

Abstract
Previous studies suggest that the lipid-lowering effect of berberine (BBR) involves actions on the low-density lipoprotein receptor and the AMP-activated protein kinase signaling pathways. However, the implication of these mechanisms is unclear because of the low bioavailability of BBR. Because the main action site of BBR is the gut and intestinal farnesoid X receptor (FXR) plays a pivotal role in the regulation of lipid metabolism, we hypothesized that the effects of BBR on intestinal FXR signaling pathway might account for its pharmacological effectiveness. Using wild type (WT) and intestine-specific FXR knockout (FXRint−/−) mice, we found that BBR prevented the development of high-fat-diet–induced obesity and ameliorated triglyceride accumulation in livers of WT, but not FXRint−/− mice. BBR increased conjugated bile acids in serum and their excretion in feces. Furthermore, BBR inhibited bile salt hydrolase (BSH) activity in gut microbiota, and significantly increased the levels of tauro-conjugated bile acids, especially tauro-cholic acid(TCA), in the intestine. Both BBR and TCA treatment activated the intestinal FXR pathway and reduced the expression of fatty-acid translocase Cd36 in the liver. These results indicate that BBR may exert its lipid-lowering effect primarily in the gut by modulating the turnover of bile acids and subsequently the ileal FXR signaling pathway. In summary, we provide the first evidence to suggest a new mechanism of BBR action in the intestine that involves, sequentially, inhibiting BSH, elevating TCA, and activating FXR, which lead to the suppression of hepatic expression of Cd36 that results in reduced uptake of long-chain fatty acids in the liver.
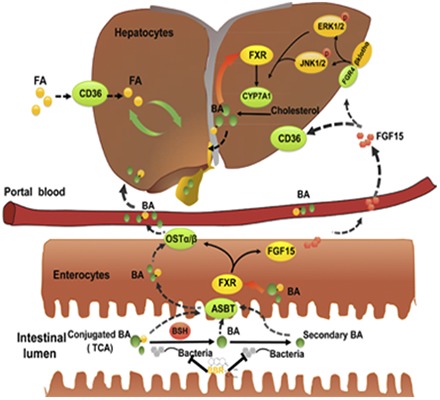
Introduction
Obesity with excess fat accumulation and extensively distorted metabolic regulation is a major risk factor for cardiovascular disease (Hubert et al., 1983; Lavie et al., 2009), type 2 diabetes (Mokdad et al., 2003), a variety types of cancer (Vaughan et al., 1995; Carroll, 1998), and nonalcoholic fatty liver disease (NAFLD) (Wanless and Lentz, 1990). The prevalence of NAFLD is high in developed countries (up to 30%) (Williams, 2006; Angulo, 2007) and is increasing in developing countries (nearly 10%) (Fan and Farrell, 2009). Nonalcoholic steatohepatitis (NASH) is a severe form of NAFLD that includes steatosis, inflammation, and fibrosis in the liver. NASH is becoming a major cause of hepatic cirrhosis and hepatocellular carcinoma. According to the “two-hit” model of NASH, two sequential injuries, lipid accumulation followed by a second insult, lead to the development of NASH. Thus, preventing the accumulation of lipids in the liver may be extremely important in the prevention of NASH (James and Day, 1998; Polyzos et al., 2009).
Berberine (BBR), which is extracted from the roots of Rhizoma Coptidis, has been used traditionally to treat diarrhea. Interestingly, BBR decreases serum lipids in humans, hamsters, mice, and rats (Kong et al., 2004; Chang et al., 2010; Wang et al., 2010, 2014). BBR was also reported to be effective in the prevention and treatment of NAFLD (Chang et al., 2010; Yuan et al., 2015; Guo et al., 2016). In a previous study using hamsters, we found that BBR was poorly absorbed into the systemic circulation but significantly accumulated in the intestine (Gu et al., 2015). Furthermore, some studies have revealed that BBR treatment could change the composition of gut microbiota (Xie et al., 2011; Zhang et al., 2012, 2015). Therefore we hypothesized that multiple mechanisms in the intestine might be responsible for the lipid-lowering effects of BBR.
Farnesoid X receptor (FXR, NRIH4) is a nuclear receptor that is mainly expressed in the liver, intestine, kidney, and adrenals (Lee et al., 2006a,b). FXR is essential in maintaining bile acid homeostasis and is important in the regulation of cholesterol metabolism (Sinal et al., 2000). Bile acids are endogenous ligands of FXR. Intestinal, followed by hepatic, FXR signaling pathways are important for suppressing bile acid synthesis. This suppression is achieved by regulation of the expression of the Cyp7a1/CYP7A1 gene, which encodes cholesterol 7 alpha-hydroxylase, the rate-limiting enzyme in the conversion of cholesterol into bile acids (Kim et al., 2007). FXR also regulates the expression of genes encoding various bile acid transporters, including sodium/tauro-cholate cotransporting polypeptide, bile salt export pump, multidrug resistance protein 2, and organic solute transporters α and β in the liver and apical sodium-dependent bile acid transporter and organic solute transporters α and β in the intestine (Laffitte et al., 2000; Jung et al., 2007). FXR has also been found to modulate triglyceride and glucose homeostasis (Watanabe et al., 2004; Trauner et al., 2010; Potthoff et al., 2011), reduce energy expenditure (Watanabe et al., 2011), and exert anti-inflammatory and antifibrotic effects (Zhang et al., 2009). However, to the best of our knowledge, the effects of BBR on intestinal FXR function have not been evaluated.
Gut microbiota, or gut flora, are bacteria that have colonized within the intestines. Interactions between these microbiota and the human host play an important role in metabolic homeostasis and human health (Serino et al., 2009; Nicholson et al., 2012). Bile acid metabolism is closely related to gut microbiota composition. It is well established that secondary bile acids are derived from the primary bile acids by the action of bacteria present in the intestine (Chiang, 2009). Bile salt hydrolase (BSH), an enzyme synthesized by gut microbiota, acts on carbon-nitrogen bonds to hydrolyze taurine- or glycine-conjugated bile acids for deconjugation (Begley et al., 2006). BSH activity can be modulated by various antibiotics and probiotics, and this modulation may affect lipid metabolism and host health (De Smet et al., 1995, 1998; Li et al., 2013).
As shown in our previous study, BBR enhanced the production of bile acids in hamsters. Moreover, CYP7A1 and CYP27A1 mRNA and protein expression was induced by BBR in a concentration-dependent manner (Gu et al., 2015). In the current study, we have used WT and intestine-specific FXR knockout (FXRint−/−) mice to evaluate the regulatory effects of BBR on BSH activity and bile acid turnover, with the purpose of gaining insight into the antiobesity and lipid-lowering mechanisms of BBR.
Materials and Methods
Chemicals and Reagents.
