

Abstract
Dengue disease is a global disease that has no effective treatment. The dengue virus (DENV) NS4B is a target for designing specific antivirals due to its importance in viral replication. Medicinal plants have been a savior for dengue virus as they consist of a class of phytochemicals having anti-viral activity and can pose a new approach ofstrong drug against viruses. The present study analyzes the activity of compounds against NS4B of DENV (1-4) serotypes. In this study Catechin, Cianidanol, Epicatechin, Eupatoretin, Glabranin, Laurifolin, DL-Catechin, astherapeutic agents were filtered by using Lipinski rule’s five and the drug-likeness property of these agents were used for assessment of pharmacological properties. The molecular docking results presented the 2-D structures of bioactive complex, which interacted with especially conserved residues of target domains. Interestingly, we find the Catechin, Laurifolin, Cianidanol have highest binding energy against NS4B in DENV-1,2,4 which is evident by the formation of more hydrogen bonds with the amino acid residues at the binding site of the receptor. Our results revealed that the bioactive compound, especially Catechin has significant anti-dengue activities. In addition, this study may be helpful in further experimental investigations.
Keywords: Dengue Virus, bioactive compounds, molecular docking, drug-likeness score, Lipinski Rule’s
Background
Dengue is the fastest spreading viral disease that is transmitted by Aedes aegypti mosquito worldwide [1,2]. In tropical and subtropical countries, dengue is a major public health problem due to multiple factors, such as reemergence and intense transmission, the behavior of epidemic cycles every two or three years, the increase in the frequency of severe dengue outbreaks and the simultaneous circulation of different serotypes. Approximately 50,000 people suffer from dengue annually, and relatively 10% of the total cases involved dengue hemorrhagic fever [2,3]. Despite its global spread, currently there is no drug that is available for the disease.
Dengue is a virus that belongs to the genus Flavivirus, which has five known serotypes [4]. Dengue virus (DENV) co-circulates as a complex of five closely related but antigenically distinct serotypes (DENV-1 and DENV-5) [5], all of which are etiologic agents of dengue fever, life-threatening dengue hemorrhagic fever (DHF), and dengue shock syndrome (DSS) [6]. DENV-2 and DENV-3 have historically been the most prevalent agents that have caused severe disease [6]. This disease is characterized by high fever, headache, joint pain, fatigue, swollen lymph nodes, skin rashes, and in some cases, bleeding and shock (DHF), which can cause death [2,7]. The dengue virus genome consists of a singlestranded, positive sense RNA of approximately 11 kb that is organized as 5'NCR-C-prM-E-NS1-NS2A-NS2B-NS3-NS4ANS4B- NS5-3'NCR, where NCR is the noncoding region, C is the capsid, prM is the premembrane [8], and E is the envelope, while NS corresponds to nonstructural proteins. Among these proteins, envelope glycoprotein, NS3 protease, NS3 helicase [8,9], NS5 methyltransferase and NS5 RNA-dependent RNA polymerase have been suggested to be therapeutic targets for specific antiviral searches against dengue [10].
Currently, studies to find antiviral molecules have focused on targeting essential viral enzymes in the infection process by the direct or indirect inhibition of their biological functions or by blocking the viral replication machinery [11]. One of the most used targets that is employed for this purpose is NS4B which plays a central role in the viral life cycle; it acts as an attractive therapeutic target for antiviral compounds, essential for viral replication and infectivity [12]. Previous studies have provided considerable evidence for the importance of NS4B as potential drug target [13]. NS4B is highly hydrophobic membrane protein, which appears to have two hydrophobic segments (residues 1 to 56 and residues 56–93), which are probably associated with the ER lumen side of the membrane, and three C-terminal TM segments [10,34] (residues 93 to 146, residues 146 to 190 and residues 190 to 248) [14]. NS4B is capable of interfering with phosphorylation of STAT1 blocking the IFN-α/β induced signal transduction cascade [15]. NS4B is also a negative modulator of the NS3 helicase function, being this modulation dependent on the conformation of NS4B [16] it might function cooperatively in viral replication and the anti-host response [17].
