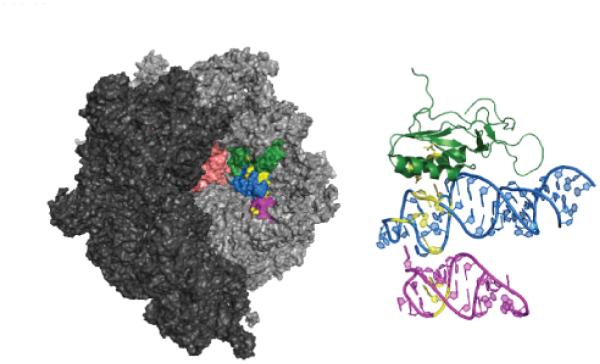
Ribosomal binding site of orthosomycin antibiotics. Small ribosomal subunit (PDB 2J00) is shown in dark grey and large subunit (PDB 2J01) is shown in lighter grey. The A and P sites are shown in salmon. Ribosomal protein L16 is shown in green (chain Q), helix 89 (chain A, residues 2454-2498) in blue, and helix 91 (chain A, residues 2520-2545) in magenta. Amino acid residues and nucleotides known to interact with everninomicin and avilamycin are highlighted in yellow.
In addition, an Enterococcus faecium strain was isolated which exhibited high-level resistance to everninomicin and avilamycin. The resistant strains contained a plasmid-borne rRNA methyltransferase called EmtA which methylates G2470 of 23S rRNA.34 rRNA footprinting as well as mutational studies indicate that everninomicin interacts with several residues in helices 89 and 91 of 23S rRNA: A2468, A2469, A2476, A2478, A2482, A2534, G2535. 32, 35 Additionally, characterization of rRNA methyltransferases AviRa and AviRb revealed their role in methylation of G2535 and U2479 in 23S rRNA indicating that these residues are important for orthosomycin binding to the ribosome.36, 37 Figure 3 highlights the amino acid residues and nucleotides which are known to interact with everninomicin and avilamycin.
More recently, Wilson and co-workers have shown that everninomicin prevents formation of the 70S initiation complex in an IF2-dependent manner. Moreover, it was demonstrated that orthosomycins do not inhibit translocation but are potent inhibitors of EF4-dependent back-translocation. Interestingly, mutations in protein L16 convey low-level everninomicin resistance while methylation or mutation of 23S rRNA results in high-level resistance. These results indicate that everninomicin interacts directly with helices 89 and 91 but indirectly with ribosomal protein L16. Nevertheless, the orthosomycins have been shown to target a unique site on the large ribosomal subunit which is approximately 50 angstroms from the peptidyl-transferase center where many commonly used antibiotics are known to bind. 38
During its development as an antimicrobial agent, the structure-activity relationship of everninomicin was investigated. Methylation or other capping of the hydroxyl group of the dichloroisoeverninic acid moiety significantly reduced activity against S. aureus; this suggests a crucial interaction between dichloroisoeverninic acid and the target. In addition permethylation of hydroxyls on rings C-G significantly reduces activity against S. aureus.39, 40 Compound SCH58777 which is composed of rings F, G, H and orsellinic acid had no antibacterial activity.15 While everninomicin A was active against both Gram-negative and Gram-positive bacteria, both everninomicin D and everninomicin B were highly active against Gram-positive bacteria but inactive against Gram-negative strains.10,26 The major difference between everninomicins A and D is the attachment of orsellinic acid to the eurekanate residue suggesting the second aromatic ring is important for conferring activity against Gram-negative strains. Additionally, reduction of the nitro group to the amino oxidation state abolished Gram-positive activity while substantially increasing Gram-negative activity.41 Everninomicin 2 has activity similar to that of everninomicin A yet does not contain the nitrosugar indicating that while L-evernitrose can modulate activity it is not necessary for antibacterial activity.10 Finally, hydrolysis of the orthoester between rings C and D resulted in a loss of activity while reported inversion of the orthoester stereochemistry also reduced activity.40
Studies of the biosynthesis of avilamycin have also revealed clear structure-activity relationships. Inversion of the stereochemistry of C2 of ring F by disruption of aviX12 resulted in the generation of a new analog with drastically reduced activity.42 Desmethyl analogs created by disruption of five methyltransferases were isolated and tested for activity against a panel of streptococcus, enterococcus, and staphylococcus strains. Removal of any of the naturally occurring methyl groups significantly lowered the activity against enterococci species but did not drastically alter activity against streptococcus and staphylococcus strains. Although the removal of methyl groups did not inhibit antibacterial activity against streptococcus and staphylococcus strains, the polarity of the compounds was changed significantly. Physiochemical studies indicated that the desmethyl analogs had up to 10-fold higher water solubility.43 These results underscore the potential for pharmacological improvements of the orthosomycins by creating new analogs.
2.2 Biosynthesis
The avilamycin A biosynthetic gene cluster from Streptomyces viridochromogenes Tü57 was first reported in 1997. Inactivation of two genes confirmed the role of this cluster in avilamycin biosynthesis.44 This large cluster appears to contain 4 glycosyltransferases, 22 sugar synthesis and tailoring genes, 2 genes for orsellinic acid biosynthesis, 1 halogenase, 3 oxidases, 5 genes involved in regulation and transport, and 2 genes responsible for avilamycin resistance (figure 4b).45
Figure 4.

Orthosomycin biosynthesis. Structure of avilamycin A and everninomicin A. Avi gene cluster from S. viridichromogenes Tü57 and Eve gene cluster from M. carbonacea var africana. Genes are color-coded according to putative functions.
Four genes from the avilamycin cluster have been implicated in dichloroisoeverninic acid biosynthesis. AviM is responsible for orsellinic acid synthesis while AviN may control the starter unit. Inactivation of aviG4 resulted in loss of a methyl group from dichloroisoeverninic acid confirming it as an O-methyltransferase. Additionally, inactivation of the halogenase aviH resulted in an avilamycin analog lacking the two chlorine atoms of ring A.45
The exact function of the remaining three O-methyltransferases was determined by gene inactivation. AviG2 methylates the C6 oxygen of ring F, AviG5 is responsible for O-methylation of ring E, and AviG6 methylates the C2 hydroxyl of ring F (Scheme 1B). Bechthold and coworkers generated double and triple mutant combinations of these methyltransferases to produce an array of avilamycin analogs termed gavibamycins.43 Disruption of the gene encoding C-methyltransferase AviG1 resulted in abolished avilamycin production. However, complementation of a C-methyltransferase eryBIII mutant with aviG1 from the erythromycin pathway in Saccharopolyspora erythraea resulted in restored erythromycin production. This experiment confirmed the role of AviG1 as a C-methyltransferase likely involved in the synthesis of ring D.46
Scheme 1.

Orthosomycin biosynthesis. A) Proposed scheme for formation of ring G, L-lyxose. AviE2 has been shown to catalyze the decarboxylation of UDP-D-glucuronic acid to UDP-D-xylose. B) Scheme for formation of 2,6-di-O-methyl-D-mannose from 6-O-methyl-D-glucose. AviX12 catalyzes a unique radical epimerization reaction while AviG6 methylates the 2-hydroxyl presumably after epimerization. C) Formation of L-evernitrose from L-epi-vancosamine. ORF36 catalyzes the oxidation of the nitrogen from the amino to the nitroso oxidation state. It is likely spontaneous oxidation of the nitroso congener which leads to the nitro form.