BBR (≥98%) was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China). Chenodeoxycholic acid (CDCA), ursodeoxycholic acid (UDCA), and hyodeoxycholic acid (HDCA) were purchased from the National Institutes for Food and Drug Control (Beijing, China). Cholic acid (CA), deoxycholic acid (DCA), tauro-cholic acid (TCA), lithocholic acid (LCA), glycochenodeoxycholic acid (GCDCA), glycocholic acid (GCA), tauro-chenodeoxycholic acid (TCDCA), tauro-deoxycholic acid (TDCA), cholic-2,2,4,4-D4 acid (d4-CA), Triton X-114, and tert-butanol were purchased from Sigma-Aldrich (St. Louis, MO); tauro-cholic-2,2,4,4-D4 acid (d4-TCA) and glycocholic-2,2,4,4-D4 (d4-GCA) were purchased from TLC PharmaChem (Concord, ON, Canada). α-muricholic acid (αMCA), β-muricholic acid (βMCA), tauro-β-muricholic acid (TβMCA), glycocholic acid, glycoursodeoxycholic acid, glycodeoxycholic acid, tauro-ursodeoxycholic acid (TUDCA), tauro-hyodeoxycholic acid, and tauro-lithocholic acid (TLCA) were purchased from Steraloids Inc. (Newport, RI). Bio-Bag and AnaeroPack (2.5-liter) were purchased from Mitsubishi Gas Chemical Company, Inc. (Kyoto, Japan).
Animal Treatment.
WT and FXRint−/− mice with a C57BL/6J background (6- to 8-weeks-old, male) were bred and maintained at Rutgers, The State University of New Jersey (Kong et al., 2012). All animals were housed under specific-pathogen–free conditions, fed ad libitum, and maintained under a 12-hour light-dark cycle. All animal experiments were performed with approval from the Institutional Animal Care and Use Committee of Rutgers University and from the Animal Ethics Committee of China Pharmaceutical University.
To determine the effects of BBR on the hepatic accumulation of triglycerides and its antiobesity activity, four groups of WT mice were used. Two groups of mice (n = 6) were fed with control diet (AIN-93M; Research Diets, New Brunswick, NJ) and treated with sodium carboxymethyl cellulose (CMC-Na; 0.5%) or BBR (150 mg/kg). Two other groups were fed with high-fat diet (HFD; 60% calories from fat and 1% cholesterol; Research Diets, New Brunswick, NJ): the HFD vehicle group (n = 6) was administered CMC-Na, and the treatment group (n = 6) was administered BBR (150 mg/kg). For the FXRint−/− study, four groups of FXRint−/− mice were fed with AIN-93M or HFD. Groups were paired and intragastrically administered either CMC-Na as a vehicle control (n = 4) or BBR (150 mg/kg, n = 4) for 8 weeks. BBR was ground into fine particles and passed through a 120-mesh sieve. The powder was then stirred and suspended with 0.5% CMC-Na aqueous solution to achieve a final concentration of 15 mg/ml. This suspension was given to mice by oral gavage at a dose of 10 ml/kg body weight. The body weights of the mice were recorded every week. The animals were fasted overnight before euthanasia. At the time of euthanasia, the mice were anesthetized by xylazine-ketamine and avertin, and samples of blood, liver, gallbladder, small intestine, and cecal content were collected and stored at –80°C for further analysis.
For the TCA treatment, WT mice (6 weeks old, male) were provided by Yangzhou University, Yangzhou, Jiangsu, China. Two groups (n = 6) of mice were intragastrically administered either vehicle (saline) or 100 mg/kg TCA for 7 days. Tissue collection was performed as described above.
Oral Glucose Tolerance Test.
Seven weeks after treatment with BBR, an oral glucose tolerance test was performed: Mice were fasted for 6 hours (9:00 AM to 3:00 PM) and oral administration of 1 g/kg glucose followed. Blood was drawn at 0, 15, 30, 60, 90, and 120 minutes after administration, and blood glucose was measured by ContourTS blood glucose meter (Bayer HealthCare LLC, Mishawaka, IN). Area under the curve was calculated.
Measurement of Hepatic Total Triglycerides.
To further evaluate the lipid-lowering effect of BBR, hepatic total triglycerides were measured. Briefly, a 100-mg sample of liver was homogenized with 1 ml of buffer I (18 mM Tris HCl, pH 7.5, 300 mM mannitol, 50 mM EGTA, and 0.1 mM phenylmethylsulfonyl fluoride), and 400 μl of homogenate was extracted with 2 ml of chloroform/methanol mixture (2:1). After overnight mixing, 1 ml of water was added to each tube and mixed, followed by centrifugation at 3000g for 5 minutes. Afterward, the lower lipid phase was collected and concentrated using a SpeedVac (ThermoFisher Scientific, Waltham, MA). The lipid pellets were resuspended with buffer II [60 μl of tert-butanol and 40 μl of Triton X-114/methanol (2:1) in 100 μl]. Triglycerides were measured using an enzymatic-colorimetric assay kit purchased from Pointe Scientific (Canton, MI) according to the protocol provided.
Oil-Red-O Staining.
Livers were frozen in optimal cutting temperature compound during tissue collection, sectioned into 8-μm-thick slices and stained with Oil Red O using a previously described protocol (Kong et al., 2009).
Reverse Transcription and Real-Time Polymerase Chain Reaction Analysis.
Total RNA was isolated using TRI-Reagent (Sigma-Aldrich). The mRNA concentration was quantified using a Nano Drop ND-1000 spectrophotometer (ThermoFisher Scientific). The diluted mRNA (0.5 μg/μl) was reverse-transcribed according to the manufacturer’s protocol, and the gene expression levels were determined by SYBR Green-based real-time PCR technique (RT-qPCR; ABI ViiA 7 Real-Time PCR System; Applied Biosystems/ThermoFisher Scientific). The mRNA levels of β-actin were used for an internal normalization. The sequences of primers used for RT-qPCR are listed in Table 1.
TABLE 1 .
Primers used for gene expression analysis
| Primer | Forward Primer Sequence (5′ - 3′) | Reverse Primer Sequence (5′ - 3′) |
|---|---|---|
| β-actin | GCGTGACATCAAAGAGAAGC | CTCGTTGCCAATAGTGATGAC |
| Cd36 | GATGACGTGGCAAAGAACAG | TCCTCGGGGTCCTGAGTTAT |
| Cyp7a1 | AACAACCTGCCAGTACTAGATAGC | GTGTAGAGTGAAGTCCTCCTTAGC |
| Fgf15 | GCCATCAAGGACGTCAGCA | CTTCCTCCGAGTAGCGAATCAG |
| Fxr | TCCGGACATTCAACCATCAC | TCACTGCACATCCCAGATCTC |
| Shp | CGATCCTCTTCAACCCAGATG | AGGGCTCCAAGACTTCACACA |
| Ibabp | GGTCTTCCAGGAGACGTGAT | ACATTCTTTGCCAATGGTGA |
| OSTβ | GACAAGCATGTTCCTCCTGAG | GATGCAGGTCTTCTGGTGTTTC |
| Srebp-1c | GGAGCCATGGATTGCACATT | GCTTCCAGAGAGGAGGCCAG |
| Pparα | CAGTGGGGAGAGAGGACAGA | AGTTCGGGAACAAGACGTTG |
| Acc | TGACAGACTGATCGCAGAGAAAG | TGGAGAGCCCCACACACA |
| Fas | GCTGCGGAAACTTCAGGAAAT | AGAGACGTGTCACTCCTGGACTT |
| Cyp4a10 | CTCCAAGTGGCCTCTGTGCT | TAAGTAGGCCTTGCTTCCCCA |
| ApoB | TGGGATTCCATCTGCCATCTCGAG | GTACAGATCCATCACAGGACAATG |
| Mtp | GACCACCCTGGATCTCCATA | AGCGTGGTGAAAGGGCTTAT |
Western Blot.
