

Abstract
Purpose
To evaluate a 15-year-old boy with MLIV (mucolipidosis type IV) and clinical abnormalities restricted to the eye who also had achlorhydria with elevated blood gastrin levels.
Methods
In addition to a detailed neuro-ophthalmic and electrophysiological assessment, his mutant mucolipin-1 was experimentally expressed in liposomes and its channel properties studied in vitro.
Results
The patient was a compound heterzygote for c.920delT and c.1615delG. Detailed neuro-ophthalmic examination including electroretinography showed him to have a typical retinal dystrophy predominantly affecting rod and bipolar cell function. In vitro expression of MCOLN1 in liposomes showed that the c.1615delG mutated channel had significantly reduced conductance compared with wild-type mucolipin-1, whereas the inhibitory effect of low pH and amiloride remained intact.
Conclusions
These findings suggest that reduced channel conductance is relatively well tolerated by the brain during development, whereas retinal cells and stomach parietal cells require normal protein function. MLIV should be considered in patients with retinal dystrophy of unknown cause and screened for using blood gastrin levels.
Retinal degeneration is a prominent cause of visual impairment worldwide. In the United States alone, there are presently more than 6 million people with retinal diseases. Studies have estimated the prevalence of hereditary retinal dystrophies at approximately 1:1490.1 Mucolipidosis type IV (MLIV) is an autosomal recessive disorder typically characterized by severe psychomotor delay evident by the end of the first year of life and slowly progressive visual impairment during the first decade as a result of a combination of corneal clouding and retinal degeneration.2–4 By the end of the first decade of life and always by the early teens, individuals with typical MLIV have severe visual impairment as a result of retinal degeneration.4 A handful of patients have been described with a mild form of MLIV, presenting with only the ocular form of the disease with essentially no neurologic involvement.5,6
MLIV is caused by mutations in MCOLN1, which codes for mucolipin-1, a 580-amino-acid protein that is a member of the transient receptor potential (TRP) family.7–9 Thus far, most of the patients have been Ashkenazi Jews with either a splice mutation c.406-2A>G, which prevents splicing of mucolipin-1 mRNA at exon 4, or g.511_6943del, which is a deletion mutation that eliminates 6434 bp of DNA, including the first five exons and part of exon 6 of MCOLN1.9 Mutations in the area of the presumed channel pore between the fifth and sixth transmembrane domain were also associated with a severe MLIV phenotype.3 However, non-Jewish patients were found to have missense mutations in the loop between the first and second transmembrane domain, in the lipase domain, or eliminating one of the four cysteines in the loop, possibly reducing the stability of mucolipin-1.3 Individuals with these mutations had a mild phenotype, an independent ataxic gait, and the ability to use their hands to feed themselves. A p.D362Y amino acid change was identified in the third transmembrane domain and was associated with a slower progression of the retinal disease and a relatively mild neurologic phenotype, although membrane preparations containing mucolipin-1 with this mutation had no channel activity.10 The mildest form of the disease was associated with p.Phe408del, and the protein construct containing this mutation was shown to function as a channel in liposome preparations and displayed only a deficient regulation.10 More recently, a patient with corneal and retinal dystrophy but no neurologic abnormalities was found to be a compound heterozygote for D362Y and A>T transition leading to the creation of a novel donor splicing site and a 4-bp deletion in exon 13.5
Here we describe the consequences of such a MCOLN1 mutation on known functional deficits identified in MLIV: stomach acid secretion, autofluorescence of cultured patient's fibroblasts, and abnormal mucolipin-1 ion-channel activity. Our findings raise the possibility that a fraction of the cases with isolated progressive retinal degeneration is caused by mutations in MCOLN1.
Methods
The Patient
The present research adhered to the tenets of the Declaration of Helsinki. The patient was examined in an NINDS Institutional Review Board approved clinical research protocol. The patient's parents gave their written informed consent.
This non-Hispanic, non-Ashkenazi Jewish Caucasian boy had normal early motor and cognitive development. Between ages 4 and 11 years he had increasing visual difficulty. His best-corrected visual acuity has always been moderately subnormal. This was considered to be due to amblyopia and was treated with monocular patching around age 6. At age 11, he had difficulties seeing in dim illumination and was found to have bilateral paracentral scotomas and corneal clouding along with a rod-cone retinal dystrophy, and MLIV was diagnosed at age 13. His visual symptoms included a moderate decline in central vision and a severe paracentral visual loss. He reported abnormal night vision and dark adaptation, but his day vision was essentially normal. He did not notice significant glare sensitivity, but had color confusion (e.g., blues with greens). The patient was examined at age 14 and again at age 15 years of age. There were no changes in his visual symptoms or in his cognitive status in the interval.
