

Abstract
Ebola virus causes severe and often fatal hemorrhagic fevers in humans. The 2014 Ebola epidemic affected multiple countries. The virus matrix protein (VP40) plays a central role in virus assembly and budding. Since there is no FDA-approved vaccine or medicine against Ebola viral infection, discovering new compounds with different binding patterns against it is required. Therefore, we aim to identify small molecules that target the Arg 134 RNA binding and active site of VP40 protein. 1800 molecules were retrieved from PubChem compound database based on Structure Similarity and Conformers of pyrimidine-2, 4-dione. Molecular docking approach using Lamarckian Genetic Algorithm was carried out to find the potent inhibitors for VP40 based on calculated ligand-protein pairwise interaction energies. The grid maps representing the protein were calculated using auto grid and grid size was set to 60*60*60 points with grid spacing of 0.375 Ǻ. Ten independent docking runs were carried out for each ligand and results were clustered according to the 1.0 Ǻ RMSD criteria. The post-docking analysis showed that binding energies ranged from -8.87 to 0.6 Kcal/mol. We report 7 molecules, which showed promising ADMET results, LD-50, as well as H-bond interaction in the binding pocket. The small molecules discovered could act as potential inhibitors for VP40 and could interfere with virus assembly and budding process.
Keywords: Ebola VP40, molecular docking, virtual screening, anti-PV40 agents, proteomics, Lamarcking GA
Background
Ebola virus (EBOV) is a member of Filoviridae virus family, which causes acute hemorrhagic fever with a high case-fatality rate in humans (up to 90%). in a recent outbreak in 2014, it affected Guinea, Liberia, and Sierra Leone, and Nigeria with high case fatality rates [1].
EBOV is an enveloped, negative-sense RNA virus which encodes seven structural proteins within its 19kb linear genome including nucleoprotein (NP), VP35, VP40, VP30, VP24, polymerase (L) and membrane-anchored glycoprotein (GP) as well as the nonstructural secreted glycoprotein (sGP) and small secreted glycoprotein (ssGP) [2]. EBOV strains consist of four different species: Zaire ebolavirus (ZEBOV), Sudan ebolavirus (SEBOV), Côte d’Ivoire ebolavirus (CIEBOV) and Reston ebolavirus (REBOV). The newly discovered Bundibugyo ebolavirus (BEBOV) has been proposed as the fifth species.
VP40 is essential for Ebov viral assembly. Crystal structure, biochemistry and cellular microscopy postulated that VP40 rearranges into different structures, each with a distinct function required for the ebolavirus life cycle [3]. Figure 1 shows the 3D structure of the matrix protein VP40 from Ebola virus of Sudan. The structure revealed that VP40 contains distinct N- and Cterminal domains, both are essential for trafficking to and interaction with the membrane. Bornholdt et al. stated that a butterfly-shaped VP40 dimer traffics to the cellular membrane. There, electrostatic interactions trigger rearrangement of the polypeptide into a linear hexamer. These hexamers construct a multi-layered, filamentous matrix structure that is critical for budding and resembles tomograms of authentic virions. A third structure of VP40, formed by a different rearrangement, is not involved in virus assembly, but instead uniquely binds RNA to regulate viral transcription inside infected cells [3]. In addition, the crystallographic structure of VP40-RNA reveals that the R-134 and F-125 of VP40 mainly interact with RNA [4]. Thus, the RNA binding pocket of VP40 could be considered as the drug target site for structure based drug design [5,6,7].
For finding lead compounds for anti VP40 drug design by virtual screening, traditional Chinese medicine (TCM) database and Asinex database were used in one study [5], Zinc database was used in another study [6], and TCM database alone was used in a more recent study [7].
Pyrimidinediones are a class of chemical compounds characterized by a pyrimidine ring substituted with two carbonyl groups. The compound 1-[(2s,4s,5r)-4-hydroxy-5- methyloxolan-2-yl]-5-methylpyrimidine,2,4dione was one of 4 selected compounds to show possible inhibition of VP40 using virtual screening [8]. In our study however, we searched for conformers of pyrimidine-2,4-dione in PubChem database. We found 1800 conformers and used them for molecular docking of Crystal Structure of matrix protein VP40 from Sudan Ebola virus targeting AA Arg 134. Docking and ADMET studies were done according to the method previously described [9]. Seven chemical compounds were found to have promising ADMET results, LD- 50, as well as H-bond interaction in the binding pocket. These seven molecules are waiting to be studied in vitro.