Total protein in the liver was extracted and Western blot analysis was performed as previously described (Kong et al., 2012; Cao et al., 2013). Antibodies against phospho-ERK (p-ERK)1/2 and total ERK (T-ERK)1/2 were purchased from Cell Signaling Technology, Inc. (Beverly, MA), antibodies against Cd36 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (the loading control) were purchased from Wuhan Boster Biologic Technology Co., Ltd. (Wuhan, China). The intensity of bands on the Western blots was quantified by Image Laboratory statistical software (Bio-Rad Laboratories, Hercules, CA).
Determination of Bile Acid Composition in Serum, Intestine, and Feces.
Bile acids in serum, liver, intestine and feces were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) as previously reported with some revisions (Zhou et al., 2014). Briefly, bile acids were extracted with 70% ethanol at 55°C for 4 hours and then subjected to solid-phase extraction using solid-phase extraction technique [UCT-Clean-Up C18 (Chromatographic Specialties Inc., Brockville, ON, Canada); Oasis-HLB and Oasis-MAX cartridges (Waters Corporation, Milford, MA)]. The resultant residue was redissolved in 100 μl of methanol and centrifuged at 18,000 rpm for 10 minutes. Chromatography was performed on a Shimadzu HPLC system (Kyoto, Japan) coupled to an AB SCIEX 4000 mass spectrometer (Applied Biosystems, MDS; SCIEX, Framingham, MA). A Waters Atlantis T3 column (2.1 × 100 mm, 3 μm) protected by a Security Guard (Phenomenex Inc., Torrance, CA) was used for chromatographic separation. The mobile phase consisted of 0.1% formic acid in water and methanol. d4-CA and d4-GCA acid were used as internal standards.
Measurement of BSH Activity.
After 8 weeks of treatment, the cecal contents of mice were recovered and pooled in sterile tubes. The gut contents were boiled with an aqueous solution containing 20% glycerol and 1.8% sodium chloride at a 1:3 ratio (g/ml). The samples were stored at –80°C for further analysis. All experimental procedures were performed in a sterile environment.
Gut flora were diluted 10-fold in a peptone-yeast extract (PY) medium prepared as previously described (Zhao et al., 2013) and then cultured at 37°C and 200 rpm for 12 hours to allow anaerobic bacteria to proliferate. For analysis, 10 μl of PY medium (containing 50 μg/ml of d4-TCA) and 90 μl of the fermented gut content were mixed in a 1.5-ml Eppendorf tube and placed into a 2.5-liter sealed can with a compostable liner. The sealed can was then placed into an incubator and shaken at 200 rpm and 37°C for 20 minutes. After incubation, 300 μl of methanol (containing 100 ng/ml of internal standard, d4-GCA) was added to each tube, and the tubes were vigorously vortexed for 5 minutes. After centrifugation at 12,000g for 10 minutes, 200 μl of the supernatant was transferred into a LC vial for LC-MS/MS analysis of d4-CA.
To analyze the activity of BSH in gut contents of the mice fed with the control diet or the HFD, BBR was added to PY medium at concentrations of 0 and 100 μg/ml. Following this, the gut flora were diluted in PY medium 10-fold and then cultured at 37°C and 200 rpm for 12 hours to allow anaerobic bacteria to proliferate. The following procedure was the same as previously described, and the production of d4-CA was determined by LC-MS/MS.
Statistical Analysis.
All data were expressed as mean ± S.D. Differences among groups were tested by one-way analysis of variance and/or two-tailed Student’s t tests. A heatmap was constructed using the R package pheatmap (http://cran.r-project.org/web/packages/pheatmap/index.html).
Results
BBR Prevented Diet-Induced Obesity and Triglyceride Accumulation in the Liver.
To investigate the antiobesity and lipid-lowering effects of BBR, the body weight of mice were recorded weekly. After 8 weeks, the mice fed the HFD showed a significant increase in body weight (31.03 ± 1.82 g versus 27.57 ± 0.9 g, P < 0.001), whereas treatment with BBR reduced this effect (25.33 ± 1.76 g versus 31.03 ± 1.82 g, P < 0.001) (Fig. 1A). To explore the effect of BBR on glucose homoeostasis, an oral glucose tolerance test was performed, and the results revealed that after 7 weeks of HFD, compared with vehicle groups, mice treated with BBR displayed significantly reduced blood glucose levels after glucose loading, suggesting improved insulin sensitivity. For mice fed with AIN-93M, there was no change (Fig. 1, B and C). Moreover, fasting serum glucose levels from BBR-treated groups decreased significantly (Fig. 1D). Triglyceride levels in serum and livers of mice fed with HFD were significantly increased, by 1.2 fold and 4.4 fold, respectively. After BBR treatment, there was a significant decrease in serum and hepatic triglycerides by 19% and 47%, respectively (Fig. 1, E and F). Lipid droplets in the liver also appeared to be reduced in size as revealed by Oil-Red-O staining (Fig. 1G).
Fig. 1.

BBR prevented HFD-induced weight gain and ameliorated hepatic triglyceride accumulation in WT mice. Male WT mice were treated with BBR [150 mg/kg, intragastric (i.g.)] or vehicle (CMC-Na) for 8 weeks. (A) Weekly body weight changes. (B) Oral glucose tolerance test. (C) Area under the curve (AUC) of blood glucose. (D) Blood glucose after a 6-hour fast. (E) Serum triglycerides at the end of the treatment. (F) Hepatic triglycerides at the end of the experiment. (G) Oil-Red-O staining (20×). WT AIN-93M vehicle, mice fed with control diet and treated with CMC-Na; WT AIN-93M BBR, mice fed with control diet and treated with BBR; WT HFD vehicle, mice fed with HFD and treated with CMC-Na; WT HFD BBR, mice fed with control diet and treated with BBR. Error bars show S.D. of replicates (n = 6). *P < 0.05; **P < 0.01, ***P < 0.001, compared with normal control group; #P < 0.05; ##P < 0.01, compared with high-fat control group.