Therefore, creating an alarming situation to develop an effective anti-dengue compounds to combat this epidemic infection [18, 19]. The use of bioinformatics tools, molecular modeling programs and high performance computing has been leading the process of designing and in silico searching for therapeutically useful molecules [20]. This same approach has been applied to the discovery or development of compounds that could target several diseases. One of the strategies at the forefront of drug discovery is the virtual screening of databases [21], which can identify powerful new protein ligands [22,23]. Using the structural information on the dengue virus protease and molecules published in PubChem (http://pubchem.ncbi.nlm. nih.gov) [22,23], virtual screening was performed to search for low molecular weight molecules that could inhibit this protease and that could be potentially employed in the treatment of dengue infection [24,25]. The bioactive compounds are a different group of metabolite derivative familiar for having a range of human health promoting actions, large group of naturally occurring polyphenol compounds constitute the most abundant class of dietary natural products and are present in fruits, different part of the herbal plants and various dietary supplements [26]. Several researches based on bioactive compound gives the details about their valuable biological activities, such as anti-allergenic, anti-cancer, anti-inflammatory, anti-oxidant, anti-viral and vasodilating properties [26,27]. However, in silico approach of the bioactive compounds on NS4B protein for dengue has not been characterized. The current study is to predict the In silico evaluation of NS4B protein inhibitory activity [28] and also in this study the stereochemistry binding of the bioactive compounds on NS4B protein in dengue has been carried out, which may be helpful in the development of potent NS4B protein inhibitors [28,29].
Catechin is a type of natural phenol and antioxidant, present in green tea in excessive quantity [29,30]. It is a plant secondary metabolite. It belongs to the group of flavan-3-ols (or simply flavanols), part of the chemical family of flavonoids [31]. The use of green tea as a cancer chemopreventive or for other health benefits has been confounded by the low oral bioavailability of its active polyphenolic catechins, molecular formula C15H14O6. Epicatechin, molecular formula C15H14O6 and Catechin are epimers, with (-)-epicatechin and (+)-catechin being the most common optical isomers found in nature, such as its presence in Chocolate.Mixtures of the different enantiomers can be also be known as (+/-)-catechin or DL-catechin and (+/-)-epicatechin or DL-epicatechin. Cianidanol, molecular formula C15H14O6 is an antioxidant flavonoid, occurs especially in woody plants inboth (+)-catechin and (-)-epicatechin (cis) forms. Eupatoretin is a flavonoid and its molecular formula is C19H18O8, it has anticancer activity. Glabranin [32] is a flavonoid, molecular formula C20H20O4 and it has anti dengue activity [33]. Laurifolin is a flavonoid, molecular formula C20H20O6, having antimicrobial activity. The discovery of molecules that interfere with the antiviral activity of the DENV NS4B could become a potential alternative for the treatment and control of the disease [34,35].
Methodology
An investigation was carried out at laboratories, which have permitted the authors to work there to identify potential bioactive compounds using chemoinformatics and to study the binding action of compounds on molecular target and inhibitory effect on DENV 1-4.
Identification of Bioactive Compounds:
For the identification of active compounds, authors selected the available compounds from Pubchem server tools, Drug Bank and the following procedures were undertaken.
Sketch the structures of Compounds:
The structure of selected bioactive compounds was drawn with the help of Pubchem project which are freely available to use. 2-D and 3-D structures were generated by using the software and were further used for docking [16].
Using Open Babel Converting File Format:
Open Babel is free software which is used for converting the file formats.
Receptor 3D structure
The three dimensional structure of NS4B cofactor is available in the protein data bank (PDB) in the authors previous study. The three dimensional structure of NS4B of DENV 1-4 was retrieved from the protein data bank. All water molecules were removed and on the final stage hydrogen atoms were added to the target protein molecule [13]. Collections of active compounds were selected. The 2D structure of seven ligands were retrieved from the NCBI PubChem database, in addition to it all the ligand molecules were converted into 3D structure using the 3D converter module program and then energy minimized. Figure 1 shows the 3D structure of the bioactive compounds [14].
Figure 1.