In vitro characterization of AviE2 revealed that it is a UDP-D-glucuronic acid decarboxylase involved in conversion of UDP-D-glucuronic acid to UDP-D-xylose. This results indicate that the pentose L-lyxose is originally derived from UDP-D-glucose. Two additional epimerization steps are necessary to convert D-xylose to L-lyxose (Scheme 1A). The authors hypothesize that aviQ1, aviQ2, or aviQ3 may encode the necessary chemistries for these epimerizations. This is the first description of a UDP-glucuronic acid decarboxylase involved in secondary metabolism.47
Inactivation of aviX12 resulted in formation of an avilamycin analog containing D-glucose rather than D-mannose (ring F) which possess different stereochemistries at C2. Additionally the C2 hydroxyl was not methylated suggesting that epimerization precedes methylation of this position. As mentioned above, epimerization of the hydroxyl at C2 results in complete loss of antibiotic activity. Therefore, AviX12 is necessary for formation of an active avilamycin. However, this epimerization is notable as it takes place at an unactivated carbon (Scheme 1B). Upon characterization of its [Fe-S] cluster, AviX12 was determined to be a member of the radical AdoMet family, and AviX12 appears to be the first reported member of the radical AdoMet family involved in epimerization of a sugar.42
Gene inactivation experiments suggest that AviO2 and AviB1 are involved in eurekanate biosynthesis. Loss of aviO2 and aviB1 resulted in an avilamycin derivative proposed to have lost the acetyl residue at position C4 of ring H. It was hypothesized that AviB1 and AviB2 are part of an incomplete pyruvate decarboxylase complex that catalyzes the conversion of pyruvate to an acetyl carbanion which is subsequently attached to the saccharide chain through the action of AviO2.48
However, it has been previously proposed that AviO1, AviO2, and AviO3 were oxidases involved in orthoester and methylenedioxy bridge formation. Their original analysis of the avilamycin gene cluster found that these three genes had homology to non-heme iron, α-ketoglutarate dependent oxidases which are not likely involved in deoxysugar biosynthesis.45 Inactivation of aviO1 and aviO3 resulted in abolished production although, as detailed above, inactivation of aviO2 resulted in a putative de-acetylated avilamycin analog.48 These results are curious in light of inspection of the everninomicin gene clusters from M. carbonacea var africana49 (GenBank accession number AX195929) (figure 4C) and M. carbonacea var aurantiaca (GenBank accession numbers AX574200-2). Although everninomicin contains orsellinic acid attached to eurekanate rather than an acetyl group, its gene cluster still contains a close homolog of aviO2. Based on translated sequence similarities, putative functions for the genes have been proposed (see figure 4C). Additionally all known class I orthosomycins gene clusters contain three oxidases with striking homology to the three from the avi cluster. The class II orthosomycin hygromycin B gene cluster also contains a putative non-heme iron, α-ketoglutarate dependent oxidase, HygX. Based on this evidence, we hypothesize that the family of α-ketoglutarate dependent oxidases is responsible for orthoester and methylenedioxy bridge formation.
Gene inactivation of aviGT4 resulted in an avilamycin derivative which lacked the terminal eurekanate moiety. Interestingly, eurekanate is attached to the saccharide chain via an orthoester linkage in all orthosomycins. The lack of this linkage suggests that either AviGT4 alone is responsible for orthoester formation or, more likely, glycosylation precedes orthoester formation.47
The everninomicin gene cluster from M. carbonacea var africana ATCC39149 was reported in 2001. Insertional inactivation of everJ, everF, and everW resulted in abolished everninomicin production confirming the role of this gene cluster in everninomicin biosynthesis.49 Although few biosynthetic studies of the everninomicin gene cluster have been reported, the nitrososynthase ORF36 from M. carbonacea var africana has been well characterized. Analysis of two everninomicin gene clusters and two avilamycin gene clusters accompanied by subtractive analysis identified a cassette of genes involved in L-evernitrose formation (Figure 4C, genes N1-M7). Of particular interest is ORF36 (N1) a flavin-dependent monooxygenase which has been shown to oxidize the amino sugar L-TDP-epi-vancosamine to the nitroso form (Scheme 1C).50 Fermentation under aphotic conditions also results in accumulation of the nitroso compound indicating that full oxidation to the nitro may not be enzymatically catalyzed. A five-enzyme in vitro pathway was constructed to test the catalytic competence of ORF36. ORF36 was able to convert TDP-L-epi-vancosamine progenitors to the hydroxylamine oxidation state. 18O2 labelling experiments revealed that molecular oxygen is incorporated into the hydroxylamine and nitroso products. Additionally, an X-ray crystal structure of ORF36 was solved revealing a tetrameric enzyme with a fold similar to that of class D flavin-containing monooxygenases. The structure also revealed an unusually open active site which may explain their promiscuity.51 Inactivation of aviP, a putative phosphatase, did not influence avilamycin production. However, inactivation of aviD, aviO1, aviO3, aviE2, aviG1, everJ, everF, and everW resulted in abolished orthosomycin production.
3. Moenomycins
First described in 1965, the moenomycins comprise a novel class of phosphoglycolipids natural products produced by at least four Streptomyces species.52,53 The moenomycins were first isolated as a complex of antibiotics from S. ghanaensis with moenomycin A being the predominant congener and founding member.52, 54 Moenomycin A can be divided into three structurally distinct regions: a pentasaccharide, a phosphoglycerate, and a C25 isoprenyl (moenocinyl) lipid tail. As shown in Figure 5, all moenomycins contain a core tetrasaccharide attached to a lipid chain through phosphoglycerate. Approximately half of structurally characterized moenomycins contain the D-ring sugar residue forming the full pentasaccharide.53 Other points of variability include rings C and E, which can be either N-acetyl glucosamine or the 6-deoxy derivative chinovosamine, and the C4 position of ring F. A new subfamily of the moenomycins termed nosokomycins have been isolated from Streptomyces sp. K04-0144. All nosokomycins lack the A ring common to other moenomycins and instead contain either a carboxylate or carbamate at this position.55, 56 Two additional structurally characterized members include pholipomycin57-60 and AC326-α61 produced by S. lividoclavatus and the unidentified Actinomyces sp. AC326 respectively. AC326-α is the only moenomycin that contains a diumycinol lipid tail rather than moenocinol.61 At least 25 potential moenomycin family members have been reported although the structures of most have yet to be fully elucidated.56, 58 A natural mixture of moenomycins (also known as bambermycins) marketed as Flavomycin® and Flavophospholipol have been used as animal growth promoters for at least forty years.62-64 Despite their long use, no significant resistance to moenomycins has been described.62, 65, 66
Figure 5.

Naturally occurring moenomycins. Adapted from Ostash 2010.53
The moenomycins are the only known direct inhibitors of peptidoglycan glycosyltransferases (PGTs) and are active against a range of Gram-positive bacteria with a potency of up to 1000-fold that of vancomycin.53, 67, 68 Despite their novel mode of action and potency, the moenomycins have poor pharmacokinetic properties, including poor oral bioavailability and a long half-life, which have inhibited their clinical use.69 However, interest in this class of antibiotics was renewed in the late 1990s and early 2000s, and significant efforts have been made to create moenomycin analogs with improved properties and retained potency. An excellent review has been written recently on the moenomycin family of antibiotics including chemical and biological efforts to produce novel derivatives, and we direct readers to that article for a thorough history of the moenomycins.53 In this review, we aim to highlight their unique mode of action, structural features contributing to their activity, and the biosynthetic pathway.
3.1 Mode of Action
Moenomycin was first determined to be an inhibitor of cell wall biosynthesis of S. aureus in 1968 by Huber and Nesemann.70 Bacterial cell wall synthesis begins with the cytosolic synthesis of UDP-N-acetyl muramyl-pentapeptide which is subsequently transferred to a C55 undecaprenol phosphate carrier lipid by MraY to generate MurNAc-pentapeptide-pyrophosphoyl-undecaprenol or lipid I. N-acetylglucosamine is then transferred to lipid I by MurG to produce N-acetylglucosamine-β-1,3-MurNAc-pentapeptide-pyrophosphoryl-undecaprenol or lipid II. Transglycosylases, such as the bifunctional penicillin-binding proteins (PBPs) or the monofunctional MtgA, catalyze the polymerization of lipid II into peptidoglycan.69, 71 Moenomycins exert their effects by inhibiting the transglycosylation step of cell wall biosynthesis leading to cell lysis and death.67
Although moenomycin was originally thought to be a competitive inhibitor of lipid II,69 more recent studies have shown the inability of lipid II to displace moenomycin in E. coli PBP1b indicating that moenomycin does not bind in the same area as lipid II. The authors proposed that moenomycin could be blocking elongation by binding in the polysaccharide binding site rather than the lipid II site.72 Supporting this experiment, the structure of bifunctional PBP2 in complex with moenomycin reveals that it binds in an extended conformation that probably mimics the growing polysaccharide-chain substrate.73 Subsequently, additional structures of moenomycin bound to various PGTs have been reported. Figure 6 illustrates two S. aureus monofunctional glycosyltransferase (MtgA) in complex with moenomycin. Moenomycin interacts with highly conserved residues in the active site of the glycosyltransferase providing the basis for its interaction across a broad range of PGTs.74 Figure 6B depicts the binding of moenomycin and a lipid II analog to a S. aureus membrane-bound MGT.75 As demonstrated in earlier experiments, moenomycin does not bind in the lipid II site but a separate portion of the active site where the extending saccharide likely binds. More recent studies have revealed that moenomycin A binds at the same site as lipid IV. The authors therefore suggest that moenomycin blocks the initiation step rather than elongation or termination. It is proposed that the phosphoglycerate and lipid portion of moenomycin mimics the diphospholipid leaving group of the donor substrate.76
Figure 6.