BBR Activated the Intestinal FXR Pathway and Reduced Cd36 Gene Expression in Liver.
For genes involved in fatty-acid metabolism and transportation, the expression of Fas was decreased by BBR treatment, and the expression of Cyp4a10 was upregulated significantly. The expression of Mtp and ApoB were not changed (Fig. 2, A and B). The intestinal FXR pathway plays a key role in the regulation of cholesterol metabolism and bile acid homeostasis. The 8-week treatment with BBR may have activated the intestinal, but not hepatic, FXR pathway, as revealed by increased extracellular signal-regulated kinase (ERK)1/2 phosphorylation in the liver and the upregulation of Fgf15 and Ibabp mRNA levels in the distal ileum (Fig. 2, C–F). Furthermore, oral administration of BBR reduced Cyp7a1 mRNA levels in mice (Fig. 2D), which is consistent with the upregulation of intestinal Fgf15. Because Cd36 is critical to the uptake of long-chain fatty acids into the liver and has been shown to be reduced by FXR activation, we also examined its mRNA expression and protein level. We found that treatment with BBR markedly downregulated the mRNA expression of Cd36 in the liver by approximately 48% (P = 0.045, Fig. 2B). The protein level of Cd36 was also reduced by BBR treatment (Fig. 2, E and F).
Fig. 2.

BBR activated intestinal but not hepatic FXR signaling pathway and reduced Cd36 expression in WT mice. Total RNA were isolated from the liver and intestine from male WT mice after treatment with BBR (150 mg/kg, i.g.) or vehicle (CMC-Na) for 8 weeks and RT-qPCR was performed to measure the expression of genes. (A) Relative mRNA levels of Pparα, Srebp-1c, Acc, and Fas in liver. (B) Relative mRNA levels of Cyp4a10, Mtp, ApoB, and Cd36 gene in the liver. (C) Relative mRNA levels of Fxr, Fgf15, Shp, and Ibabp gene in the distal ileum. (D) Relative mRNA levels of Fxr, Shp, and Cyp7a1 gene in the liver. (E) Semiquantitative analysis of the protein levels in liver. (F) Protein level of Cd36, p-ERK1/2, and T-ERK1/2 in liver. Error bars show S.D. of replicates (n = 6). *P < 0.05; **P < 0.01, compared with normal control group; #P < 0.05; ##P < 0.01, compared with high-fat control group.
BBR Treatment Caused Modifications in Bile Acid Composition.
Bile acid composition is an indicator of the homeostasis and turnover of cholesterol as well as of bile acid metabolism activity. There are species differences in bile acid composition, and muricholic acid (MCA) only exists in rodent. In mice, with respect to individual bile acid, TCA and TβMCA were agonist and antagonist of FXR, respectively (Sayin et al., 2013). Therefore, we measured bile acid profiles in serum, liver, intestine, and feces using LC-MS/MS. The compositions of bile acids are shown in Figs. 3A, 4A, 5A, and 6A.
Fig. 3.

Changes in bile acid composition in the serum after BBR treatment. Bile acids in serum of male WT mice after treatment with BBR (150 mg/kg, i.g.) or vehicle (CMC-Na) for 8 weeks were measured using LC-MS/MS. (A) Heatmap detailing the bile acid composition of serum from WT mice. The color key represents the calibrated contents of bile acids, and the dendrogram on the left showed the clustering of bile acids with the similar changes. (B) TβMCA levels in the serum. (C) TCA levels in the serum. Error bars show S.D. of replicates (n = 6). *P < 0.05, compared with normal control group; #P < 0.05; ##P < 0.01, compared with high-fat control group.
Fig. 4.

Changes in bile acid composition in the liver after BBR treatment. Bile acids in liver of male WT mice after treatment with BBR (150 mg/kg, i.g.) or vehicle (CMC-Na) for 8 weeks were measured using LC-MS/MS. (A) Heatmap detailing the bile acid composition of liver. The color key represents the calibrated contents of bile acids, and the dendrogram on the left showed the clustering of bile acids with the similar changes. (B) TβMCA levels in the liver. (C) TCA levels in the liver. Error bars show S.D. of replicates (n = 6). **P < 0.01, ***P < 0.001, compared with normal control group; #P < 0.05, compared with high-fat control group.
Fig. 5.

Changes in bile acid composition in the intestine after BBR treatment. Bile acids in the intestine of male WT mice after treatment with BBR (150 mg/kg, i.g.) or vehicle (CMC-Na) for 8 weeks were measured. (A) Heatmap detailing the bile acid composition of intestine. The color key represents the calibrated contents of bile acids, and the dendrogram on the left showed the clustering of bile acids with the similar changes. (B) TβMCA levels in the intestine. (C) TCA levels in the intestine. Error bars show S.D. of replicates (n = 6). **P < 0.01, compared with normal control group; #P < 0.05, compared with high-fat control group.
Fig. 6.

Changes in bile acid composition in feces after BBR treatment. Bile acids were extracted from feces of male WT mice after treatment with BBR (150 mg/kg, i.g.) or vehicle (CMC-Na) for 8 weeks and measured. (A) Heatmap detailing the bile acid composition of feces. The color key represents the calibrated contents of bile acids, and the dendrogram on the left showed the clustering of bile acids with similar changes. (B) TβMCA levels increased in feces after BBR treatment. (C) TCA levels increased in feces after BBR treatment. Error bars show S.D. of replicates (n = 6). ***P < 0.001, compared with normal control group; ###P < 0.001, compared with high-fat control group.
In serum, HFD increased the levels of GCA, TCA, and TUDCA, whereas it decreased the levels of other bile acids. Treatment with BBR increased the levels of TCA and LCA but decreased the levels of other bile acids (Fig. 3A). Specifically, for TβMCA and TCA, we found that the consumption of the HFD did not change TCA levels, whereas TβMCA levels increased in serum. Treatment with BBR reduced TβMCA in the serum of mice fed with both AIN-93M and HFD. For TCA, treatment with BBR had no influence on mice fed with AIN-93M but significantly increased TCA level in serum of mice fed with HFD (Fig. 3, B and C).
For bile acids in the liver, the consumption of the HFD did not change the levels of bile acids significantly. Treatment with BBR increased the level of TUDCA, TDCA, LCA, and TCA but not the levels of GCA and CA. The other bile acids trended toward reduction (Fig. 4A). Specifically, for TβMCA and TCA, treatment with BBR decreased TβMCA levels in mice fed with both diets, and BBR also decreased TCA levels in mice fed with AIN-93M, whereas there was no obvious change for TCA in mice fed with the HFD (Fig. 4, B and C).
In the intestine, HFD rarely changed the levels of individual bile acids. BBR also had no effect on tauro-hyodeoxycholic acid, LCA, and GCA; however, the levels of UDCA, αMCA, βMCA, ωMCA, CA, CDCA, HDCA, and DCA were decreased. The levels of TDCA, TCDCA, and TUDCA showed a trend of increase (Fig. 5A). Specifically, BBR showed rare regulation effects on TβMCA, and both TβMCA and TCA levels were elevated after treatment with BBR (Fig. 5, B and C).