Methodology
Ligand generation
2D equivalent structural derivatives of 1-[(2s,4s,5r)-4-hydroxy-5- methyloxolan-2-yl]-5-methylpyrimidine,2,4 dione were searched in Pub Chem. By using this compound and inbuilt similarity fingerprinting search, molecules with minimum a 0.5 Tanimoto score had been selected. The search generates a total of 3000 ligands. Only 1800 molecules were tested. Chem Sketch was used for sketching and generating MDL\Mol v2000 molecules with 2D coordinates. The ligands were converted into Protein Databank (PDB) formats.
Target protein
Crystal Structure of matrix protein VP40 from Ebola virus Sudan had been acquired from RCSB [http://www.rcsb.org/pdb/explore/explore.do?structureId=3tc q] with PDB ID: 3TCQ in PDB format. The attached ligands were removed and energy minimization performed by standard optimization parameter of Swiss PDB Viewer [10].
Active Site
The amino acid Arg 134 is where matrix protein interacts with RNA which lies in the N-terminal domain. This AA was chosen for docking.
Docking Setup
Protein-Ligand docking had been tested by Auto Dock 4 [11], which combines energy evaluation through grids of affinity potential employing various search algorithms to find the suitable binding position for a ligand on a given protein. Docking involved the addition of polar hydrogen to the ligands using the Auto Dock hydrogen module and assigning the Kollman united atom partial charges. A standard docking procedure was used. It involved randomly placed individuals with a population size of 150. Maximum number of energy evaluations were 2.5 X107 and the mutation rate was 0.2 with crossing over rate of 0.80. Elitism was 1 for every generation. The results were clustered according to the 1.0 Ǻ rmsd criterion with 10 independent docking runs for each ligand. Auto Grid was used to calculate the grid maps representing proteins. Grid size was set to 60*60*60 points with grid spacing of 0.375 Ǻ. UCSF chimera was used to visualize the co-ordinates of the docked proteins to ligands within a region of 5 Ǻ and the hydrogen bond to stabilize the molecule - protein interaction (http://www.cgl.ucsf.edu/ chimera/).
Molecules showing minimum binding energies were evaluated for drug likeness using "Lipinski Rule of Five" for detecting probable pharmacokinetics. In addition, they were all subjected to molecular properties and drug likeness scores.
LD-50 was determined using PROTOX, a webserver for the prediction of oral toxicities of small molecules in rodents (http://tox.charite.de/tox). Toxic doses are often given as LD50 values in mg/kg body weight. The LD50 is the median lethal dose meaning the dose at which 50% of test subjects die upon exposure to a compound [12].
Toxicity classes are defined according to the globally harmonized system of classification of labeling of chemicals (GHS):
(1) Class I: fatal if swallowed (LD50 ≤ 5 mg/kg);
(2) Class II: fatal if swallowed (5 ‹ LD50 ≤ 50 mg/kg)
(3) Class III: toxic if swallowed (50 ‹ LD50 ≤ 300 mg/kg)
(4) Class IV: harmful if swallowed (300 ‹ LD50 ≤ 2000 mg/kg)
(5) Class V: may be harmful if swallowed (2000 ‹ LD50 ≤ 5000 mg/kg)
(6) Class VI: non-toxic (LD50 › 5000 mg/kg)
Toxicity targets are protein targets, which have been associated with adverse drug reactions and toxic effects. They predict possible binding to toxicity targets using a collection of proteinligand- based pharmacophores. All molecules were also subjected to drug likeness, Absorption, distribution, metabolism and elimination (ADME) using the Molsoft (http://molsoft.com/ mprop/) and (http:// ilab.acdlabs.com/ilab2/) websites.