General examination was unremarkable with normal height, weight, and head circumference. His neurologic examination was normal as well with completely normal motor function, gait, and coordination. EEG and brain MRI examinations were normal.
On the Wechsler Intelligence Scale for Children-Fourth Edition (WISC-IV) he had a Verbal Comprehension Index of 142 and a Perceptual Reasoning Index of 102 in at age 15. His full-scale IQ of 114 was in the high average range of intellectual ability and exceeded that obtained by 82% of those of his age. He had deficient motor processing that led to the relatively low Perceptual Reasoning Index.
Laboratory Testing
Blood gastrin level was 576 pg/mL (normal, 0–99); the only abnormalities in the complete blood were hemoglobin of 12.5 g/dL (normal, 12.7–16.7) and mean corpuscular volume of 74.9 fL (normal, 79–98).
Electroretinogram (ERG)
A full-field (Ganzfeld) electroretinogram (ERG) was recorded after the protocol recommended by the International Society for Clinical Electrophysiology of Vision. Since all other tests of visual function and fundus features were compatible with a bilateral, symmetrical retinal disease, only responses elicited by stimulation of the right eye were recorded.
Mutation Analysis
Mutations were identified by sequencing the cDNA and genomic DNA of the patient, as described.3 Confirmation was done by sequencing genomic DNA from both parents.
Ion Channel Activities
The mutant (Δ1740G) mucolipin-1 plasmid was generated from mucolipin-1-WT-pSV-Sport1 plasmid with a site-directed mutagenesis kit (QuickChange; Stratagene, La Jolla, CA).
In Vitro Translation of WT and Mutant Mucolipin-1 Proteins
The wild-type and mutant mucolipin-1 proteins were translated in vitro with the TNT T7 Coupled Reticulocyte Lysate System (Promega, Madison, WI). Briefly, 1 μg WT and mutant MUCOLIPIN-1 plasmids were mixed with rabbit reticulocyte lysate, T7 RNA polymerase, amino acid mixture in a tube according to the protocol of the kit, followed by incubation for 90 minutes at 30°C. As a control, the plasmids were omitted in some translation processes. The translated proteins were then stored at −70°C for Western blot detection and channel reconstitution.
Detailed Method for Western Blot Analysis of In Vitro Translated Mucolipin-1
Thirty microliters in vitro translated mucolipin-1 WT and Δ1740G proteins were separated onto SDS gel (NuPage; Invitrogen, Carlsbad, CA) and transferred to PVDF membrane. The nonspecific antigens were blocked with 3% skim milk and then incubated for 1 hour with 1:100 rabbit-anti-human mucolipin-1 antibody (amino acids 106-180 mapping within an internal region of mucolipin-1 of human origin; Santa Cruz Biotechnology, Santa Cruz, CA), and another 1 hour with 1:10,000 goat-anti-rabbit HRP secondary antibodies (Santa Cruz Biotechnology). The blot was detected with chemiluminescence (Super-Signal West Pico Chemiluminescent Substrate; Pierce, Rockford, IL).
Ion Channel Reconstitution in Lipid Bilayers
A lipid mixture in n-decane (∼20 –25 mg/mL) was made with a content of 70% 1-palmitoyl-2-oleoyl phosphatidylcholine and 30% 1-palmitoyl-2-oleoyl phosphatidylethanolamine. The in vitro-translated mucolipin-1-WT and mutant proteins were mixed with the lipid solution in 1:1 ratio and then sonicated briefly to form liposomes. The liposomes were painted with a glass rod to the aperture (150 μM) of a polystyrene cuvette (CP13-150) that fits into a lipid bilayer chamber (model BCH-13; Warner Instruments Corp., Hamden, CT). The cis side of the lipid bilayer was bathed in 150 mM K aspartate, 10 mM HEPES (pH 7.4), while the trans side of the bilayer was bathed in 15 mM K aspartate, 10 mM HEPES (pH7.4). The electrical signal of single channel activities were recorded with a PC501A amplifier (Warner Instruments Corp.) and then digitized (1440A Digidata; Molecular Devices, Sunnyvale, CA) at 2 kHz. The data were acquired and analyzed (pClamp 10 software; Molecular Devices). For the display purpose, the single channel activities were further filtered at 200 Hz.