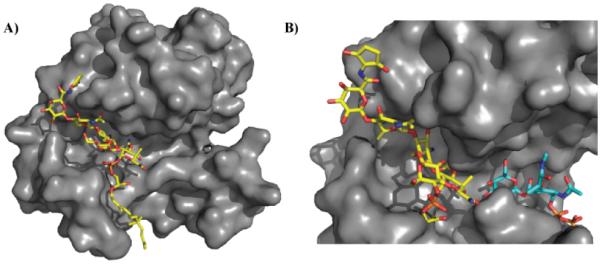
Site of moenomycin interaction with glycosyltransferases. A) Crystal structure of moenomycin (yellow) bound to S. aureus monofunctional transglycosylase (PDB 3HZS). Only 15 carbons of the lipid tail are ordered in this structure. B) Crystal structures of membrane-bound monofunctional glycosyltransferase with moenomycin (yellow; PDB 3VMR) and a lipid II analog (blue; PDB 3VMT). Note: 3VMR and 3VMT were aligned, and the surface of 3VMR was subsequently hidden for clarity. Moenomycin binds in the channel where the growing glycan chain binds thereby disrupting transglycosylation activity. Under normal biological conditions, lipid II would be transferred to the growing glycan chain forming a new β1-4 glycosidic linkage.
Although natural moenomycins are not currently suitable for clinical use, multiple studies have been conducted to identify the minimal pharmacophore so that moenomycins may be designed with improved pharmacokinetic properties. Crystal structures of neryl-moenomycin A bound to the PGT domain of Aquifex aeolicus PBP1A revealed six conserved active site residues which appear to interact with the F ring and phosphoglycerate portions. Mutations of all six residues resulted in abolished or reduced enzyme activity. The authors propose that this may explain why resistance to moenomycin has not been observed. Although the majority of contacts involve interactions with the F ring and phosphoglycerate, there are a small number of interactions with the E ring and a single interaction with the C ring. Using heterologous expression, Walker and coworkers generated a moenomycin analog which maintained the key contacts. This DEF trisaccharide was found to inhibit S. aureus PBP2 more strongly than moenomycin A.77 Previously the minimal pharmacophore was thought to be the tetrasaccharide CDEF. 69 Together these results suggest that the minimal pharmacophore includes rings E and F, the phosphoglycerate, and either ring C or D.77 Moenomycin derivatives which lack the D ring retain antibiotic activity. 73 Indeed crystal structures of bound moenomycin reveal that the D ring protrudes into the solvent, reinforcing the observation that the D ring is not necessary for activity. 77 In addition, both the carboxylate and the phosphoglycerate interact with important conserved active site residues and are necessary for activity.78
Because the lipid of moenomycin is thought to be responsible for the majority of its poor pharmacological properties, much effort has been devoted to identifying ways in which this portion can be modified. Although many studies have confirmed that the entire 25 carbon chain is not necessary for activity, the optimal length is still unknown.68,72,76,77 A lipid chain with ten carbons is sufficient for in vitro enzyme inhibition activity but is not active in vivo.68 In the crystal structure of S. aureus MTG in complex with moenomycin, fifteen carbons of the lipid chain are ordered and form hydrophobic interactions with four residues.74 A 15 carbon analog possessed biological activity although the activity was reduced compared to the parent compound. 79 Since the length of the lipid tail is necessary for biological activity, it is hypothesized that the lipid portion interacts with the membrane and is involved in anchoring.78 This theory is supported by the fact that the transmembrane portion of PBPs is necessary for moenomycin binding. 80
In 2001, Kahne and coworkers reported three E. coli imp mutants which were resistant to moenomycin as well as vancomycin analogues and teicoplanin. Notably, the mutants remained sensitive to β-lactams which inhibit the transpepdidation reaction as well as other antibiotics targeting other essential cellular functions which rules out a nonspecific mechanism of resistance such as altered permeability. The mutant strains all lacked expression of yfgL, a lipoprotein located on the inner surface of outer membrane. Although the role of YfgL in resistance is not known, the authors hypothesize that it affects regulation of lytic transglycosylases which when inappropriately activated may cause rapid cell death.81 This marks the first reported study of moenomycin resistant mutant strains and provides insights into ways in which bacteria might acquire moenomycin resistance. Although PGT inhibition had long been hypothesized to be the bactericidal activity of moenomycin, only recently have moenomycin-resistant S. aureus mutants have been generated which resulted in a point mutation in the active site of PBP2 confirming the this mechanism of action.82
Though moenomycin A has yet to realize its full clinical potential, it is already used as a tool to discover new PGT inhibitors with better pharmacological properties. For example, a fluorescently labelled moenomycin analog based on the minimal pharmacophore has been synthesized and used in a screen to identify small molecules inhibitors of PGTs in displacement assays.83 While non-carbohydrate bacterial transglycosylase inhibitors have been discovered, moenomycin remains the most potent inhibitor known to date. 84
3.2 Biosynthesis
Although the total synthesis of moenomycin A has been reported,85 the complexity of the molecule makes biosynthetic creation of analogs an attractive alternative. Walker and coworkers have characterized the entire seventeen step moenomycin pathway.79,86 The moenomycin biosynthetic genes from S. ghanaensis (ATCC14672) were identified using a whole-genome scanning approach. Surprisingly, the moe gene cluster is split into two separate clusters, a small 3 gene cassette involved in A ring biosynthesis and a larger 20 gene primary moe cluster involved in assembly of the phosphoglycolipid pentasaccharide (Figure 7). Despite moenomycin’s large size, its gene cluster is surprisingly compact due to the use of isoprenoid and sugar-nucleotide building blocks from the host’s primary metabolism.86
Figure 7.

Moenomycin biosynthetic gene cluster from Streptomyces ghanaensis. Genes are color-coded according to function.
The large moe cluster 1 contains two prenyltransferases, five glycosyltransferases, seven sugar synthesis genes, and four transporters. It also contains two non-functional genes involved in A ring assembly, moeA5 and moeB5. MoeA4 is an aminolevulinate synthase, and moeB4 is an aminolevulinate cyclase. Both of these genes as well as an amide synthetase moeC4, which attaches the A ring to moenomycin, are found in the smaller moe cluster 2. Therefore, moe cluster 2 is required for A ring assembly and attachment.79, 86
The entire moenomycin A biosynthetic pathway has been reconstituted in the heterologous host S. lividans and the function and timing of each gene was characterized (Scheme 2). MoeO5 and MoeN5 are prenyltransferases which construct the lipid tail from primary metabolites. MoeO5 catalyzes the first step in the moenomycin biosynthetic pathway to form a C15 lipid-phosphoglycerate.79 Interestingly, in addition to transferring the C15 lipid of farnesyl pyrophosphate to the 2-hydroxyl group of 3-phosphoglycerate, MoeO5 also catalyzes an unusual trans-to-cis isomerization of the C2-C3 double bond. Recently, the crystal structure of MoeO5 in complex with the product lipid-phosphoglycerate has been solved providing insights into this unique isomerization.87 Confirming the lipid-phosphoglycerate as an early intermediate in moenomycin biosynthesis, nosokophic acid (a C15 lipid-phosphoglycerate) was recently isolated from the culture broth of Streptomyces sp. K02-0144.88 MoeN5 catalyzes transfer of the C10 lipid onto the C15 lipid tail after disaccharide assembly. 79
Scheme 2.

Experimentally determined order of moenomycin biosynthesis. P = phosphate; C15 = 15-carbon lipid chain; C25 = 25-carbon lipid chain; B-F indicate ring systems of moenomycin. Enzymes are color-coded to match the gene cluster in figure 7.