With respect to bile acid composition in the feces, the consumption of a HFD increased TDCA, αMCA, ωMCA, βMCA, TCDCA, TLCA, CDCA, DCA, LCA, UDCA, and HDCA levels (Fig. 6A). Specifically, levels of both TβMCA (Fig. 6B) and TCA (Fig. 6C) were increased after the consumption of the HFD and were even further elevated after treatment with BBR.
BBR Inhibited BSH Activity.
BSH activity is critically involved in the capacity of gut microbiota to hydrolyze conjugated bile acids. A previously established in vitro metabolism system was used to evaluate the BSH activity of gut microbiota (Zhao et al., 2013). Overall, a decrease in d4-TCA level was found, and d4-CA production is a measure of BSH activity (Fig. 7A). Relative to the gut microbiota of the mice that were fed the control diet, d4-TCA was hydrolyzed at a slower rate, and there was less of an increase of d4-CA in the mice fed the HFD. Moreover, treatment with BBR reduced the hydrolyzation of d4-TCA and reduced d4-CA to undetectable levels, indicating a marked decrease in the production of d4-CA. Concordantly, coincubation of BBR (100 μg/ml) in vitro also showed an inhibition of BSH activity for gut microbiota from mice fed with both control diet and HFD (Fig. 7, B and C).
Fig. 7.

BBR suppressed the activity of BSH in gut microbiota. (A) In gut microbiota, BSH hydrolyzed d4-TCA to d4-CA and taurine. (B) The consumption of HFD increased the reduction of d4-TCA, and BBR aggravated this function. (C) The consumption of HFD also reduced the production of d4-CA, and BBR aggravated this function. Error bars show S.D. of replicates (n = 6). *P < 0.05; **P < 0.01, ***P < 0.001, compared with control group.
TCA Activated the Intestinal FXR Pathway and Reduced Cd36 Expression in the Liver.
BBR treatment decreased BSH activity and increased the levels of TCA in the small intestine (Fig. 5C). Therefore, we determined the effects of a 7-day TCA treatment on FXR signaling pathway. After TCA treatment, mRNA levels of hepatic FXR target gene Shp and intestinal FXR target genes Fgf15 and Ibabp were upregulated (Fig. 8, A and B). The expression of Fas was decreased and the expression of Cyp4a10 was increased significantly (Fig. 8C). Consistent with the effects of BBR treatment, ERK1/2 phosphorylation in the liver was activated, Cyp7a1 mRNA levels were significantly reduced, and the mRNA expression and protein level of Cd36 were both reduced (Fig. 8, A, D–F).
Fig. 8.

TCA treatment activated the intestinal FXR pathway and reduced Cd36 expression. After TCA treatment of 7 days, livers and intestines of male WT C57BL/6J mice were collected. Protein levels and gene expressions at mRNA level were measured. (A) Relative mRNA levels of Fxr, Shp, and Cyp7a1 gene in the liver. (B) Relative mRNA levels of Fxr, Fgf15, Ibabp, and OSTβ gene in the distal ileum. (C) Relative mRNA levels of Srebp-1c, Pparα, Acc, Fas and Cyp4a10 gene in the liver. (D) Relative mRNA levels of Mtp, ApoB, and Cd36 gene in the liver. (E) Semiquantitative analysis of the protein levels in the liver. (F) Protein level of CD36, p-ERK1/2 and T-ERK1/2 in the liver. Error bars show S.D. of replicates (n = 6). *P < 0.05; **P < 0.01, ***P < 0.001, compared with control group.
Antiobesity and Lipid-Lowering Effects of BBR Was Diminished in FXRint−/− Mice.
To confirm the role of the intestinal FXR signaling pathway in the lipid-lowering effect of BBR, FXRint−/− mice were used. The data showed that BBR treatment did not change either the body weights or the serum triglyceride levels of FXRint−/− mice that were fed with HFD (Fig. 9, A and B). Triglycerides in the livers were not affected by BBR treatment as well, as shown by measurement of lipids and Oil-Red-O staining (Fig. 9, C and D). The expression of the genes involved in fatty-acid metabolism and transportation, including Srebp-1c, Pparα, Acc, Fas, Mtp, and ApoB, was not changed significantly by BBR treatment, whereas Cyp4a10 was induced (P = 0.049) (Fig. 10, A and B). In contrast to an increasing trend of intestinal mRNA levels of Fgf15 that were observed, the mRNA levels of Ibabp and Ostβ were significantly reduced, which is an opposite trend from that observed in WT mice (Fig. 10C). Moreover, the expression of the genes encoding Cyp7a1 (P = 0.005) and Cd36 (not significantly) was increased after BBR treatment (Fig. 10, B and D). These results indicate that the lipid-lowering effect of BBR relies on an intact intestinal FXR signaling pathway, and regulation of Cd36 expression by increased Fgf15 signaling in the liver might be the key mechanism.
Fig. 9.

BBR treatment did not change body weight or hepatic triglyceride in FXRint−/− mice. To further evaluate the role of the intestinal FXR pathway in the pharmacological effects of BBR, intestinal-specific FXR knockout (FXRint−/−) mice were fed an HFD and treated with either BBR (150 mg/kg per day, i.g.) or vehicle (CMC-Na) for 8 weeks. Livers and intestines were collected, and mRNA was extracted and measured by RT-qPCR. (A) The ratio change of mouse body weight at the time of euthanasia to the body weight at the time of the beginning of the experiment revealed that BBR treatment only slightly reduced the body weight gain of FXRint−/− mice. (B) Serum triglycerides at the end of the treatment. (C) Hepatic triglycerides at the end of the experiment. (D) Oil-Red-O staining (20×). FXRint−/− AIN-93M vehicle, mice fed the control diet and treated with CMC-Na; FXRint−/− AIN-93M BBR, mice fed the control diet and treated with BBR; FXRint−/− HFD vehicle, mice fed the HFD and treated with CMC-Na; FXRint−/− HFD BBR, mice fed the HFD and treated with BBR. Error bars show S.D. of replicates (n = 4).
Fig. 10.

BBR inhibited intestinal FXR signaling pathway but did not change the expression of Cd36 in FXRint−/− mice. Total RNA were isolated from the liver and intestine of male FXRint−/− mice after treatment with BBR (150 mg/kg, i.g.) or vehicle (CMC-Na) for 8 weeks, and RT-qPCR was performed to measure the expression of genes. (A) Relative mRNA levels of Pparα, Srebp-1c, Acc, and Fas gene in the liver. (B) Relative mRNA levels of Cyp4a10, Mtp, ApoB, and Cd36 gene in the liver. (C) Relative mRNA levels of Fgf15, Ibabp, and OSTβ gene in the distal ileum. (D) Relative mRNA levels of Fxr, Shp, and Cyp7a1 gene in the liver. Error bars show S.D. of replicates (n = 4). *P < 0.05; **P < 0.01, compared with normal control group; #P < 0.05; ##P < 0.01, compared with high-fat control group.