Results
Ophthalmic Examination
The patient was seen for two examinations separated by 1 year.
Visual Function
Visual acuity was measured with ETDRS distance charts (Lighthouse, New York, NY). At age 14 years, the results were as follows: Snellen equivalent 20/63 OD (score: 60 letters) and 20/32 OS (score: 76 letters). The correction used for this measurement was +0.50 +3.25 cylinder axis 118° OD and pl +2.00 cyl axis 060° OS.
A retest at age 15 yielded the following values: Snellen equivalent 20/32 OD (score: 75 letters) and 20/25 OS (score: 82 letters). The correction used for this measurement was pl +2.75 cyl axis 130° OD; pl +1.75 cyl axis 060° OS.
The external eye examination was within normal limits. Ocular motility was normal; ductions were full, saccadic eye movements had normal velocity, and smooth pursuit movements were present. Given the presence of a paracentral visual field defect, pursuit movements became inaccurate if target velocity increased, as the target moved into the scotoma area. Binocular balance was orthophoric for distant and near targets. Pupils were equal, round, and reactive to light and accommodation stimuli.
Slit lamp examination revealed normal conjunctivae and sclerae. Corneal clouding was conspicuous; fine granular deposits were present in the corneal epithelium and the subepithelial stroma, giving the cornea a ground-glass appearance. In the inferior quadrant, the deposits adopted a whorl-like configuration. These deposits were less abundant in the corneal margin adjacent to the limbus. The anterior chambers were normal, with no cells or flare; the chamber angle was open but narrow. The irides, lenses and vitreous body were normal in structure. A trace amount of cells was seen in the vitreous body of the right eye, none were detected in the left eye, though corneal haze obscured fine details of vitreous structure. Intraocular pressure was normal.
Ophthalmoscopic examination showed abnormal pigmentation in the central retina (Fig. 1). The foveal area had moderate hyperpigmentation; no foveolar reflex was visible. In contrast, the parafovea had a depigmented background with large pigment granules. This gave the macular area a bull's-eye appearance. In the parafovea, background pigmentation was moderately reduced, with fine pigment granules. The pigmentation of the posterior peripheral retina resembled that of the parafovea; very scanty intraretinal pigment spicules were seen in OD (Fig. 1). In contrast, the pigmentation of the intermediate and anterior peripheral retina was normal. Retinal arterioles were mildly attenuated; retinal venules had normal diameter. Optic discs showed mild pallor and distinct margins; the nerve fiber layer had a normal appearance OU (Fig. 1). These fundus findings indicate the presence of a bilateral and symmetric retinal dystrophy that has compromised the central retina and adjacent retinal periphery. The fovea, the midperiphery, and the anterior periphery of the retina did not seem compromised by the dystrophic process.
Figure 1.

Montage of fundus color photography: (A) right eye; (B) left eye. Note bull's-eye maculopathy and depigmentation of the central retina (both eyes) and peripheral pigment spicules (right eye).
On the second visit, two additional imaging modalities were used. Autofluorescence imaging showed a large area of patchy hypofluorescence throughout the central retina. The extension of this area corresponded with the area of the ring scotoma identified with kinetic perimetry. The mechanism underlying hypofluorescence is likely to be a loss of retinal pigment epithelium fluorophores (primarily lipofuscin) (Fig. 2). Optical coherence tomography imaging showed that retinal thickness was normal in the foveal area, where an inner region with decreased signal strength, possibly due to increased intraretinal fluid. In the parafovea, retinal thickness was mildly reduced, and retinal layers were not very conspicuous. This last finding may be due to the effect of corneal haze on optical imaging.
Figure 2.

Montage of fundus autofluorescence photography: (A) right eye; (B) left eye. Note darker area of patchy hypofluorescence throughout the central retina in both eyes, sparing the fovea.