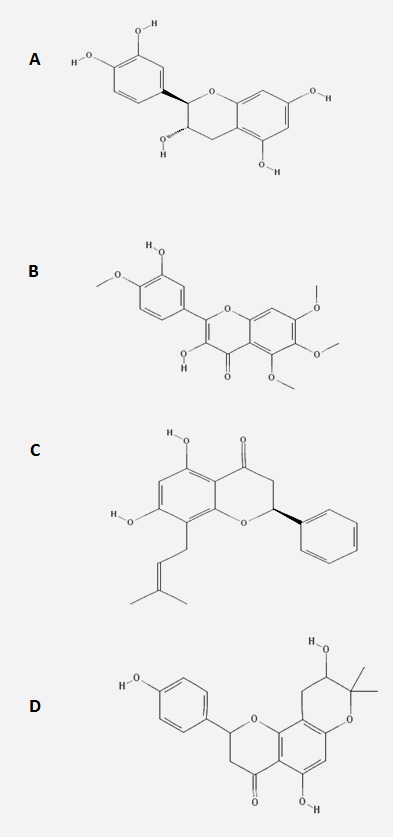
3D structure of bioactive compounds (A) Catechin, (B) Eupatoretin, (C) Glabranin and (D) Laurifolin
Molecular properties Prediction
Molecular Formula, Molecular Weight, Isoelectric point, Log P, H-bond acceptor sites, H-bond donor sites, Atom count, Bond count, Polar surface area, Vander Waals surface area, Polarizability, Lipinski’s Rule were calculated by using Marvin Sketch 5.11.4 software to predict the molecular properties of bioactive compounds [15].
Drug-likeness Prediction
To predict the drug-likeness value molsoft web tool is used (http://www.molsoft.com/mprop/). The graphical image and value shows that the drug is either non-toxic or toxic [2].
Molecular docking
To determine the binding energy of active compounds characterized by a molecular docking calculation was done in Docking Web Server. The docking calculations were carried out on protein model [16].
Results and discussion
This section is divided into three parts where first the results of molecular docking are revealed followed by Lipinski’s Rule analysis and Drug-Likeness Prediction in later section.
Molecular docking
The docking poses were obtained according to their docking parameters and their corresponding binding pockets. This evaluation of the bioactive compounds was based upon their binding parameters with the target enzyme. The docked pose of NS4B with the ligands Catechin, Cianidanol, Epicatechin, Eupatoretin, Glabranin, Laurifolin, DL-Catechin clearly demonstrated the binding positions of the ligand with the target. The potential binding sites of Catechin was found to be (-5.25 kcal/mol) in DENV-4 and Cianidanol was found (-5.20 kcal/mol) in DENV-4. The potential binding sites of Epicatechin was found to be (-3.49 kcal/mol) in DENV-1. The binding sites of Eupatoretin was found to be (-3.31 kcal/mol) and that of Glabranin was (-5.87 kcal/mol) in DENV-2. The binding positions of Laurifolin was found to be (-5.38 kcal/mol) and DLCatechin was (-5.73 kcal/mol) in DENV-4. The 2D docking image as revealed in Figure 2 shows the interaction between proteinligand.
Figure 2.