Results:
Figure 1 shows 3D of Crystal Structure of matrix protein VP40 from Ebola virus Sudan acquired from RCSB. Figure 2 shows chemical structures of seven chosen molecules with the least minimum docking energy for VP40, best drug likeness and LD-50 scores. They are all conformers of pyrimidine, 2,4 dione. All chemical properties of the seven chosen molecules are shown in Table 1. Figure 3 shows docking interaction of the top seven chosen compounds with matrix protein VP40 argenin 134.Molecules 1 to 6 shows interaction with Arg134 in the form of hydrogen bonds. However, molecule 7 was very interesting since it gave a hydrogen bond with ASN 136, which gave a chance of another hydrogen bonding of Arg 134 with this AA leading to almost the same effect where Arg 134 was engaged in a hydrogen bond and will not be available for RNA binding. Table 2 shows minimum binding energy of the 7 chosen molecules that ranged from -5.21 to -4.08. All agreed to Lipinsk’s rule but number 5. All have low Ames probability. LD-50 ranged from 16877 to 8400 and the 7 molecules are thus nontoxic of class 6. They are arranged from the highest to the lowest LD-50.
Figure 1.
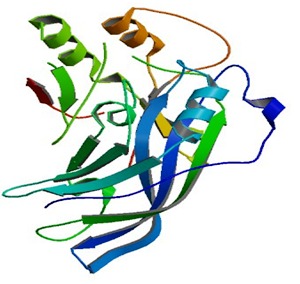
A 3D structure cartoon representation of the matrix protein VP40 from Ebola virus of Sudan (PDB ID: 3TCQ) as viewed in JSmol (JavaScript). (http://www.rcsb.org)
Figure 2.
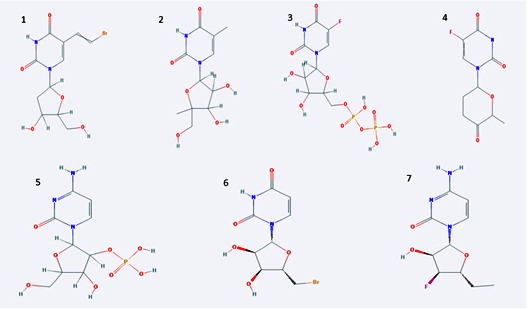
Chemical structures of the top seven molecules chosen as best docked for VP40 protein. All were conformers of pyrimidine-2, 4-dione. Each is characterized by its unique functional group. Describe the salient features (class) of the compounds in 2 statements.
Table 1. Chemical properties of the top 7 chosen compounds as best docked molecules for VP40 protein.
| No. | PubChem CID | Compound IUPAC name | Properties | ||||
| Mol formula | Mol wt (g/mol) | Log P | H bond donor | H bond acceptor | |||
| 1 | 416724 | [5-(5-fluoro-2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphono hydrogen phosphate | C9H13FN2O12P2 | 422.151 | -4.7 | 6 | 13 |
| 2 | 374108 | 1-[3,4-dihydroxy-5-(hydroxymethyl)-5-methyloxolan-2-yl]-5- methylpyrimidine-2,4-dione | C11H16N2O6 | 272.254 | -1.7 | 4 | 6 |
| 3 | 3851453 | phenyl 2-[3,4-dihydroxy-5-(5-methyl-2,4-dioxopyrimidin-1- yl)oxolan-2-yl]acetate | C17H18N2O7 | 362.334 | -0.4 | 3 | 7 |
| 4 | 256623 | [2-(4-amino-2-oxopyrimidin-1-yl)-4-hydroxy-5-(hydroxymethyl)oxolan-3-yl] dihydrogen phosphate | C10H11FN2O4 | 242.203 | -0.4 | 1 | 5 |
| 5 | 44149862 | 1-[(2R,3R,4S,5S)-5-(bromomethyl)-3,4-dihydroxyoxolan-2- yl]pyrimidine-2,4-dione | C9H11BrN2O5 | 307.098 | -1.1 | 3 | 5 |
| 6 | 254616 | 3,4-dihydroxy-5-(5-nitro-2,4-dioxopyrimidin-1-yl)oxolane-2-carboxylic acid | C9H9N3O9 | 303.182 | -2 | 4 | 9 |
| 7 | 3183 | 5-(2-bromoethenyl)-1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione | C11H13BrN2O5 | 333.135 | -0.4 | 3 | 5 |
Figure 3.