Visual fields were assessed with Goldmann kinetic perimetry (Fig. 3). In both eyes, isopters measured with large bright stimuli (V 4e, III 4e) were normal (horizontal diameter > 140° OU). In contrast, the isopters measured with a small bright stimulus (I 4e) were moderately constricted (horizontal diameter: 105° OD and 95° OS, normal, >130°). A large, absolute ring scotoma, (horizontal diameter with the III 4e stimulus: 105° OD and 95° OS) was detected. A very small circular central island of vision (5° in diameter with the III 4e stimulus) was preserved in both eyes. A modest progression of this defect was observed between age 14 and 15 years (Fig. 3). Color vision was measured with the Farnsworth D-15 panel. This test detected a marked blue-yellow color vision deficiency, with color confusion errors along the tritan axis (Fig. 4). There was a modest increase in the number of errors between the first (4 errors OD, 3 OS) and the second examination (5 errors OD, 6 OS). This fluctuation may be within expected test-retest variability.
Figure 3.

Visual fields (Goldmann kinetic perimetry). (A) left eye; (B) right eye. Isopters plotted with V4e and I4e stimuli are depicted. Note large bilateral absolute ring scotoma (V4e stimulus).
Figure 4.

Color vision (Farnsworth D-15 panel). In both eyes, multiple color confusion errors are oriented along the tritan axis, indicating a blue-yellow color vision deficiency.
His absolute central luminance threshold after 30 minutes of dark adaptation was measured with a Goldmann-Weekers adaptometer. This was −39 log cd/m2 OD and −3.8 log cd/m2 OS at 14 years of age. The results obtained during his second visit were essentially unchanged. These results indicate that he had a moderate threshold elevation OU (normal, range: − 5.2 to −4.3 log cd/m2).
Electroretinogram
The standard ERG responses are shown in Table 1. These ERG abnormalities indicate that the visual field defect was due to abnormal retinal function. The reduction in a-wave amplitude observed in the mixed (combined rod- and cone-mediated response) implies that photoreceptor function was compromised. The b-wave/a-wave amplitude ratio was reduced (1.16; normal, >1.85; Fig. 5).
Table 1. Standard ERG Responses.
| Amplitude (μV) | Implicit Time (ms) | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| 2006 | 2007 | Normal | 2006 | 2007 | Normal | |
| Dark-adapted scotopic ERG | ||||||
| Rod response | 77 | 43 | >167 | 126 | 129 | <120 |
| Mixed a-wave | 112 | 109 | >188 | 26 | 28 | <23.5 |
| Mixed b-wave | 130 | 126 | >373 | 50 | 52 | <59.5 |
| Oscillatory potential | 49 | 51 | >63 | |||
| Light-adapted photopic ERG | ||||||
| Cone response | 91 | 93 | >101 | 34 | 35 | <34.0 |
| Flicker response | 108 | 117 | >66 | 34 | 34 | <31.5 |
Figure 5.

Full-field (Ganzfeld) electroretinogram (ERG). Left: patient; right: age-matched control subject. Two superimposed responses are shown for each recording condition. Scotopic ERG: (A) rod response; (B) combined (rod and cone) response. Photopic ERG: (C) cone response; (D) flicker response. Refer to interpretation in text.
Cortical visual evoked potentials (VEP) elicited by a full-field (Ganzfeld) stimulus were recorded according to after the protocol recommended by the International Society for Clinical Electrophysiology of Vision. The main positive peak (P2) recorded at Oz showed normal latency-to-peak (OD, 88 ms; OS, 92 ms) and borderline amplitude (OD, 8.1 μV, OS, 9.6 μV) at age 14. Results obtained 1 year later did not show clinically significant differences (latency-to-peak: OD, 95 ms; OS, 90 ms; amplitude OD, 6.8 μV; OS, 7.1 μV).
MCOLN1 Mutations in the Patient
MCOLN1 was sequenced, and two mutations were identified c.920delT and c.1615delG. The first mutation was identified in the father's DNA and the second in the mother's DNA. The cDNA from the patient did not contain the first mutation, suggesting that the mRNA with this mutation was not spliced properly and was degraded, possibly by a nonsense-mediated decay process. The RNA with the second mutation appeared homozygous, and since there is a stop codon in the second frame, it encoded a predicted protein with an altered C-terminal as follows: 539 PTSHSARTAPPPASSAAGAARPAAFSAAAEG-TPRRSIRCW 578.
The mutant sequence resembles a section of the sequence of an avian glucose transporter but has no known function.