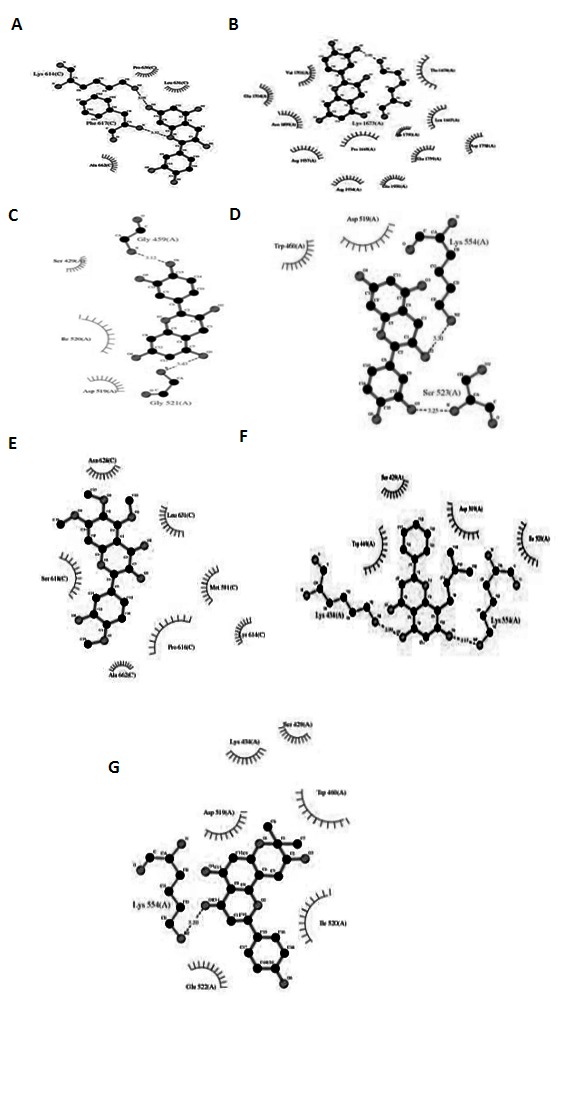
2D docking image show the interaction between protein-ligand (A) (-)-Catechin- DENV-1, (B) Cianidanol-DENV- 4, (C) DL-Catechin-DENV-2, (D) Epicatechin-DENV-2, (E) Eupatoretin-DENV-1, (F) Glabranin-DENV and (G) Laurifolin- DENV-2.
These results show that the effective binding sites are present in the selected bioactive compounds and the ability of inhibiting NS4B by the selected ligands.To have a better understanding of the estimation of free energy of binding, estimation of inhibition constant, eelectrostatic energy, total internal energy are tabulated in Table 1,Table 2, Table 3,Table 4. Evaluation of docking results depends upon the number of hydrogen bonds and interactions formed between the ligand and the protein as shown in Table 5.
Table 1. Molecular docking result of DENV-1.
| Protein-Ligand DENV 1 | Estimation of free energy of binding | Estimation of inhibition constant Ki (mM) | Electrostatic energy | Total Internal energy |
| (-)-Catechin | -3.09 | 5.43 mM | -0.31 | -4.14 |
| Cianidanol | -3.57 | 2.42 mM | -0.12 | -4.66 |
| Epicatechin | -3.49 | 2.76 mM | -0.08 | -4.31 |
| Eupatoretin | -2.17 | 25.77 mM | 0.08 | -3.72 |
| Glabranin | -4.38 | 612.92 microM | -0.11 | -4.53 |
| Laurifolin | -4.67 | 377.63 microM | -0.11 | -5.22 |
| DL-Catechin | -3.45 | 2.95 mM | -0.41 | -4.5 |
Table 2. Molecular docking result of DENV-2.
| Protein-Ligand DENV 2 | Estimation of free energy of binding | Estimation of inhibition constant Ki | Electrostatic energy | Total Internal energy |
| (-)-Catechin | -4.06 | 1.06 mM | -0.24 | -4.93 |
| Cianidanol | -4.04 | 1.10 mM | -0.19 | -5.01 |
| Epicatechin | -3.56 | 2.48 mM | -0.56 | -4.51 |
| Eupatoretin | -3.31 | 3.72 mM | 0.11 | -4.8 |
| Glabranin | -5.87 | 49.46 microM | -0.51 | -6.18 |
| Laurifolin | -5.38 | 113.31 mM | -0.35 | -5.89 |
| DL-Catechin | -4.16 | 887.49 microM | -0.27 | -5.09 |
Table 3. Molecular docking result of DENV-3.
| Protein-Ligand DENV 3 | Estimation of free energy of binding | Estimation of inhibition constant Ki | Electrostatic energy | Total Internal energy |
| (-)-Catechin | 312.63 | - | 311.14 | 0.21 |
| Cianidanol | 86.6 | - | -0.18 | 85.14 |
| Epicatechin | 7.81 | - | -0.25 | 6.41 |
| Eupatoretin | 517.21 | - | 0.23 | 503.75 |
| Glabranin | 306.64 | - | 0.01 | 302.12 |
| Laurifolin | 975.48 | - | 0.07 | 974.29 |
| DLCatechin | 269.31 | - | -0.11 | 267.51 |
Table 4. Molecular docking result of DENV-4.