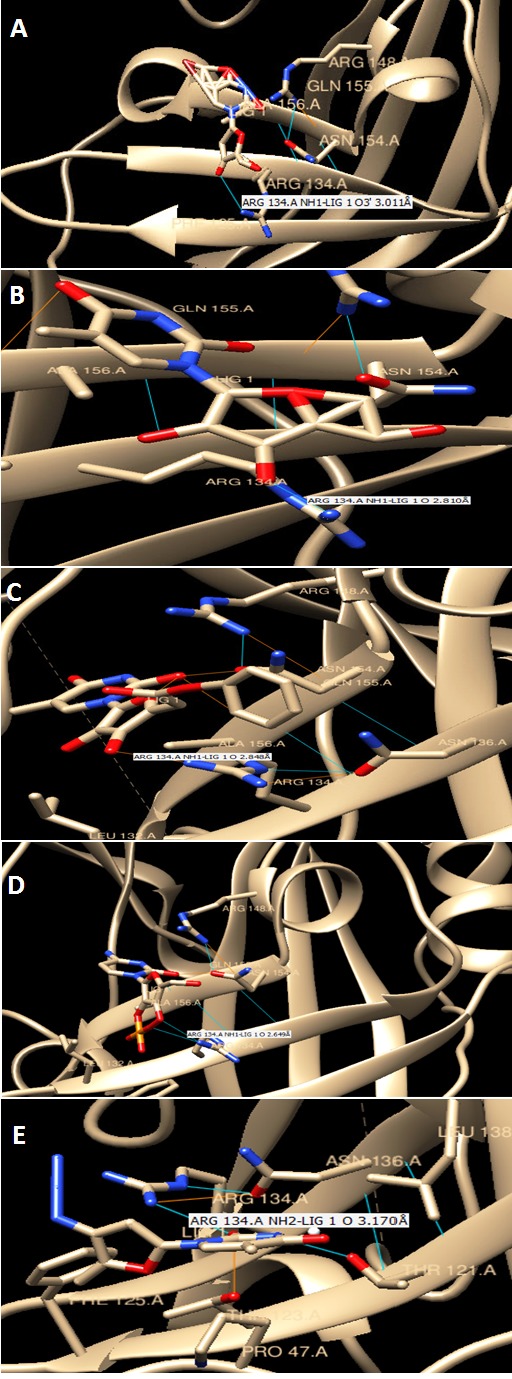
Docking interaction of the top seven chosen compounds with matrix protein VP40 argenin 134 from A-E showing H bonding with the protein within 5 Ǻ.
Discussion:
According to the CDC guidelines, no FDA-approved vaccine or medicine (e.g., antiviral drug) is available for Ebola up until now [13]. Management of Ebola is done by treating its Symptoms and its complications as they appear. Thus there is an urgent need to develop new drugs using virtual screening and experiment on those most suitable as would-be-drugs. Many studies have already addressed this issue [[5,6, 7,8], [14]] along with a review article on the subject [15].
Palamthodi et al. (2012) discussed the discovery of 4 ligand hits against VP40, one of them was our lead compound (1-[(2s,4s,5r)- 4-hydroxy-5-methyloxolan-2-yl]-5-methylpyrimidine,2,4 dione) [8]. Tamilvanan and Hopper, 2013 discussed the discovery of 5 synthetic and 5 natural compounds by virtual screening targeting Ebola VP40 [5]. Abazari et al. 2015 reported 4 chemicals that could bind VP40 to prevent its oligomerization [6]. Raj and Varadwaj 2015 showed that two top-ranking falvonoids showed better docking scores and binding energies than a reported lead compound (BCX4430) for Ebola viral protein targets [14]. Whereas, Karthick et al., 2016 showed that a lead compound (emodin-8-beta-D-glucoside) from the Traditional Chinese Medicine Database (TCMD) could be active against the Ebola virus [7]. Our reported lead chemical compounds are new and were not reported anywhere else. We are reporting seven molecules using virtual screening and molecular docking against VP40 Arg 134 with hydrogen bonding to the active site. All agreed to Lipinsk’s rule but number 5. All have low Ames probability. Their LD-50 ranged from 16877 to 8400 and are thus predicted to be nontoxic and could act as potential drugs. They can negatively influence binding of RNA to VP40 and potentially inhibit Ebola virus replication. More experimentation is mandatory in order to test these compounds in vitro.