Autofluorescence of Fibroblasts
Autofluorescence of various degrees was noticed in all skin fibroblasts derived from all MLIV patients so far.11 It was important to know whether the minimal functional deficit in this patient would eliminate this phenotype. Fibroblasts from the patient were cultured and fused with control and MLIV fibroblasts to detect complementation of the phenotype. The current patient's fibroblasts were as fluorescent as the cells from the most severe cases, although there was a greater cell-to-cell variability (Fig. 6). There was complementation of the phenotype when the cells were fused with control cells, but not in the fusion product with MLIV fibroblasts from a different patient (data not shown). This result indicates that autofluorescence is a sensitive indicator for even a minor dysfunction of mucolipin-1.
Figure 6.
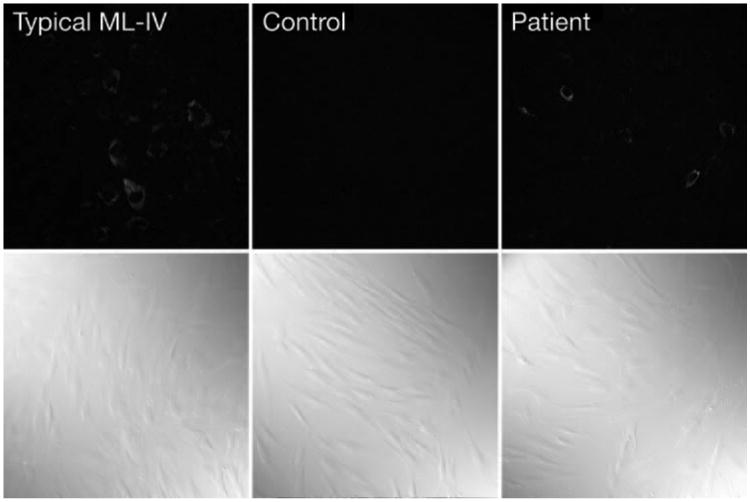
Autofluorescence of live cultured skin fibroblasts from normal and MLIV patients. Autofluorescence was viewed under UV with a 488-nm filter. The bottom panels show the cells under phase microscope. Magnification, ×20.
Ion Channel Function of Mutated Mucolipin-1
Western blot showed the presence of wild-type, mutated mu-colipin-1, and wild-type-flagged forms of the expressed channel (data not shown). There was no difference in the percentage of channel activity between the mutated and the wild-type channel (Table 2). Mutant mucolipin-1 required higher voltage to open compared to wild-type (Figs. 7A, 7B), and allowed lower current across a wide range of cross membrane electrical potentials (Figs. 7C, 8). On the other hand, the activity of the mutated mucolipin-1 channel decreased significantly when the pH was lowered from 6.4 to 5.0 (Fig. 9) and in the presence of amiloride (Fig. 10).
Table 2. Channel Activities of WT and Mutant MUCOLIPIN-1 Protein.
| Sample | n | Percentage (%) |
|---|---|---|
| Control | 0/20 | 0 |
| MUCOLIPIN-1-WT | 14/65 | 21.5 |
| MUCOLIPIN-1-Mutant | 14/72 | 19.4 |
Percentage of channel activity found in the various in vitro– translated tissues. Channel activity was assessed as the average current density for the first 12 seconds after testing the membrane at a given holding potential.
Figure 7.

Single channel currents of wild-type and mutant mucolipin-1 proteins. Representative single channel for wild-type (A) and D1740G mutant mucolipin-1 (B). Tracings were obtained by reconstituting the in vitro-translated protein, in a lipid bilayer exposed to a KCl chemical gradient. Holding potentials are indicated above the tracings. (C) Current-to-voltage relationships for wild-type and D1740G mucolipin-1. Values are the average single-channel currents of seven experiments for the wild-type, and six for the mutant mucolipin-1.
Figure 8.

Mean currents of wild-type and mutant mucolipin-1 proteins. Mean currents of wild-type and D1740G mutant mucolipin-1 are shown at holding potentials indicated in the abscissa. Data were obtained by average 15 seconds of tracing starting at the change in holding potential, for several of the tracings indicated on each bar.
Figure 9.

Effect of lowering pH on mutant mucolipin-1 channel function. Mutant D1749G mucolipin-1 was reconstituted in the presence of a KCl chemical gradient. Addition of HCl to the cis chamber, to shift pH from 7.4 to 5.0, completely blocked mucolipin-1 channel function. Data are representative of three experiments.
Figure 10.

Effect of amiloride on mutant mucolipin-1 channel function. Mutant D1749G mucolipin-1 was reconstituted in the presence of a KCl chemical gradient. Addition of amiloride (1 mM) to the trans chamber induced a complete inhibition of mucolipin-1 channel function. Data are representative of two experiments.