| Protein-Ligand DENV 4 | Estimation of free energy of binding | Estimation of inhibition constant Ki | Electrostatic energy | Total Internal energy |
| (-)-Catechin | -5.25 | 141.69 uM | -0.51 | -6.45 |
| Cianidanol | -5.2 | 153.62 uM | -0.7 | -6.56 |
| Epicatechin | -5.32 | 125.01 uM | -0.72 | -6.66 |
| Eupatoretin | 1.75 | - | 0.61 | 0.14 |
| Glabranin | -3.49 | 2.75 mM | -0.22 | -4.46 |
| Laurifolin | -4.84 | 284.57 uM | -0.23 | 5.23 |
| DL-Catechin | -5.73 | 62.69 uM | -0.52 | -6.94 |
Table 5. Interaction table after molecular docking.
| Interaction | DENV1 | DENV2 | DENV3 | DENV4 |
| (-)-Catechin | 614:K,617:F, 631:L,636:P, 662:A | 429:S, 519:D,520:I, 521:G,522:E | 408:V, 410:K, 415:H,444:L, 451:W, 453:L,506:P, 514:V, 517:L,536:Q | 1667:L, 1669:P, 1673:K, 1674:T, 1701:V,1704:E, 1758:D,1759:E, 1790:A, 1890:N,1930:Q, 1934:R |
| Cianidanol | 581:M, 618:S,628:N | 429:S, 519:D,520:I, 522:E,554:K | 417:M, 443:D, 444:L,491:I, 520:N, 534:A,535:G | 1667:L, 669:P, 1673:K, 1674:T,1701:V,1704:E, 1758:D, 1759:E, 1790:A,1890:N, 1930:E, 1934:R, 1937:R |
| Epicatechin | 581:M,614:K,616:P,617:F,618:S, 631:L,659:V, 662:A | 460:W,519:D, 523:S,554:K | 404:V, 419:H, 500:P,501:G, 502:T, 503:S,518:Y,529:Y | 1667:L, 1669:P, 1673:K, 1674:T, 1701:V,1704:E, 1758:D, 1759:D, 1790:A, 1890:D,1930:Q, 1934:R |
| Eupatoretin | 581:M, 614:K,616:P,618:S,628:N,631:L,662:A | 477:T, 520:I,523:S,554:K,588:T,590:E | 417:L,418 :M, 443:D,444:L, 451:W, 453:L,514:V, 517:L, 520:N,534:A, 535:Q | 1669:P, 1673:K, 1674:T, 1675:K, 1701:V,1704:E, 1705;M, 1707:E, 1758;N, 1759:E,1890:N, 1937:R |
| Glabranin | 581:M, 614:K,616:P, 617:F,618:S, 628:N,631:L, 636:P | 429:S, 434:K,460:W,519:D, 520:I,554:K | 417:M, 442:K, 443:D,444:L, 451:W, 491:ILE,517:L, 520:N, 534:A,535:Q | 1667:L, 1669:P, 1673:K, 1674:T, 1675:K,1701:V, 1704:E, 1705:M, 1708:A,1758:D, 1759:E, 1890:N |
| Laurifolin | 581:M, 614:K,616:P, 617:F,618:S, 628:N,631:L, 636:P,662:A | 429:S, 434:K,460:W,519:D, 520:I,522:E, 554:K | 390:V, 408:V, 415:H,444:L, 451:W, 453:L,466:I, 506:P, 507:I,506:P, 507:I, 514:V,516:G, 517:L | 1673:K, 1674:T, 1675:K, 1704:E, 1708:A,1872:K, 1890:N, 1892:R, 1937:R, 1938:N |
| DL-Catechin | 581:M, 614:K,616:P, 617:F, 636:P | 429:S, 459:G,519:D, 520:I,521:G | 390:V, 410:K, 415:H,444;L, 451:W, 453:L,451:W, 453:L, 466:I,506:P, 514:V, 517:L,535:Q | 1667:L, 1669:P, 1673:K, 1674:T, 1701:V,1704:G, 1790:N, 1890:N, 1930:Q, 1934:R |
Lipinski’s rule analysis
Small molecules obtained from the plant material are always screened for the potential drug like activity predictions, therefore in this study all the compounds were screened for the drug likeness calculation and the molecular properties of these compounds are analysed on the basis of “Lipinski’s Rule of Five” using Marvin Sketch 5.11.4 software. The Marvin Sketch 5.11.4 Software predicts the molecular properties of the active compounds for different types of models and all these compounds have shown positive results as in Table 6. All the bioactive compounds have follow Lipinski’s Rule depending on the molecular properties of compounds.