Discussion
Antiobesity and Lipid-Lowering Effects of Orally Administered BBR.
BBR is reported to possess antiobesity and lipid-lowering effects in different species, and previous studies have suggested that the lipid-lowering mechanisms exerted by BBR include the upregulation of LDL receptor and the activation of the 5′-AMP-activated protein kinase pathway (Kong et al., 2004; Brusq et al., 2006). Other studies have reported that BBR can ameliorate NAFLD by modulating hepatic mRNA and long-noncoding-RNA expression profiles (Yuan et al., 2015). However, the activation of 5′-AMP-activated protein kinase was not observed in an in vivo study (Guo et al., 2016). In addition, the in vitro concentrations of BBR used in the referenced studies were two orders of magnitude higher than the blood concentrations achieved following the oral administration, and when BBR was present in the liver at low concentrations, it failed to produce pharmacological effects (Kong et al., 2004; Liu et al., 2010). In contrast, high levels of BBR accumulate in the gastrointestinal tract after the oral administration. Therefore, we hypothesized that the pharmacological effects of orally administered BBR are exerted through BBR-mediated modulation of intestinal microbiota. We first tested this hypothesis in WT mice. We found that BBR indeed exerted an antiobesity effect, improved insulin resistance, and ameliorated the accumulation of triglycerides in the liver (Fig. 1). Moreover, we hypothesized the intestinal, but not hepatic, FXR signaling pathway was involved in these beneficial effects (Fig. 2, C and D).
Intestinal FXR Plays a Pivotal Role in the Pharmacological Effects of BBR.
The intestinal FXR signaling pathway plays an important role in the development of NAFLD. It has been documented that FXR agonists inhibit inflammation, repress macrophage activation, reduce adipose differentiation-related protein levels, increase cholesteryl ester transfer protein expression, reduce Cd36 gene expression, and promote the conversion of adipose tissue from white adipose tissue to brown adipose tissue (BAT). These actions reduce obesity and improve insulin resistance (Zhang et al., 2009; Ma et al., 2013; Yao et al., 2014; Fang et al., 2015). As the intestinal FXR signaling pathway appeared to be activated in WT mice following treatment with BBR (Fig. 2C), FXRint−/− mice were used to further test the role of intestinal FXR in the pharmacological effects of BBR. We found that BBR did not produce antiobesity and lipid-lowering effects in FXRint−/− mice (Fig. 9), which further suggests that intestinal FXR may be the critical mediator of the pharmacological effects of BBR.
According to previous tissue distribution studies in the liver and small intestine, the concentration of BBR is approximately 100 ng/g tissue (Tan et al., 2013; Hu et al., 2014). Owing to the low bioavailability of BBR, the pharmacological effects of BBR may be a secondary effect following its modulation of gut microbiota and, subsequently, bile acid metabolism in the gut.
BBR Modulated Bile Acid Composition and BSH Activity.
It is well known that different bile acids differentially regulate the activity of FXR, and the modulation of FXR activity by endogenous bile acids creates complicated and comprehensive effects. CDCA, CA, and TCA are endogenous agonists of FXR (Makishima et al., 1999; Parks et al., 1999), whereas ursodeoxycholic acid (UDCA) (Mueller et al., 2015) and TβMCA (Li et al., 2013; Sayin et al., 2013) are endogenous antagonists of FXR. We measured the compositions of bile acids in the sera and feces of mice treated with BBR. In the sera of the mice that were treated with BBR, the majority of conjugated bile acids, especially TCA, were present at elevated levels, whereas the level of TβMCA was unchanged, and the majority of unconjugated bile acids were present at reduced levels. A similar phenomenon was also observed in the feces, in which tauro-conjugated bile acid levels increased following BBR treatment. Bile acid metabolism is closely related to gut microbiota composition; therefore, the modulation of gut microbiota should be considered in the context of BBR treatment.
Both obesity and NAFLD are considered to be closely related to changes in gut microbiota composition, indicating the importance of modulation of gut microbiota (Abu-Shanab and Quigley, 2010; Musso et al., 2011; Compare et al., 2012; Nicholson et al., 2012; Tremaroli and Bäckhed, 2012). BSH, an enzyme produced by gut microbiota, serves as one of the mechanisms by which microbes and hosts interact, and such interactions regulate lipid and cholesterol metabolism. Previous reports have shown that the expression of exogenous BSH in mouse intestines or treatment with probiotics can significantly reduce weight gain and plasma and liver lipids, as well as improve insulin resistance (Degirolamo et al., 2014; Joyce et al., 2014). In the current study, we found that BBR significantly inhibited BSH activity both in vivo and in vitro, and the levels of tauro-conjugated bile acids, especially TCA, became substantially elevated following the inhibition of BSH activity. However, BSH has been found to be expressed by all major bacterial divisions and Archaea species in the gut on the basis of metagenomic analyses (Jones et al., 2008), and BBR was found to reduce the proportions of both fecal Firmicutes and fecal Bacteroidetes in HFD-fed mice (Xie et al., 2011). On the basis of these findings, the modulation of BSH activity by BBR may result from an indiscriminate inhibition of gut microbiota.
Reduction of Cd36 Expression Ameliorated Hepatic Lipid Accumulation.
The activation of the intestinal FXR signaling pathway induces the expression of Fgf15 (human homolog, Fgf19), which acts on the liver and inhibits the expression of Cd36. Cd36 (cluster of differentiation 36), also known as fatty-acid translocase, belongs to the class B scavenger receptor family, which includes receptors for selective cholesteryl ester uptake and is important for lipid transportation and metabolism (Febbraio et al., 2001; Bieghs et al., 2010). The FXR agonist GW4064 showed an inhibitory effect on Cd36 expression and reduced the accumulation of triglycerides in the liver without affecting expression of genes that are directly involved in lipogenesis (Ma et al., 2013). Furthermore, the treatment of FXR knockout mice with fibroblast growth factor 19 protein reduced liver expression of Cd36, reduced the transportation of free fatty acids, and inhibited the synthesis of triglycerides (Miyata et al., 2011).
As TCA was elevated in both serum and feces after BBR treatment, we treated a group of mice with TCA (100 mg/kg) for a week. We found that the TCA treatment produced results consistent with those achieved following BBR treatment: activated intestinal FXR signaling pathway and reduced expression of Cd36. Thus, the antiobesity and lipid-lowering effects of BBR are mainly owing to the activation of intestinal FXR and a reduction in Cd36 expression. In FXRint−/− mice treated with BBR, the expression of FXR target genes in the intestine was inhibited, and Cd36 expression was not changed obviously. Furthermore, the pharmacological effects of BBR were no longer evident, which further validates our hypothesis.