Discussion
MLIV is a rather unique disease that combines a developmental brain disorder that usually causes a static encephalopathy with a degenerative retinal process that leads to a progressive visual impairment and acquired blindness.3 We described a 15-years-old patient with MLIV and normal neurodevelopmental and cognitive function as well as a normal brain MRI who had only corneal retinal disease and achlorhydria. The only indication of a developmental brain abnormality was the relative weakness of his motor-related performance on cognitive testing compared with to his superior perceptive and verbal abilities.
The excellent cooperation from the patient allowed us to perform, for the first time in an MLIV patient, a very comprehensive physiological examination. The examination confirmed that the MLIV retinopathy has all the characteristics of a typical retinal dystrophy including a central ring scotoma and blue-yellow color blindness. There was a small but definite deterioration in his visual function over 1 year of follow-up. The patient's electroretinogram demonstrated a photoreceptor abnormality. The rod-mediated components show a marked reduction in amplitude, whereas cone-mediated responses were only modestly reduced; this implies that rod function is considerably more affected than cone function. The reduced b-wave/a-wave amplitude ratio suggests that depolarizing bipolar cells (or synaptic transmission from photoreceptors to depolarizing bipolar cells) were also compromised. The rod response showed reduction in amplitude with respect to the baseline value. In contrast, no significant differences were seen in all other responses. His VEP results imply that the conduction velocity of retinocortical pathways was normal. The borderline amplitude value was probably due to the missing contribution to the VEP from the retinal ganglion cells that receive input from the retinal area corresponding to the ring scotoma.
The patient was indistinguishable from other MLIV patients in other aspects of the disease such as autofluorescence of cultured fibroblasts and achlorhydria (which causes elevated gastrin levels).11,12 These deficits, which are specific to MLIV, indicate that the mutation in MCOLN1 is indeed the cause for his eye disease and not an unrelated defect.
We have demonstrated that wild-type mucolipin-1 is a cation channel that is regulated by pH.10 The pH regulation was missing in patients with V446L and ΔF408, although the channel function remained. The only abnormality in mutation of the present patient was a reduction in conductance while the inhibitory effect of low pH and amiloride remained intact. This finding suggests that normal channel conductance of mucolipin-1 is essential for the maintenance of retinal neurons, whereas it is less important for normal brain development. The change in pH is thought to modify the multimeric aggregation of mucolipin-1 ion channels.10 Preservation of pH sensitivity may be less important for brain development. The molecular phenotype likely also resulted from the elimination of important regulatory phosphorylation sites in the C-terminal end of our patient's mutated mucolipin-1.13
Although the precise role of mucolipin-1 is not completely understood, it was suggested to function as a lysosomal Ca2+ channel,14 an outwardly rectifying monovalent cation channel regulated by either Ca2 +15 or pH.10 More recent work suggested that mucolipin-1 limits lysosomal acidification by providing a lysosomal H+ leak pathway.16 MLIV lysosomes were found to be overacidified, a finding that explains hypersensitivity to chloroquine of cultured skin fibroblasts from MLIV patients.11 It is likely that abnormal membrane trafficking in MLIV is secondary to accumulation of undigested material in lysosomes.17 Another study suggests that mucolipin-1 is involved in calcium release from lysosomes in rat liver.18 Cleavage of mucolipin-1 may be another mechanism to limit its activity to a selective subset of organelles in the lysosomal degradation pathway.16 Loss-of-function mutations of CUP-5 the MCOLN1 homologue in the C. elegans lead to endocytic defects, the formation of large lysosomal vacuoles, and increased apoptosis. Interestingly, mutation of the ABC transporter MRP-4 rescues the lysosomal degradation defect and the corresponding lethality in this model.19 These insights do not yet explain the universal achlorhydria seen in MLIV.12
In summary, different channel properties may be necessary for different functions of mucolipin-1 in different cell types. Reduction in channel conductance seems to be well tolerated by the brain during development, whereas retinal cells and parietal cells require intact protein function to maintain their viability and normal function. This study makes the case for screening patients with idiopathic retinal disease for defective mucolipin-1. This task can be easily achieved by determining serum gastrin levels.
Acknowledgments
The authors thank the Electrophysiology Core at the Massachusetts General Hospital for performing electrophysiological analysis of the mutant channel and the patient for his dedication and help.
Supported by the Intramural Program of the National Institutes of Health.