Table 6. Molecular properties of bioactive compounds.
| Calculated Properties | (-)-Catechin | Cianidanol | Epicatechin | Eupatoretin | Glabranin | Laurifolin | DL-Catechin |
| Molecular Formula | C15H14O6 | C15H14O6 | C15H14O6 | C19H18O6 | C20H20O4 | C20H20O6 | C15H14O6 |
| Molecular Weight | 290.268 | 290.268 | 290.268 | 374.341 | 324.37 | 356.369 | 290.268 |
| Isoelectric point | 2.81 | 2.81 | 1.8 | 2.98 | 1.96 | 3.08 | 2.81 |
| Log P | 1.8 | 1.8 | 1.8 | 1.79 | 4.87 | 3.09 | 1.8 |
| H-bond acceptor sites | 6 | 6 | 6 | 9 | 5 | 7 | 6 |
| H-bond donor sites | 5 | 5 | 5 | 2 | 2 | 3 | 5 |
| Atom count | 35 | 35 | 35 | 45 | 44 | 46 | 35 |
| Bond count | 37 | 37 | 37 | 47 | 46 | 49 | 37 |
| Polar surface area | 110.38 | 110.38 | 110.38 | 103.68 | 66.76 | 96.22 | 110.38 |
| Vander Waals surface area | 370.62 | 244.18 | 244.13 | 317.81 | 297.88 | 312.43 | 244.18 |
| Polarizability | 28.42 | 28.42 | 28.42 | 36.66 | 35.61 | 36.39 | 28.42 |
| Lipinski’sRule | Suitable | Suitable | Suitable | Suitable | Suitable | Suitable | Suitable |
Drug-likeness prediction
Using molsoft web tool, we can predict the drug-likeness value. The drug-likeness score of Catechin is 0.92, Eupatoretin is 0.70, Glabranin is 1.08 and Laurifolin is 1.10. These score indicate that all the compounds are nontoxic and can be used as drugs (Figure 3).
Figure 3.
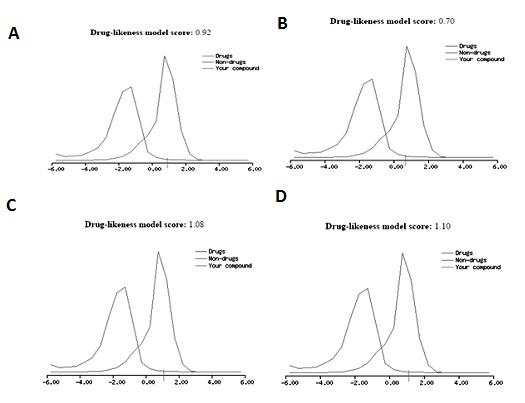
Drug-likeness score of (A) Catechin (B) Eupatoretin (C) Glabranin and (D) Laurifolin
The docking parameters such as hydrogen bond interactions, estimation of free energy of binding, estimation of inhibition constant, electrostatic energy, total internal energy and orientation of the docked ligand within the active site has been generated for the evaluation. From our detailed analysis, we found (-)-Catechin as best inhibitors as they show better ligandenzyme interactions and stability in DENV-1, 2, 4. In terms of hydrogen bond in DENV-3, the compounds did not show best binding affinity. Eupatoretin shows poor values in case of ligandreceptor atom pair interaction in DENV-2 (Figure 2).