Investigation of the five glycosyltransferases revealed the substrate and timing of each.79 Interestingly, the glycosyltransferases of the moenomycin pathway are more closely related to those involved in primary metabolism than to other secondary metabolism glycosyltransferases.86 MoeGT1 transfers the first sugar to the lipid phosphoglycerate. MoeGT4 transfers the next sugar to form the disaccharide. The authors suggest a branched pathway from the disaccharide with either MoeGT5 or MoeGT4 acting first followed by the remaining glycosyltransferase. Finally MoeGT2 attaches the B ring sugar to form the full pentasaccharide. 79
Only three genes in the moe cluster are involved in the synthesis of NDP-sugars. MoeE5 has been shown to catalyze the epimerization of UDP-glucuronic acid to UDP-galacturonic acid (UDP-GalUA). UDP-GalUA is the natural substrate for MoeGT1; therefore epimerization occurs prior to glycosyl transfer. MoeR5 and MoeS5, a 4,6-dehydratase/ketoreductase pair, are responsible for the conversion of UDP-GlcNAc to its 6-deoxy version, UDP-chinovosamine.79
Four genes are involved in final tailoring of the sugar residues of moenomycin. MoeF5 is responsible for F ring carboxamidation while MoeH5 catalyzes amidation of the B ring. MoeK5 methylates ring F before epimerization of the C4 hydroxyl. The final sugar tailoring enzyme, MoeM5 is a carbamoyltransferase which installs the carbamate group on the F ring primarily during the trisaccharide stage.79
Because moenomycins are produced at low levels in heterologous hosts, recent studies have focused on improving moenomycin production. In particular, recent studies have shown that rational manipulation of the regulators and increased moe gene dosage could be useful for improving moenomycin biosynthesis in heterologous hosts.89 In addition, deletion of wblAgh, encoding a homologue of the WhiB-family of proteins, blocked aerial mycelium sporulation and led to a 230% increase in moenomycin production.90 Disruption of putative ABC transporters moeX5 and moeP5 resulted in reduced moenomycin production.91 While much is known about the biosynthetic steps leading to production of moenomycin, less is known about its regulation and the regulation of natural product gene clusters in general.
Although the moenomycin cluster is already surprisingly compact, Walker and co-workers have shown that only seven genes (moeE5, moeO5, moeGT1, moeF5, moeGT4, moeM5, and either moeGT3 or moeGT5) are required to make bioactive analogs. In addition, the biosynthetic pathway for moenomycin production appears to be highly malleable as only four gene disruptions (moeGT1, moeF5, moeE5, moeO5) resulted in abolished production of moenomycin analogs.79 The mutability of the moenomycin gene cluster provides ample opportunity for creation of analogs.
4. Saccharomicins
In 1998, a new family of antibacterial polysaccharides were discovered and termed the saccharomicins. The saccharomicins are novel heptadecaglycoside antibiotics produced by the rare actinomycete Saccharothrix espanaensis LL-C19004 originally isolated from a soil sample in Spain.92,93 To date, only two saccharomicins have been reported, saccharomicins A and B which differ only by the identity of the tenth sugar residue (see Figure 8). The saccharomicins are large oligosaccharides composed of seventeen 6-deoxy sugars and the aglycone N-(m,p-dihydroxycinnamoyl)taurine. The sugar residues can be separated into eight amino sugars and eight non-amino sugars. Half of the amino sugars are 4-epi-vancosamines and half are saccharosamines, a unique amino sugar found only in the saccharomicins. In addition there are four fucoses and a variant number of rhamnoses and digitoxoses adding to a total of four. Finally, the aglycone is linked to the sugar chain via a sulphated fucose residue.92 While the absolute stereochemistry of all sugar residues has not been determined, synthetic efforts have determined that the sulphated fucose as well as the fucose-saccharosamine disaccharide units are all D-configuration.94,95 In addition to the novel amino sugar saccharosamine, the saccharomicins also contain a unique β-β glycosidic linkage between the fucoses and saccharosamines allowing them to survive harsh acidic conditions.92
Figure 8.

Naturally occurring saccharomicins and corresponding gene cluster from Saccharothrix espanaensis LL-C19004. Genes are color-coded according to putative function.
Both saccharomicins A and B possess activity against a panel of Gram-positive organisms including MRSA and VRE but are less potent against Gram-negative organisms. In addition, the saccharomicins had promising activity against all multiply-resistant staphylococci strains tested. These results provide evidence that the saccharomicins likely do not have cross-resistance with other families of antibiotics. Despite their promising antibacterial activities, the compounds exhibit toxic effects at high doses (LD50 = 16mg/kg) rendering them currently unsuitable for general use in the clinic.92
4. 1 Saccharomicins Mode of Action
While the precise mode of action has yet to be determined, various experiments have indicated that the primary target of the saccharomicins is associated with the bacterial cellular membrane. Initial experiments revealed that the saccharomicins completely inhibit DNA, RNA, and protein synthesis within ten minutes of treatment. Further experiments indicated that the saccharomicins caused cell death and lysis by disrupting the cell membrane. Adding the divalent cations Ca2+ or Mg2+ substantially decreased the activity against Gram-negative by up to sixteen-fold but had no effect on the activity against Gram-positive organisms. Other divalent cations such as Fe2+ or Cu2+ had no effect on the saccharomicins’ activity indicating that they are not general chelators but form interactions with the outer membrane-bound cations and self-promote their uptake like aminoglycosides and quinolones. Using an E. coli imp strain which has increased cell membrane permeability, the saccharomicins had an eight-fold higher activity compared to a regular E. coli strain. These results indicate that the poor activity against Gram-negative organisms is most likely due to the saccharomicins’ inability to penetrate the outer membrane. In addition, there was extensive potassium leakage when the E. coli imp strain was treated with saccharomicins consistent with a sudden disruption of the cell membrane.93
4. 2 Biosynthesis of the Saccharomicins
In 2012, genomic analysis of the complete sequence of S. espanaensis DM44229T revealed the presence of twenty-six biosynthetic gene clusters, including the cluster responsible for biosynthesis of the saccharomicins, designated the sam cluster. The gene cluster contains genes responsible for aglycone biosynthesis as well as ten glycosyltransferases and eight sugar synthesis genes. While saccharomicins contain seventeen sugar residues, only ten putative glycosyltransferases, sam11-20, are present in the cluster suggesting that some of the glycosyltransferases may work iteratively or be recruited from primary metabolism. In addition, there are only eight putative NDP-sugar biosynthesis genes which are responsible for formation of all seventeen sugars. Sam6 is a putative glucose-1-phosphate nucleotidyltransferase. Two putative methyltransferases are found in the gene cluster. Sam9 appears to be a SAM-dependent methyltransferase while Sam34 has homology to NDP-hexose C-3 methyltransferases and is likely responsible for adding the C-3 methyl group to form L-epi-vancosamine and/or the saccharosamine residues. Sam21 which has homology to NDP-4-keto-6-deoxyhexose-4-ketoreductases, Sam30 a putative transaminase, Sam31 a probable NDP-hexose 2,3-dehydratase, Sam32 which has homology to a dTDP-4-dehydrorhamnose 3,5-epimerase, and Sam33 which is a putative NDP-hexose 3-ketoreductase are responsible for the biosynthesis of the five types of sugars found in the saccharomicins. Sam23 and Sam24 encode putative transport proteins while Sam1, 3, and 38 are most likely regulatory elements.96
Investigation of the biosynthesis of the aglycone portion of the saccharomicins was first reported in 2006 by Bechthold and coworkers who cloned and identified two genes from S. espanaensis which are involved in caffeic acid biosynthesis. Heterologous expression of sam8 and sam5 led to production of trans-caffeic acid (Scheme 3). Assays identified Sam8 as a tyrosine ammonia-lyase and Sam5 as a 4-coumarate 3-hydroxylase, the first of both of these enzymes to be reported in an actinomycete.97 Sam7 has similarity to acyl-CoA synthases indicating that it may be involved in the synthesis of caffeoyl-CoA which could be used to link taurine to form the aglycone, and Sam36 has homology to penicillin amidases and may be responsible for forming the amide bond between caffeic acid and taurine. The synthesis of taurine has yet to be described in bacteria; however, the authors speculate that sam29 may encode a sulfinic acid decarboxylase.96
Scheme 3.