Conclusion:
Based on the molecular docking and ADMET studies of 1800 small molecules derivatives of pyrimidine-2, 6-dione we chose 7 molecules with good minimum binding energy and drug likeness. All seven chosen molecules showed hydrogen bonding with amino acid Arg 134 of Ebola virus matrix protein VP40 suggesting that they hinder binding with viral RNA and hence can abort budding process. These seven reported molecules are promising as drugs against EBOV. They wait to be verified experimentally by testing them in vitro and in vivo to reach for candidate drugs.
Author Contributions:
Alam El-Din HM wrote the manuscript and revised it. Alam El- Din HM, Lotfy SA, Fathy N, Elberry M, Mayla AM, and Kassem S did the molecular docking and ADMET studies of 300 molecules each. Naqvi A tutored the rest with the methods, chose the protein, its active site and chose the lead compound and its conformers.
Table 2. The overall properties of the top seven chosen compounds from virtual screening drug likeness score and ADMET studies.
| Property | Lig1 | Lig2 | Lig3 | Lig4 | Lig5 | Lig6 | Lig7 |
| Maximum binding energy | -4.48 | -4.36 | -4.08 | -5.21 | -5.16 | -5.1 | -4.32 |
| Lipinski’s rule of 5 | Yes | Yes | Yes | No | Yes | Yes | Yes |
| Drug likeness model score | 0.83 | -0.05 | -0.17 | 0.96 | 1.11 | 0.66 | 0.93 |
| Probability of +ve Ames | 0.03 | 0.11 | 0.06 | 0.09 | 0.36 | 0.51 | 0.01 |
| Human oral Bioavailability | ‹30% | 30-70% | 30-70% | ‹30% | 30-70% | ‹30% | 30-70% |
| Maximum binding energy | 1% | 32% | 93% | 1% | 87% | 2% | 81% |
| LD 50 (mg/kg) | 16877 | 15000 | 14665 | 12000 | 8400 | 8400 | 8400 |
| Toxicity class | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
Edited by P Kangueane
Citation: Alam El-Din et al. Bioinformation 12(3): 192-196 (2016)
References
- 1.Moghadam S, et al. Asian Pac J Trop Biomed. 2015;5(4):260. [Google Scholar]
- 2.Mehedi M, et al. J Virol. 2011;85(11):5406. doi: 10.1128/JVI.02190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bornholdt ZA, et al. Cell. 2013;154(4):763. doi: 10.1016/j.cell.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomis-Rüth FX, et al. Structure. 2003;11(4):423. doi: 10.1016/S0969-2126(03)00050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamilvanan T, Hopper W. Bioinformation. 2013;9(6):286. doi: 10.6026/97320630009286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abazari D, et al. Bioinformation. 2015;11(5):243. doi: 10.6026/97320630011243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karthick V, et al. Infect Dis Poverty. 2016;5(1):12. doi: 10.1186/s40249-016-0105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palamthodi S, et al. Journal of Computational Methods in Molecular Design. 2012;2(2):76. [Google Scholar]
- 9.Ahire V, et al. European Journal of Biotechnology and Bioscience. 2014;1(6):1. [Google Scholar]
- 10.Guex N, Peitsch MC. Electrophoresis. 1997;18(15):2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 11.Morris GM, et al. J Comput Chem. 2009;30(16):2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drwal MN, et al. Nucleic Acids Res. 2014;42:W53. doi: 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. http://www.cdc.gov/vhf/ebola/treatment/
- 14.Raj U, Varadwaj PK. Interdiscip Sci. 2015;7:1. [Google Scholar]
- 15.Janeba Z, Med Res Rev. 2015;35(6):1175. doi: 10.1002/med.21355. [DOI] [PMC free article] [PubMed] [Google Scholar]