Based on the in silico evaluation and stereochemistry binding of the active compounds, NS4B inhibitory activity of the selected compounds was found to be decreased in the order of Catechin, Cianidanol, Glabranin, Laurifolin, Epicatechin, Eupatoretin and DL-Catechin. Therefore on the basis of the results of above study, the selected active compounds possess potential NS4B inhibitory excellent binding sites. This may be attributed due to the differences in the position of the functional groups in the compounds (Figure 1). Prediction of drug-likeness score is very important as to tell whether the compounds are nontoxic or toxic. The scores show that all the active compounds can be used as drugs. In addition to it, Catechin can be the most suitable drug in dengue research antiviral therapy (Figure 3). Thus we can say that the results of this study clearly demonstrated by graphical representations that Catechin, Cianidanol, Epicatechin, Eupatoretin, Glabranin, Laurifolin, DL-Catechinare excellent binding sites and have interactions with NS4B in DENV-1, DENV-2, DENV-4 (Figure 4).
Figure 4.
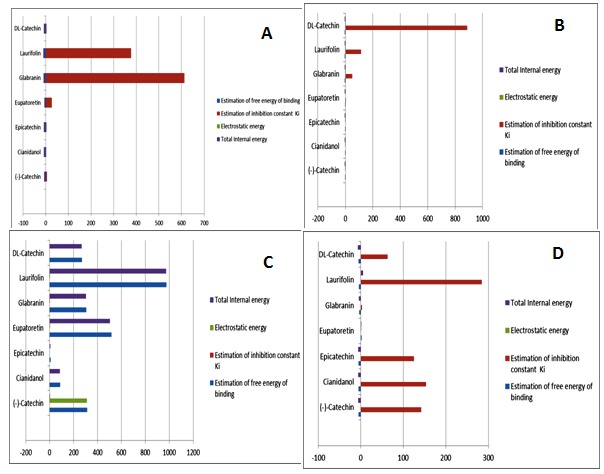
Graphical representation (Bar Diagram) of docking results (A) DENV-1; (B) DENV-2; (C) DENV-3 and (D) DENV-4
Conclusion:
This research is itself novel in nature as authors can say this based on their literature review Hence, suggesting that (-)- Catechin, Epicatechin, DL-Catechin are good candidates for the development of effective anti-dengue compounds in DENV-1, 2, 4. Whereas, the future directions and limitations of the study are concerned, we can say that further investigations on the above compounds and in vivo studies are necessary and can be done to develop potential chemical entities for the prevention and treatment of dengue. The limitation is that to develop an accurate computational model it requires a reliable and widespread experimental data. On the whole, we conclude that the Catechin can be further investigated and it can possibly be developed as an effective anti-dengue compound.
Acknowledgments
AP is grateful to Mrs. Jayashree Shaw for providing necessary guidance to carry out this work in her laboratory.
Edited by P Kangueane
Citation: Paul et al. Bioinformation 12(3): 140-148 (2016)
References
- 1.Cabarcas-Montalvo M, et al. Eur J Med Chem. 2016;110:87. doi: 10.1016/j.ejmech.2015.12.030. [DOI] [PubMed] [Google Scholar]
- 2.Manvar D, et al. Biochem Biophys Res Commun. 2016;469(3) :743. doi: 10.1016/j.bbrc.2015.12.042. [DOI] [PubMed] [Google Scholar]
- 3.Tambunan US, et al. Drug Target Insights. 2015;9:33. doi: 10.4137/DTI.S31566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allonso D, et al. J Virol. 2015;89(23) :11871. doi: 10.1128/JVI.01342-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu DW, et al. Acta Pharmacol Sin. 2015;36(9) :1126. doi: 10.1038/aps.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bastos Lima A, et al. Bioorg Med Chem. 2015;23(17) :5748. doi: 10.1016/j.bmc.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Lei Y, et al. PLoS One. 2015;24;10(7) :e0132122.. doi: 10.1371/journal.pone.0132122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vincetti P, et al. J Med Chem. 2015;58(12) :4964. doi: 10.1021/acs.jmedchem.5b00108. [DOI] [PubMed] [Google Scholar]