Footnotes
Disclosure: E. Goldin, None; R.C. Caruso, None; W. Benko, None; C.R. Kaneski, None; S. Stahl, None; R. Schiffmann, None
References
- 1.Puech B, Kostrubiec B, Hache JC, Francois P. Epidemiology and prevalence of hereditary retinal dystrophies in the Northern France (in French) J Fr Ophtalmol. 1991;14:153–164. [PubMed] [Google Scholar]
- 2.Berman ER, Livni N, Shapira E, Merin S, Levij IS. Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. J Pediatr. 1974;84:519–526. doi: 10.1016/s0022-3476(74)80671-2. [DOI] [PubMed] [Google Scholar]
- 3.Altarescu G, Sun M, Moore DF, et al. The neurogenetics of mucolipidosis type IV. Neurology. 2002;59:306–313. doi: 10.1212/wnl.59.3.306. [DOI] [PubMed] [Google Scholar]
- 4.Smith JA, Chan CC, Goldin E, Schiffmann R. Noninvasive diagnosis and ophthalmic features of mucolipidosis type IV. Ophthalmology. 2002;109:588–594. doi: 10.1016/s0161-6420(01)00968-x. [DOI] [PubMed] [Google Scholar]
- 5.Dobrovolny R, Liskova P, Ledvinova J, et al. Mucolipidosis IV: report of a case with ocular restricted phenotype caused by leaky splice mutation. Am J Ophthalmol. 2007;143:663–671. doi: 10.1016/j.ajo.2006.11.049. [DOI] [PubMed] [Google Scholar]
- 6.Casteels I, Taylor DS, Lake BD, Spalton DJ, Bach G. Mucolipidosis type IV. Presentation of a mild variant. Ophthalmic Paediatr Genet. 1992;13:205–210. doi: 10.3109/13816819209105168. [DOI] [PubMed] [Google Scholar]
- 7.Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet. 2000;67:1110–1120. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bargal R, Avidan N, Ben-Asher E, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26:118–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 9.Sun M, Goldin E, Stahl S, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- 10.Raychowdhury MK, Gonzalez-Perrett S, Montalbetti N, et al. Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Hum Mol Genet. 2004;13:617–627. doi: 10.1093/hmg/ddh067. [DOI] [PubMed] [Google Scholar]
- 11.Goldin E, Cooney A, Kaneski CR, Brady RO, Schiffmann R. Mucolipidosis IV consists of one complementation group. Proc Natl Acad Sci USA. 1999;96:8562–8566. doi: 10.1073/pnas.96.15.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiffmann R, Dwyer NK, Lubensky IA, et al. Constitutive achlorhydria in mucolipidosis type IV. Proc Natl Acad Sci USA. 1998;95:1207–1212. doi: 10.1073/pnas.95.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vergarajauregui S, Oberdick R, Kiselyov K, Puertollano R. Mucolipin-1 channel activity is regulated by protein kinase A mediated phosphorylation. Biochem J. 2008;410:417–427. doi: 10.1042/BJ20070713. [DOI] [PubMed] [Google Scholar]
- 14.LaPlante JM, Ye CP, Quinn SJ, et al. Functional links between mucolipin-1 and Ca2+-dependent membrane trafficking in mucolipidosis IV. Biochem Biophys Res Commun. 2004;322:1384–1391. doi: 10.1016/j.bbrc.2004.08.045. [DOI] [PubMed] [Google Scholar]
- 15.Cantiello HF, Montalbetti N, Goldmann WH, et al. Cation channel activity of mucolipin-1: the effect of calcium. Pflugers Arch. 2005;451:304–312. doi: 10.1007/s00424-005-1448-9. [DOI] [PubMed] [Google Scholar]
- 16.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, et al. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–7301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 17.Kiselyov K, Soyombo A, Muallem S. TRPpathies. J Physiol. 2007;578:641–653. doi: 10.1113/jphysiol.2006.119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, Li PL. Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J Biol Chem. 2007;282:25259–25269. doi: 10.1074/jbc.M701614200. [DOI] [PubMed] [Google Scholar]
- 19.Schaheen L, Patton G, Fares H. Suppression of the cup-5 mucolipidosis type IV-related lysosomal dysfunction by the inactivation of an ABC transporter in C. elegans. Development. 2006;133:3939–3948. doi: 10.1242/dev.02575. [DOI] [PubMed] [Google Scholar]