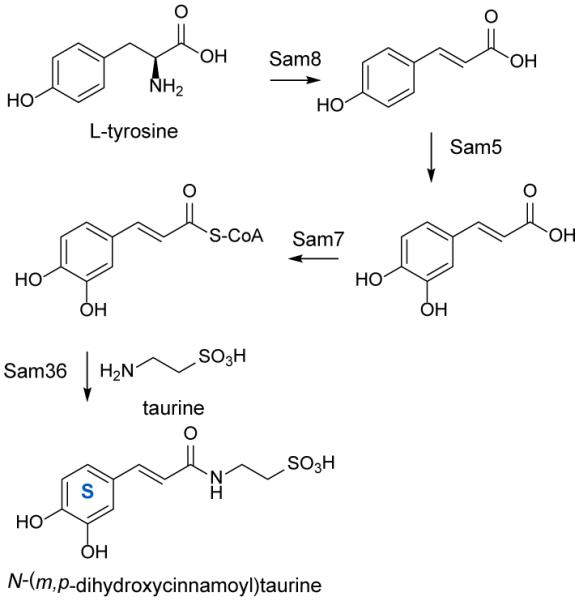
Biosynthesis of the saccharomicin aglycone beginning with L-tyrosine. Four enzymes have been shown to be sufficient to catalyze the formation of the saccharomicin aglycone.
Although due to their toxicity the saccharomicins are not currently in clinical use, understanding the biosynthesis of the largest oligosaccharide antibiotic will further understanding of these bioactive oligosaccharides in general and may lead to potential new clinical candidates to treat the growing problem of antibacterial resistance.
5. Acarviostatins
In 2008, a class of novel pseudo-oligosaccharides termed the acarviostatins were reported from cultures of S. coelicoflavus ZG0656 (also known as S. coelicoflavus var. nankaiensis).98 Six acarviostatins were originally identified and structurally characterized (Figure 9).99, 100 Based on tandem mass spectrometry fragmentation patterns of the six aforementioned acarviostatins, a total of 80 acarviostatin analogs have been identified. Fragmentation as well as multiple reaction monitoring was necessary to identify all of the acarviostatins as many are positional isomers having the same m/z ratio.101
Figure 9.
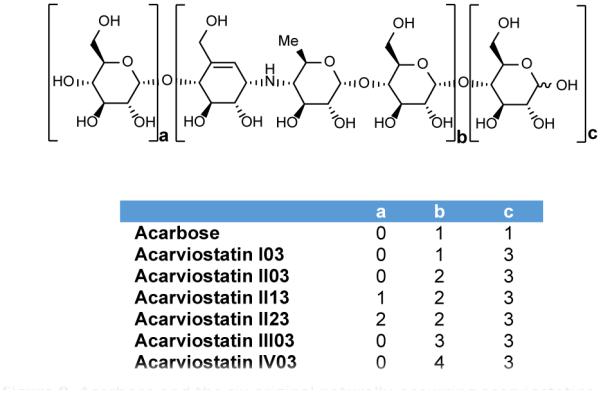
Acarbose and the six original naturally-occurring acarviostatins.
Although numerous acarviostatins have been identified, the majority can be described by three variables on which a systematic naming system has been based. Each acarviostatin is composed of a pseudotrisaccharide core flanked by a variable number of D-glucose units. Acarviostatins can contain up to 5 pseudotrisaccharides which are denoted by Roman numerals. The middle digit of the acarviostatin name corresponds to the number of D-glucose units at the non-reducing end, and the last digit describes the number of D-glucose residues at the reducing end. For instance, acarviostatin II03 contains two pseudotrisaccharide units and three glucose residues at the reducing end.99, 101
The pseudotrisaccharide is composed of valienamine, 4-amino-4,6-dideoxy-D-glucose, and D-glucose. Valienamine and 4-amino-4,6-dideoxy-D-glucose form acarviosine which is the core structure of acarbose, a potent α-amylase inhibitor. Valienamine is a unique C7N cyclitol also found in the antifungal validamycin A. Mode of action and biosynthesis of both acarviostatins and acarbose will be considered as acarbose is a building block of the acarviostatins.
5.1 Mode of Action
Human pancreatic α-amylase (HPA) is a major therapeutic target for the treatment of type II diabetes as it catalyzes the hydrolysis of α-D-(1,4)-glycosidic linkages found in starch. Acarbose, a drug successfully used for treatment of type II diabetes, was the first α-glucosidase inhibitor to be approved by the US Food and Drug Administration.102, 103 Many of the acarviostatins, however, are more potent α-amylase inhibitors than acarbose. In fact, acarviostatin III03 is 260 times more potent than acarbose and is the most effective α-amylase inhibitor known to date.99 Despite containing valienamine, validamycin A is not an inhibitor of α-amylase suggesting that the sugar residues attached to the aminocyclitol are necessary for inhibition.102
Recently, crystal structures of HPA in complex with a series of acarviostatins provided greater insights into their specific mode of action. Acarbose and acarviostatins all bind in the active site of HPA (Figure 10). Like acarbose, acarviostatin I03 which is only one glucose longer undergoes a series of hydrolysis, condensation, and transglycosylation reactions resulting in a substantially rearranged product. However, crystal structures of HPA in complex with larger acarviostatins surprisingly revealed the same seven-ring hydrolysis product in each active site regardless of the original number of sugar residues (Figure 10b). The authors propose these larger acarviostatins undergo hydrolysis at either one or both ends to generate acarviostatin II01. These results indicate that an inhibitor containing seven sugar residues would provide the most efficient α-amylase inhibition while occupying the full active site.103 In all cases, acarviosine inhibits the hydrolysis reaction by acting as a transition-state analog.98,104 The nitrogen bond between valienamine and 4,6-dideoxyglucose cannot be cleaved rendering the acarviosine containing compounds competitive inhibitors of human pancreatic amylase.103
Figure 10.
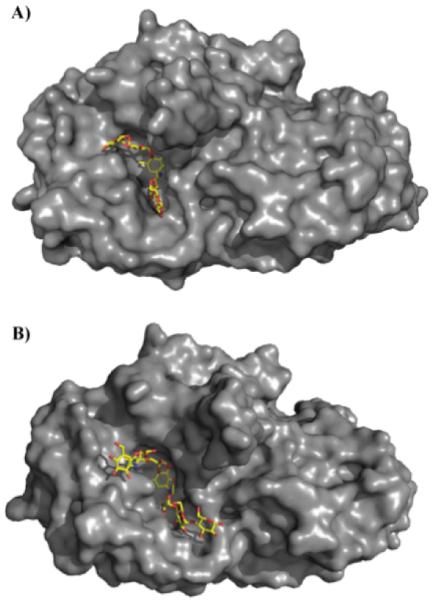
Human pancreatic amylase in complex (A) acarbose (PDB 1B2Y) and (B) the hydrolysis product of acarviostatin III03 (PDB 3OLG). Both acarbose and the acarviostatins are competitive inhibitors of human pancreatic amylase by acting as transition-state analogs.
5.2. Biosynthesis
The biosynthesis of acarbose has been well researched and we point readers to two excellent reviews of the biosynthesis of acarbose as well as other aminocyclitol natural products.105, 106 This review will briefly summarize the biosynthesis of acarbose while drawing parallels with the proposed acarviostatin biosynthetic pathway.
Reported in 2012, the genome sequence of S. coelicoflavus ZG0656 reveals the presence of a putative acarviostatin gene cluster. The gene cluster is approximately 40 kilobases and has significant similarity to two gene clusters known to be responsible for acarbose synthesis: gac from S. glaucescens107 and acb from Actinoplanes sp. SE50/110104. This putative acarviostatin gene cluster is termed the sct-cluster and contains 16 genes which are proposed to encode enzymes for the synthesis of the pseudotrisaccharide core. In addition there are five α-glucosidic hydrolases and/or glycosyltransferases (sctW, sctE1, sctE2, sctZ1, sctZ2), two putative ATP-dependent transporter systems (sctWXY and sctFGH) and two putative regulators (scrC1 and scrC2). Because the sct cluster is most similar to the acarbose gac cluster, the biosynthetic pathways may be predicted to be highly similar (Figure 11). Therefore, the acarviostatin biosynthetic pathway that the authors proposed draws heavily from knowledge of acarbose biosynthesis.
Figure 11.

Acarbose biosynthetic gene cluster from S. glaucescens and putative acarviostatin biosynthetic gene cluster from S. coelicoflavus ZG0656. Genes are color-coded according to putative function.