- 9.Aslam B, et al. Genet Mol Res. 2015;14(2) :4215. doi: 10.4238/2015.April.28.4. [DOI] [PubMed] [Google Scholar]
- 10.Vernekar SK, et al. J Med Chem. 2015;58(9) :4016. doi: 10.1021/acs.jmedchem.5b00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asnet Mary J, et al. J Biomol Struct Dyn. 2016;34(3) :676. doi: 10.1080/07391102.2015.1042914. [DOI] [PubMed] [Google Scholar]
- 12.Panya A, et al. Chem Biol Drug Des. 2015;86(5) :1093. doi: 10.1111/cbdd.12576. [DOI] [PubMed] [Google Scholar]
- 13.Jadav SS, et al. Bioorg Med Chem Lett. 2015;25(8) :1747. doi: 10.1016/j.bmcl.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 14.Wu H, et al. Bodem J. 2015;59(2):1100. [Google Scholar]
- 15.Viswanathan U, et al. J Chem Inf Model. 2014;54(10) :2816. doi: 10.1021/ci500531r. [DOI] [PubMed] [Google Scholar]
- 16.Qamar MT, et al. Bioinformation. 2014;10(7) :460. doi: 10.6026/97320630010460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Idrees S, Ashfaq UA. Asian Pac J Trop Med. 2014;7(7) :513. doi: 10.1016/S1995-7645(14)60085-7. [DOI] [PubMed] [Google Scholar]
- 18.Qamar TU, et al. Bioinformation. 2014;10(3) :115. doi: 10.6026/97320630010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panya A, et al. Chem Biol Drug Des. 2014;84(2) :148. doi: 10.1111/cbdd.12309. [DOI] [PubMed] [Google Scholar]
- 20.Parikesit AA, et al. Pak J Biol Sci. 2013;16(24) :1836. doi: 10.3923/pjbs.2013.1836.1848. [DOI] [PubMed] [Google Scholar]
- 21.Parida P, et al. Curr Pharm Biotechnol. 2013;14(11) :995. doi: 10.2174/1389201014666131226160025. [DOI] [PubMed] [Google Scholar]
- 22.Zhou GC, et al. Bioorg Med Chem Lett. 2013;23(24) :6549. doi: 10.1016/j.bmcl.2013.10.071. [DOI] [PubMed] [Google Scholar]
- 23.Saokaew N, et al. Lett Appl Microbiol. 2014;58(3) :270. doi: 10.1111/lam.12186. [DOI] [PubMed] [Google Scholar]
- 24.Angleró-Rodríguez YI, et al. Antiviral Res. 2013;100(2) :500. [Google Scholar]
- 25.de Almeida H, et al. PLoS One. 2013;8(8) :e72402.. doi: 10.1371/journal.pone.0072402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin MH, et al. J Biomol Struct Dyn. 2014;32(10) :1552. doi: 10.1080/07391102.2013.826599. [DOI] [PubMed] [Google Scholar]
- 27.Velmurugan D, et al. Protein Pept Lett. 2014;21(8) :815. doi: 10.2174/09298665113209990062. [DOI] [PubMed] [Google Scholar]
- 28.Ichiyama K, et al. PLoS Negl Trop Dis. 2013;7(4) :e2188.. doi: 10.1371/journal.pntd.0002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah M, et al. PLoS One. 2013;8(3) :e59211.. doi: 10.1371/journal.pone.0059211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, et al. Microbes Infect. 2013;15(4) :310. doi: 10.1016/j.micinf.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Austin SK, et al. PLoS Pathog. 2012;8(10) :e1002930. doi: 10.1371/journal.ppat.1002930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hidari KI, et al. Biochem Biophys Res Commun. 2012;424(3) :573. doi: 10.1016/j.bbrc.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Knehans T, et al. J Comput Aided Mol Des. 2011;25(3) :263. doi: 10.1007/s10822-011-9418-0. [DOI] [PubMed] [Google Scholar]
- 34.Kampmann T, et al. Antiviral Res. 2009;84(3) :234. doi: 10.1016/j.antiviral.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Malet H, et al. J Biol Chem. 2007;282(14) :10678. doi: 10.1074/jbc.M607273200. [DOI] [PubMed] [Google Scholar]