In summary, in mice fed with HFD, BBR shows antiobesity and lipid-lowering effects in WT mice but not in FXRint−/− mice. BBR inhibits the BSH activity of gut microbiota, leading to increased levels of conjugated bile acids. Our data suggest, for the first time, that the lipid-lowering effect of BBR is achieved via sequential events involving the inhibition of BSH, elevation of TCA, activation of the intestinal FXR signaling pathway, and reduction of Cd36 expression in the liver, which result in the reduced hepatic uptake of long-chain fatty acids. This underlying mechanism helps explain the observed discrepancy between the low oral bioavailability of BBR and its effective lipid-lowering and antiobesity effects.
Acknowledgments
The authors thank Ting Ma, Wenjuan Xia, Yong Mao, and Jian Shi for assistance and support in this study. The authors are particularly grateful to the Department of Pharmacology and Toxicology, Department of Chemical Biology, Ernest Mario School of Pharmacy at Rutgers for technical support.
Abbreviations
- αMCA
α-muricholic acid
- BBR
berberine
- βMCA
β-muricholic acid
- BSH
bile salt hydrolase
- CA
cholic acid
- Cd36
cluster of differentiation 36
- CDCA
chenodeoxycholic acid
- CMC-Na
sodium carboxymethyl cellulose
- CYP7A1
cholesterol 7α-hydroxylase
- d4-CA
cholic-2,2,4,4-d4 acid
- d4-GCA
glycocholic-2,2,4,4-D4 acid
- d4-TCA
tauro-cholic-2,2,4,4-D4 acid
- DCA
deoxycholic acid
- ERK
extracellular signal-regulated kinase
- FXR
farnesoid X receptor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GCA
glycocholic acid
- HDCA
hyodeoxycholic acid
- HFD
high-fat diet
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- LCA
lithocholic acid
- MCA
muricholic acid
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- p-ERK
phospho-ERK
- PY medium
peptone-yeast extract medium
- T-ERK
total ERK
- TCA
tauro-cholic acid
- TCDCA
tauro-chenodeoxycholic acid
- TDCA
tauro-deoxycholic acid
- TUDCA
tauro-ursodeoxycholic acid
- TβMCA
tauro-β-muricholic acid
- UDCA
ursodeoxycholic acid
- ωMCA
ω-muricholic acid
- WT
wild type
Authorship Contributions
Participated in research design: Sun, G.L. Guo, C. S. Yang, Aa, G. Wang.
Conducted experiments: Sun, N. Yang, Kong, Cao, Feng, Yu, Ge, Huang, Shen, P. Wang, S. Feng, Fei, J. Guo, He, Aa, Chen, Pan.
Performed data analysis: Sun, N. Yang, Kong, G.L. Guo, and Aa.
Wrote or contributed to the writing of the manuscript: Sun, N. Yang, Kong, Cao, Schumacher, C. S. Yang, G.L. Guo, Aa, and G. Wang.
Footnotes
This study was financially supported by the National Natural Science Foundation of the People’s Republic of China [81573495, 81530098], the Key Technology Projects of China “Creation of New Drugs” [2015ZX09501001], the Project for Jiangsu Province Key Laboratory of Drug Metabolism and Pharmacokinetics [BM2012012], and in part by the National Institutes of Health [R01GM104037]. The authors received financial support from the China Scholarship Council [201407060027].
References
- Abu-Shanab A, Quigley EM. (2010) The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7:691–701. [DOI] [PubMed] [Google Scholar]
- Angulo P. (2007) GI epidemiology: nonalcoholic fatty liver disease. Aliment Pharmacol Ther 25:883–889. [DOI] [PubMed] [Google Scholar]
- Begley M, Hill C, Gahan CG. (2006) Bile salt hydrolase activity in probiotics. Appl Environ Microbiol 72:1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieghs V, Wouters K, Van Gorp PJ, Gijbels MJ, De Winther MP, Binder CJ, Lütjohann D, Febbraio M, Moore KJ, Van Bilsen M. (2010) Role of scavenger receptor A and Cd36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology 138: 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusq J-M, Ancellin N, Grondin P, Guillard R, Martin S, Saintillan Y, Issandou M. (2006) Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J Lipid Res 47:1281–1288. [DOI] [PubMed] [Google Scholar]
- Cao B, Li M, Zha W, Zhao Q, Gu R, Liu L, Shi J, Zhou J, Zhou F, Wu X, et al. (2013) Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 9:960–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KK. (1998) Obesity as a risk factor for certain types of cancer. Lipids 33:1055–1059. [DOI] [PubMed] [Google Scholar]
- Chang X, Yan H, Fei J, Jiang M, Zhu H, Lu D, Gao X. (2010) Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a high-fat diet in rats. J Lipid Res 51:2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY. (2009) Bile acids: regulation of synthesis. J Lipid Res 50:1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Cartenì M, Nardone G. (2012) Gut--liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 22:471–476. [DOI] [PubMed] [Google Scholar]
- De Smet I, De Boever P, Verstraete W. (1998) Cholesterol lowering in pigs through enhanced bacterial bile salt hydrolase activity. Br J Nutr 79:185–194. [DOI] [PubMed] [Google Scholar]
- De Smet I, Van Hoorde L, Vande Woestyne M, Christiaens H, Verstraete W. (1995) Significance of bile salt hydrolytic activities of lactobacilli. J Appl Bacteriol 79:292–301. [DOI] [PubMed] [Google Scholar]
- Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A. (2014) Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Reports 7:12–18. [DOI] [PubMed] [Google Scholar]
- Fan J-G, Farrell GC. (2009) Epidemiology of non-alcoholic fatty liver disease in China. J Hepatol 50:204–210. [DOI] [PubMed] [Google Scholar]
- Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al. (2015) Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 21:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Hajjar DP, Silverstein RL. (2001) Cd36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 108:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Cao B, Sun R, Tang Y, Paletta JL, Wu X, Liu L, Zha W, Zhao C, Li Y, et al. (2015) A metabolomic and pharmacokinetic study on the mechanism underlying the lipid-lowering effect of orally administered berberine. Mol Biosyst 11:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Woo S-L, Guo X, Li H, Zheng J, Botchlett R, Liu M, Pei Y, Xu H, Cai Y, et al. (2016) Berberine Ameliorates Hepatic Steatosis and Suppresses Liver and Adipose Tissue Inflammation in Mice with Diet-induced Obesity. Sci Rep 6:22612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YL, Chen C, Zou ZY, Li XG, Ye XL. (2014) Comparative study of pharmacokinetics and tissue distribution of 8-cetylberberine and berberine in rats. Acta Pharm Sin B 49:1582–1587. [PubMed] [Google Scholar]
- Hubert HB, Feinleib M, McNamara PM, Castelli WP. (1983) Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 67:968–977. [DOI] [PubMed] [Google Scholar]
- James OF, Day CP. (1998) Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol 29:495–501. [DOI] [PubMed] [Google Scholar]
- Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. (2008) Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA 105:13580–13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce SA, MacSharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, Hill C, Gahan CG. (2014) Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc Natl Acad Sci USA 111:7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Elferink MG, Stellaard F, Groothuis GM. (2007) Analysis of bile acid-induced regulation of FXR target genes in human liver slices. Liver Int 27:137–144. [DOI] [PubMed] [Google Scholar]