Labelling studies have shown that valienamine of acarbose is derived from pentose phosphate pathway rather than the shikimate pathway.108 Hence, the biosynthesis of acarbose as well as the acarviostatins is proposed to begin with cyclization of sedoheptulose 7-phosphate by GacC/SctC (gac corresponds to the acarbose enzymes; sct corresponds to the acarviostatin enzymes) to give 2-epi-5-epi-valiolone. GacJ/SctJ then epimerizes C-2 of 2-epi-5-epi-valiolone to give 5-epi-valiolone. The next step is presumably phosphorylation of C7 by GacM/SctM followed by GacO/SctO catalyzed dehydration and reduction to generate 1-epi-valienol-7-phosphate. GacU/SctU is proposed to catalyze a second phosphorylation to generate 1-epi-valienol-1,7-diphosphate which is subsequently converted to NDP-1-epi-valienol-7-phosphate by GacR/SctR (Scheme 4).107, 109
Scheme 4.

Proposed biosynthetic pathway to the acarviostatins beginning with sedo-heptulose-7-phosphate and D-glucose-1-phosphate. Sct genes correspond to those from the putative acarviostatin pathway while Gac genes are their corresponding homologs found in the acarbose gene cluster from S. glaucescens. Adapted from Guo 2012.109
A second pathway is presumed to produce dTDP-4-amino-4,6-dideoxy-glucose through the sequential action of GacA/SctA (dTDP-glucose synthase), GacB/SctB (4,6-dehydratase) and GacV/SctV(4-aminotransferase). The pathways then are proposed to converge via GacS/SctS to generate the pseudodisaccharide dTDP-acarviose-7-phosphate. The final step in formation of the pseudotrisaccharide core is proposed to be glycosylation by GacI/SctI (Scheme 4).109
While these enzymes generate the pseudotrisaccharide core, glycosyl transferases and/or pseudotrisaccharide transferases are still necessary to complete the biosynthesis of the acarviostatins. SctE2 has moderate homology to AcbD of the Actinoplanes acarbose pathway. AcbD is an acarviose transferase which adds different maltooligosaccharides to acarviosine. 101, 110 Based on this homology, the authors speculate that SctE2 as well as SctZ1 and SctZ2 might be responsible for joining pseudotrisaccharide units as well as attaching additional glucose units to the reducing and non-reducing ends.109 Further characterization of these genes is needed to definitively identify the gene(s) responsible for full construction of the acarviostatins.
The putative transporters SctWXY and SctFGH have homology to the ABC transporters in both acarbose gene clusters. It has been proposed that these transporters are involved in a recycling mechanism in which one secretes acarviostatin I00-7-phosphate out of the cell where hydrolases and transferases work to attach maltooligosaccharides to the acarviostatin core. The acarviostatins are then imported back into the cell where SctQ and/or SctE1 hydrolyze the glucoses to regenerate acarviostatin I00-7-phosphate. The host may use this mechanism to transport hydrolyzed starch into the cell.109 This recycling mechanism or ‘carbophor’ may also explain the vast assortment of acarviostatins which have been identified. The acarviostatins represent a unique biological activity for oligosaccharide natural products. Fully understanding the biosynthesis of these compounds can lead to new derivatives with improved efficacy that may aid in the fight against metabolic disorders, including diabetes.
6. Conclusions
With the recent expansion in ‘genome mining’ of natural products, understanding the distinguishing biosynthetic features of oligosaccharide secondary metabolites will likely enable the discovery of many more of this family of compounds. As evidenced by the examples above, many analogs of oligosaccharides can be generated by a single organism and conservative mutagenesis studies have been able to generate a variety of non-natural analogs with modulated biological properties. Moreover, with the improvement and widespread use of mass spectrometry to identify and characterize oligosaccharides, many family members can be rapidly identified. Finally, this class of natural products provides new insights into the biology of their targets by providing large molecule probes. Despite non-optimized pharmacological properties of native secondary metabolites, the gene clusters of oligosaccharides appear to be highly mutable allowing the generation of many analogs for structure-activity relationship studies.
7. Acknowledgements
We would like to acknowledge support from the Office of Naval Research (Grant N00014-09-1-012) and the Vanderbilt Institute of Chemical Biology.
8. References
- 1.Thibodeaux CJ, Melancon CE, 3rd, Liu HW. Angew Chem Int Ed Engl. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kren V, Martinkova L. Curr Med Chem. 2001;8:1303–1328. doi: 10.2174/0929867013372193. [DOI] [PubMed] [Google Scholar]
- 3.Thibodeaux CJ, Melancon CE, Liu HW. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- 4.WeymouthWilson AC. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 5.Singh S, Phillips GN, Jr., Thorson JS. Nat Prod Rep. 2012;29:1201–1237. doi: 10.1039/c2np20039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 7.Moremen KW, Tiemeyer M, Nairn AV. Nat Rev Mol Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seeberger PH, Werz DB. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 9.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright DE. Tetrahedron. 1979;35:1207–1237. [Google Scholar]
- 11.Mann RL, Bromer WW. J Am Chem Soc. 1958;80:2714–2716. [Google Scholar]
- 12.Kudo F, Eguchi T. J Antibiot (Tokyo) 2009;62:471–481. doi: 10.1038/ja.2009.76. [DOI] [PubMed] [Google Scholar]
- 13.Weinstein MJ, Luedemann GM, Oden EM, Wagman GH. Antimicrob Agents Chemother (Bethesda) 1964;10:24–32. [PubMed] [Google Scholar]
- 14.Ganguly AK, Sarre OZ, Greeves D, Morton J. J Am Chem Soc. 1975;97:1982–1985. doi: 10.1021/ja00840a078. [DOI] [PubMed] [Google Scholar]
- 15.Chu M, Mierzwa R, Jenkins J, Chan T, Das P, Pramanik BN, Patel M, Gullo V. J Nat Prod. 2002;65:1588–1593. doi: 10.1021/np020093t. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Pramanik BN, Bartner PL, Saksena AK, Gross ML. J Am Soc Mass Spectrom. 2002;13:1313–1321. doi: 10.1016/S1044-0305(02)00624-4. [DOI] [PubMed] [Google Scholar]
- 17.Herzog HL, Meseck E, Delorenzo S, Murawski A, Charney W, Rosselet JP. Appl Microbiol. 1965;13:515–520. doi: 10.1128/am.13.4.515-520.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganguly AK, Szmulewicz S. J Antibiot (Tokyo) 1975;28:710–712. doi: 10.7164/antibiotics.28.710. [DOI] [PubMed] [Google Scholar]
- 19.Nakashio S, Iwasawa H, Dun FY, Kanemitsu K, Shimada J. Drugs Exp Clin Res. 1995;21:7–16. [PubMed] [Google Scholar]
- 20.Ganguly AK, McCormick JL, Saksena AK, Das PR, Chan TM. Bioorg Med Chem Lett. 1999;9:1209–1214. doi: 10.1016/s0960-894x(99)00163-8. [DOI] [PubMed] [Google Scholar]
- 21.Ganguly AK, Saksena AK. J Antibiot (Tokyo) 1975;28:707–709. [PubMed] [Google Scholar]
- 22.Chu M, Mierzwa R, Patel M, Jenkins J, Das P, Pramanik BN, Chan T. Tetrahedron Letters. 2000;41:6689–6693. [Google Scholar]
- 23.Mertz JL, Peloso JS, Barker BJ, Babbitt GE, Occolowitz JL, Simson VL, Kline RM. J Antibiot (Tokyo) 1986;XXXIX:877–887. doi: 10.7164/antibiotics.39.877. [DOI] [PubMed] [Google Scholar]
- 24.Jones DJ, Mowrey DH, Anderson DB, Wellenreiter RH. J Anim Sci. 1987;65:881–885. doi: 10.2527/jas1987.654881x. [DOI] [PubMed] [Google Scholar]
- 25.Shryock TR. Emerging Infectious Diseases. 2001;7:488–489. doi: 10.3201/eid0703.010336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster DR, Rybak MJ. Pharmacotherapy. 1999;19:1111–1117. doi: 10.1592/phco.19.15.1111.30576. [DOI] [PubMed] [Google Scholar]
- 27.Souli M, Thauvin-Eliopoulos C, Eliopoulos GM. Antimicrob Agents Chemother. 2000;44:2733–2739. doi: 10.1128/aac.44.10.2733-2739.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boucher HW, Thauvin-Eliopoulos C, Loebenberg D, Eliopoulos GM. Antimicrob Agents Chemother. 2001;45:208–211. doi: 10.1128/AAC.45.1.208-211.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McNicholas PM, Najarian DJ, Mann PA, Hesk D, Hare RS, Shaw KJ, Black TA. Antimicrob Agents Chemother. 2000;44:1121–1126. doi: 10.1128/aac.44.5.1121-1126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Champney WST. Antimicrob Agents Chemother. 2000;44:1413–1417. doi: 10.1128/aac.44.6.1413-1417.2000. C. L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adrian PV, Zhao W, Black TA, Shaw KJ, Hare RS, Klugman KP. Antimicrob Agents Chemother. 2000;44:732–738. doi: 10.1128/aac.44.3.732-738.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zarazaga M, Tenorio C, Del Campo R, Ruiz-Larrea F, Torres C. Antimicrob Agents Chemother. 2002;46:3657–3659. doi: 10.1128/AAC.46.11.3657-3659.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNicholas PM, Mann PA, Najarian DJ, Miesel L, Hare RS, Black TA. Antimicrob Agents Chemother. 2001;45:79–83. doi: 10.1128/AAC.45.1.79-83.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mann PA, Xiong L, Mankin AS, Chau AS, Mendrick CA, Najarian DJ, Cramer CA, Loebenberg D, Coates E, Murgolo NJ, Aerestrup FM, Goering RV, Black TA, Hare RS, McNicholas PM. Mol Microbiol. 2001;41:1349–1356. doi: 10.1046/j.1365-2958.2001.02602.x. [DOI] [PubMed] [Google Scholar]
- 35.Belova L, Tenson T, Xiong L, McNicholas PM, Mankin AS. Proc Natl Acad Sci U S A. 2001;98:3726–3731. doi: 10.1073/pnas.071527498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treede I, Jakobsen L, Kirpekar F, Vester B, Weitnauer G, Bechthold A, Douthwaite S. Mol Microbiol. 2003;49:309–318. doi: 10.1046/j.1365-2958.2003.03558.x. [DOI] [PubMed] [Google Scholar]
- 37.Mosbacher TG, Bechthold A, Schulz GE. J Mol Biol. 2003;329:147–157. doi: 10.1016/s0022-2836(03)00407-8. [DOI] [PubMed] [Google Scholar]
- 38.Mikolajka A, Liu H, Chen Y, Starosta AL, Marquez V, Ivanova M, Cooperman BS, Wilson DN. Chem Biol. 2011;18:589–600. doi: 10.1016/j.chembiol.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ganguly AK, McCormick JL, Saksena AK, Das PD, Chan T. Bioorg Med Chem. 1999;9:1209–1214. doi: 10.1016/s0960-894x(99)00163-8. [DOI] [PubMed] [Google Scholar]
- 40.Ganguly AK. J Antibiot (Tokyo) 2000;53:1038–1044. doi: 10.7164/antibiotics.53.1038. [DOI] [PubMed] [Google Scholar]
- 41.Waitz JA, Horan AC. Micromonospora carbonacea var. africana. Assignee: Schering Corporation; Kenilworth, NJ: [Google Scholar]
- 42.Boll R, Hofmann C, Heitmann B, Hauser G, Glaser S, Koslowski T, Friedrich T, Bechthold A. J Biol Chem. 2006;281:14756–14763. doi: 10.1074/jbc.M601508200. [DOI] [PubMed] [Google Scholar]
- 43.Weitnauer G, Hauser G, Hofmann C, Linder U, Boll R, Pelz K, Glaser SJ, Bechthold A. Chem Biol. 2004;11:1403–1411. doi: 10.1016/j.chembiol.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 44.Gaisser S, Trefzer A, Stockert S, Kirschning A, Bechthold A. J Bacteriol. 1997;179:6271–6278. doi: 10.1128/jb.179.20.6271-6278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weitnauer G, Muhlenweg A, Trefzer A, Hoffmeister D, SuBmuth RD, Jung G, Welzel K, Vente A, Girreser U, Bechthold A. Chem Biol. 2001;8:569–581. doi: 10.1016/s1074-5521(01)00040-0. [DOI] [PubMed] [Google Scholar]
- 46.Weitnauer G, Gaisser S, Kellenberger L, Leadlay PF, Bechthold A. Microbiology. 2002;148:373–379. doi: 10.1099/00221287-148-2-373. [DOI] [PubMed] [Google Scholar]
- 47.Hofmann C, Boll R, Heitmann B, Hauser G, Durr C, Frerich A, Weitnauer G, Glaser SJ, Bechthold A. Chem Biol. 2005;12:1137–1143. doi: 10.1016/j.chembiol.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 48.Treede I, Hauser G, Muhlenweg A, Hofmann C, Schmidt M, Weitnauer G, Glaser S, Bechthold A. Appl Environ Microbiol. 2005;71:400–406. doi: 10.1128/AEM.71.1.400-406.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hosted TJ, Wang TX, Alexander DC, Horan AC. Journal of Industrial Microbiology and Biotechnology. 2001;27:386–392. doi: 10.1038/sj.jim.7000189. [DOI] [PubMed] [Google Scholar]
- 50.Hu Y, Al-Mestarihi A, Grimes CL, Kahne D, Bachmann BO. J Am Chem Soc. 2008;130:15756–15757. doi: 10.1021/ja8051415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vey JL, Al-Mestarihi A, Hu Y, Funk MA, Bachmann BO, Iverson TM. Biochemistry. 2010;49:9306–9317. doi: 10.1021/bi101336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallhausser KH, Nesemann G, Prave P, Steigler A. Antimicrob Agents Chemother (Bethesda) 1965;5:734–736. [PubMed] [Google Scholar]
- 53.Ostash B, Walker S. Nat Prod Rep. 2010;27:1594–1617. doi: 10.1039/c001461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fehlhaber HW, Girg M, Seibert G, Hobert K, Welzel P, Vanheijenoort Y, Vanheijenoort J. Tetrahedron. 1990;46:1557–1568. [Google Scholar]
- 55.Uchida R, Iwatsuki M, Kim YP, Ohte S, Omura S, Tomoda H. J Antibiot (Tokyo) 2010;63:151–155. doi: 10.1038/ja.2010.9. [DOI] [PubMed] [Google Scholar]
- 56.Uchida R, Iwatsuki M, Kim YP, Omura S, Tomoda H. J Antibiot (Tokyo) 2010;63:157–163. doi: 10.1038/ja.2010.10. [DOI] [PubMed] [Google Scholar]
- 57.Arai M, Nakayama R, Yoshida K, Takeuchi M, Teramoto S, Torikata A. J Antibiot (Tokyo) 1977;30:1055–1059. doi: 10.7164/antibiotics.30.1055. [DOI] [PubMed] [Google Scholar]
- 58.Arai M, Torikata A, Enokita R, Fukatsu H, Nakayama R, Yoshida K. J Antibiot (Tokyo) 1977;30:1049–1054. doi: 10.7164/antibiotics.30.1049. [DOI] [PubMed] [Google Scholar]
- 59.Torikata A, Yoshikawa H, Katayama T, Arai M, Nakahara M, Kitano N. J Antibiot (Tokyo) 1977;30:1060–1063. doi: 10.7164/antibiotics.30.1060. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi S, Serita K, Arai M, Seto H, Furihata K, Otake N. Tetrahedron Letters. 1983;24:499–502. [Google Scholar]
- 61.He H, Shen B, Korshalla J, Siegel MM, Carter GT. J Antibiot (Tokyo) 2000;53:191–195. doi: 10.7164/antibiotics.53.191. [DOI] [PubMed] [Google Scholar]
- 62.Pfaller MA. Diagn Microbiol Infect Dis. 2006;56:115–121. doi: 10.1016/j.diagmicrobio.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 63.Eichhorn P, Aga DS. Rapid Commun Mass Spectrom. 2005;19:2179–2186. doi: 10.1002/rcm.2044. [DOI] [PubMed] [Google Scholar]
- 64.Zehl M, Pittenauer E, Rizzi A, Allmaier G. J Am Soc Mass Spectrom. 2006;17:1081–1090. doi: 10.1016/j.jasms.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 65.Butaye P, Devriese LA, Haesebrouck F. Clin Microbiol Rev. 2003;16:175–188. doi: 10.1128/CMR.16.2.175-188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butaye P, Devriese LA, Haesebrouck F. Antimicrob Agents Chemother. 2001;45:1374–1378. doi: 10.1128/AAC.45.5.1374-1378.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goldman RC, Baizman ER, Branstrom AA, Longley CB. Bioorg Med Chem Lett. 2000;10:2251–2254. doi: 10.1016/s0960-894x(00)00443-1. [DOI] [PubMed] [Google Scholar]