- Kim I, Ahn S-H, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. (2007) Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48:2664–2672. [DOI] [PubMed] [Google Scholar]
- Kong B, Luyendyk JP, Tawfik O, Guo GL. (2009) Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther 328:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. (2012) Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, Wang Y, Wang Z, Si S, Pan H, et al. (2004) Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med 10:1344–1351. [DOI] [PubMed] [Google Scholar]
- Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD, Edwards PA. (2000) Identification of the DNA binding specificity and potential target genes for the farnesoid X-activated receptor. J Biol Chem 275:10638–10647. [DOI] [PubMed] [Google Scholar]
- Lavie CJ, Milani RV, Ventura HO. (2009) Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol 53:1925–1932. [DOI] [PubMed] [Google Scholar]
- Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. (2006a) FXR, a multipurpose nuclear receptor. Trends Biochem Sci 31:572–580. [DOI] [PubMed] [Google Scholar]
- Lee H, Zhang Y, Lee FY, Nelson SF, Gonzalez FJ, Edwards PA. (2006b) FXR regulates organic solute transporters α and β in the adrenal gland, kidney, and intestine. J Lipid Res 47:201–214. [DOI] [PubMed] [Google Scholar]
- Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. (2013) Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun 4:2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-T, Hao H-P, Xie H-G, Lai L, Wang Q, Liu C-X, Wang G-J. (2010) Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab Dispos 38:1779–1784. [DOI] [PubMed] [Google Scholar]
- Ma Y, Huang Y, Yan L, Gao M, Liu D. (2013) Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res 30:1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284:1362–1365. [DOI] [PubMed] [Google Scholar]
- Miyata M, Sakaida Y, Matsuzawa H, Yoshinari K, Yamazoe Y. (2011) Fibroblast growth factor 19 treatment ameliorates disruption of hepatic lipid metabolism in farnesoid X receptor (Fxr)-null mice. Biol Pharm Bull 34:1885–1889. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS. (2003) Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 289:76–79. [DOI] [PubMed] [Google Scholar]
- Mueller M, Thorell A, Claudel T, Jha P, Koefeler H, Lackner C, Hoesel B, Fauler G, Stojakovic T, Einarsson C, et al. (2015) Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol 62:1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Gambino R, Cassader M. (2011) Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med 62:361–380. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. (2012) Host-gut microbiota metabolic interactions. Science 336:1262–1267. [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284:1365–1368. [DOI] [PubMed] [Google Scholar]
- Polyzos SA, Kountouras J, Zavos Ch. (2009) The multi-hit process and the antagonistic roles of tumor necrosis factor-alpha and adiponectin in non alcoholic fatty liver disease. Hippokratia 13:127–, author reply 128.. [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, Suino-Powell K, Xu HE, Gerard RD, Finck BN, et al. (2011) FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab 13:729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17:225–235. [DOI] [PubMed] [Google Scholar]
- Serino M, Luche E, Chabo C, Amar J, Burcelin R. (2009) Intestinal microflora and metabolic diseases. Diabetes Metab 35:262–272. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744. [DOI] [PubMed] [Google Scholar]
- Tan X-S, Ma J-Y, Feng R, Ma C, Chen W-J, Sun Y-P, Fu J, Huang M, He C-Y, Shou J-W, et al. (2013) Tissue distribution of berberine and its metabolites after oral administration in rats. PLoS One 8:e77969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. (2010) Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis 28:220–224. [DOI] [PubMed] [Google Scholar]
- Tremaroli V, Bäckhed F. (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249. [DOI] [PubMed] [Google Scholar]
- Vaughan TL, Davis S, Kristal A, Thomas DB. (1995) Obesity, alcohol, and tobacco as risk factors for cancers of the esophagus and gastric cardia: adenocarcinoma versus squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 4:85–92. [PubMed] [Google Scholar]
- Wang Y, Jia X, Ghanam K, Beaurepaire C, Zidichouski J, Miller L. (2010) Berberine and plant stanols synergistically inhibit cholesterol absorption in hamsters. Atherosclerosis 209:111–117. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yi X, Ghanam K, Zhang S, Zhao T, Zhu X. (2014) Berberine decreases cholesterol levels in rats through multiple mechanisms, including inhibition of cholesterol absorption. Metabolism 63:1167–1177. [DOI] [PubMed] [Google Scholar]
- Wanless IR, Lentz JS. (1990) Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology 12:1106–1110. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, Mataki C, Sato H, Tanigawara Y, Schoonjans K, et al. (2011) Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem 286:26913–26920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. (2006) Global challenges in liver disease. Hepatology 44:521–526. [DOI] [PubMed] [Google Scholar]
- Xie W, Gu D, Li J, Cui K, Zhang Y. (2011) Effects and action mechanisms of berberine and Rhizoma coptidis on gut microbes and obesity in high-fat diet-fed C57BL/6J mice. PLoS One 6:e24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhou C-S, Ma X, Fu B-Q, Tao L-S, Chen M, Xu Y-P. (2014) FXR agonist GW4064 alleviates endotoxin-induced hepatic inflammation by repressing macrophage activation. World J Gastroenterol 20:14430–14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Wang J, Tang X, Li Y, Xia P, Gao X. (2015) Berberine ameliorates nonalcoholic fatty liver disease by a global modulation of hepatic mRNA and lncRNA expression profiles. J Transl Med 13:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang J, Liu Q, Harnish DC. (2009) Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol 51:380–388. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, Zhang X, Zhao L. (2015) Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci Rep 5:14405 DOI:10.1038/srep14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhao Y, Zhang M, Pang X, Xu J, Kang C, Li M, Zhang C, Zhang Z, Zhang Y, et al. (2012) Structural changes of gut microbiota during berberine-mediated prevention of obesity and insulin resistance in high-fat diet-fed rats. PLoS One 7:e42529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Sun R, Cao B, Gu S, Zhao J, Liu L, Wang X, Zha W, Yu X, Xiao W. (2013) An in vitro metabolic system of gut flora and the metabolism of ginsenoside Rg3 and cholic acid. Eur J Drug Metab Pharmacokinet 39:1–9. [DOI] [PubMed] [Google Scholar]
- Zhou X, Cao L, Jiang C, Xie Y, Cheng X, Krausz KW, Qi Y, Sun L, Shah YM, Gonzalez FJ. (2014) PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat Commun 5:4573 DOI: 10.1038/ncomms5573. [DOI] [PMC free article] [PubMed] [Google Scholar]