- 68.Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE. J Am Chem Soc. 2006;128:14012–14013. doi: 10.1021/ja065905c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goldman RC, Gange D. Curr Med Chem. 2000;7:801–820. doi: 10.2174/0929867003374651. [DOI] [PubMed] [Google Scholar]
- 70.Huber G, Nesemann G. Biochem Biophys Res Commun. 1968;30:7–13. doi: 10.1016/0006-291x(68)90704-3. [DOI] [PubMed] [Google Scholar]
- 71.Baizman ER, Branstrom AA, Longley CB, Allanson N, Sofia MJ, Gange D, Goldman RC. Microbiology. 2000;146:3129–3140. doi: 10.1099/00221287-146-12-3129. Pt 12. [DOI] [PubMed] [Google Scholar]
- 72.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Proc Natl Acad Sci U S A. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lovering AL, de Castro LH, Lim D, Strynadka NC. Science. 2007;315:1402–1405. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- 74.Heaslet H, Shaw B, Mistry A, Miller AA. J Struct Biol. 2009;167:129–135. doi: 10.1016/j.jsb.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 75.Huang CY, Shih HW, Lin LY, Tien YW, Cheng TJ, Cheng WC, Wong CH, Ma C. Proc Natl Acad Sci U S A. 2012;109:6496–6501. doi: 10.1073/pnas.1203900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gampe CM, Tsukamoto H, Wang TS, Walker S, Kahne D. Tetrahedron. 2011;67:9771–9778. doi: 10.1016/j.tet.2011.09.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yuan Y, Fuse S, Ostash B, Sliz P, Kahne D, Walker S. ACS Chem Biol. 2008;3:429–436. doi: 10.1021/cb800078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuse S, Tsukamoto H, Yuan Y, Wang TS, Zhang Y, Bolla M, Walker S, Sliz P, Kahne D. ACS Chem Biol. 2010;5:701–711. doi: 10.1021/cb100048q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ostash B, Doud EH, Lin C, Ostash I, Perlstein DL, Fuse S, Wolpert M, Kahne D, Walker S. Biochemistry. 2009;48:8830–8841. doi: 10.1021/bi901018q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheng TJ, Sung MT, Liao HY, Chang YF, Chen CW, Huang CY, Chou LY, Wu YD, Chen YH, Cheng YS, Wong CH, Ma C, Cheng WC. Proc Natl Acad Sci U S A. 2008;105:431–436. doi: 10.1073/pnas.0710868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eggert US, Ruiz N, Falcone BV, Branstrom AA, Goldman RC, Silhavy TJ, Kahne D. Science. 2001;294:361–364. doi: 10.1126/science.1063611. [DOI] [PubMed] [Google Scholar]
- 82.Rebets Y, Lupoli T, Qiao Y, Schirner K, Villet R, Hooper D, Kahne D, Walker S. ACS Chem Biol. 2013 doi: 10.1021/cb4006744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gampe CM, Tsukamoto H, Doud EH, Walker S, Kahne D. J Am Chem Soc. 2013;135:3776–3779. doi: 10.1021/ja4000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cheng TJ, Wu YT, Yang ST, Lo KH, Chen SK, Chen YH, Huang WI, Yuan CH, Guo CW, Huang LY, Chen KT, Shih HW, Cheng YS, Cheng WC, Wong CH. Bioorganic & medicinal chemistry. 2010;18:8512–8529. doi: 10.1016/j.bmc.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 85.Taylor JG, Li X, Oberthur M, Zhu W, Kahne DE. J Am Chem Soc. 2006;128:15084–15085. doi: 10.1021/ja065907x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ostash B, Saghatelian A, Walker S. Chem Biol. 2007;14:257–267. doi: 10.1016/j.chembiol.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ren F, Ko TP, Feng X, Huang CH, Chan HC, Hu Y, Wang K, Ma Y, Liang PH, Wang AH, Oldfield E, Guo RT. Angew Chem Int Ed Engl. 2012;51:4157–4160. doi: 10.1002/anie.201108002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koyama N, Tokura Y, Takahashi Y, Tomoda H. Bioorg Med Chem Lett. 2013;23:860–863. doi: 10.1016/j.bmcl.2012.11.044. [DOI] [PubMed] [Google Scholar]
- 89.Makitrynskyy R, Rebets Y, Ostash B, Zaburannyi N, Rabyk M, Walker S, Fedorenko V. J Ind Microbiol Biotechnol. 2010;37:559–566. doi: 10.1007/s10295-010-0701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rabyk M, Ostash B, Rebets Y, Walker S, Fedorenko V. Biotechnol Lett. 2011;33:2481–2486. doi: 10.1007/s10529-011-0728-z. [DOI] [PubMed] [Google Scholar]
- 91.Ostash B, Doud E, Walker S. Arch Microbiol. 2012;194:915–922. doi: 10.1007/s00203-012-0827-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kong F, Zhao N, Siegel MM, Janota K, Ashcroft JS, Koehn FE, Borders DB, Carter GT. J Am Chem Soc. 1998;120:13301–13311. [Google Scholar]
- 93.Singh MP, Petersen PJ, Weiss WJ, Kong F, Greenstein M. Antimicrob Agents Chemother. 2000;44:2154–2159. doi: 10.1128/aac.44.8.2154-2159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pletcher JM, McDonald FE. Org Lett. 2005;7:4749–4752. doi: 10.1021/ol051971s. [DOI] [PubMed] [Google Scholar]
- 95.Balthaser BR, McDonald FE. Org Lett. 2009;11:4850–4853. doi: 10.1021/ol901923x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strobel T, Al-Dilaimi A, Blom J, Gessner A, Kalinowski J, Luzhetska M, Puhler A, Szczepanowski R, Bechthold A, Ruckert C. BMC Genomics. 2012;13:465. doi: 10.1186/1471-2164-13-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berner M, Krug D, Bihlmaier C, Vente A, Muller R, Bechthold A. J Bacteriol. 2006;188:2666–2673. doi: 10.1128/JB.188.7.2666-2673.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geng P, Bai G, Shi Q, Zhang L, Gao Z, Zhang Q. J Appl Microbiol. 2009;106:525–533. doi: 10.1111/j.1365-2672.2008.04021.x. [DOI] [PubMed] [Google Scholar]
- 99.Geng P, Bai G. Carbohydr Res. 2008;343:470–476. doi: 10.1016/j.carres.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 100.Geng P, Qiu F, Zhu Y, Bai G. Carbohydr Res. 2008;343:882–892. doi: 10.1016/j.carres.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 101.Geng P, Meng X, Bai G, Luo G. Anal Chem. 2008;80:7554–7561. doi: 10.1021/ac801117s. [DOI] [PubMed] [Google Scholar]
- 102.Mahmud T. Nat Prod Rep. 2003;20:137–166. doi: 10.1039/b205561a. [DOI] [PubMed] [Google Scholar]
- 103.Qin X, Ren L, Yang X, Bai F, Wang L, Geng P, Bai G, Shen Y. J Struct Biol. 2011;174:196–202. doi: 10.1016/j.jsb.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 104.Wehmeier UF, Piepersberg W. Appl Microbiol Biotechnol. 2004;63:613–625. doi: 10.1007/s00253-003-1477-2. [DOI] [PubMed] [Google Scholar]
- 105.Flatt PM, Mahmud T. Nat Prod Rep. 2007;24:358–392. doi: 10.1039/b603816f. [DOI] [PubMed] [Google Scholar]
- 106.Mahmud T, Flatt PM, Wu X. J Nat Prod. 2007;70:1384–1391. doi: 10.1021/np070210q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rockser Y, Wehmeier UF. J Biotechnol. 2009;140:114–123. doi: 10.1016/j.jbiotec.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 108.Degwert U, van Hulst R, Pape H, Herrold RE, Beale JM, Keller PJ, Lee JP, Floss HG. J Antibiot (Tokyo) 1987;40:855–861. doi: 10.7164/antibiotics.40.855. [DOI] [PubMed] [Google Scholar]
- 109.Guo X, Geng P, Bai F, Bai G, Sun T, Li X, Shi L, Zhong Q. Lett Appl Microbiol. 2012;55:162–169. doi: 10.1111/j.1472-765X.2012.03274.x. [DOI] [PubMed] [Google Scholar]
- 110.Hemker M, Stratmann A, Goeke K, Schroder W, Lenz J, Piepersberg W, Pape H. J Bacteriol. 2001;183:4484–4492. doi: 10.1128/JB.183.15.4484-4492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]