

Abstract
Staphylococcus aureus is one of the most common hospital acquired infections. It colonizes immunocompromised patients and with the number of antibiotic resistant strains increasing, medicine needs new treatment options. Understanding more about the proteins this organism uses would further this goal. Hypothetical proteins are sequences thought to encode a functional protein but for which little to no evidence of that function exists. About half of the genomic proteins in reference strain S. aureus NCTC 8325 are hypothetical. Since annotation of these proteins can lead to new therapeutic targets, a high demand to characterize hypothetical proteins is present. This work examines 35 hypothetical proteins from the chromosome of S. aureus NCTC 8325. Examination includes physiochemical characterization; sequence homology; structural homology; domain recognition; structure modeling; active site depiction; predicted protein-protein interactions; protein-chemical interactions; protein localization; protein stability; and protein solubility. The examination revealed some hypothetical proteins related to virulent domains and protein-protein interactions including superoxide dismutase, O-antigen, bacterial ferric iron reductase and siderophore synthesis. Yet other hypothetical proteins appear to be metabolic or transport proteins including ABC transporters, major facilitator superfamily, S-adenosylmethionine decarboxylase, and GTPases. Progress evaluating some hypothetical proteins, particularly the smaller ones, was incomplete due to limited homology and structural information in public repositories. These data characterizing hypothetical proteins will contribute to the scientific understanding of S. aureus by identifying potential drug targets and aiding in future drug discovery.
Keywords: Hypothetical proteins, Staphylococcus aureus NCTC 8325
Background
While Staphylococcus aureus is a natural bacterial inhabitant of nasal passages, it is a major cause of nosocomial infections of surgical wounds particularly involving indwelling medical devices [1]. It can also present as superficial skin lesions or localized abscesses turning into deep-seated infections such as furunculosis if left untreated. S. aureus causes toxic shock syndrome when it goes septic, a huge concern considering the rise of antibiotic resistance the organism has experienced. Other health issues related to internalized infections are heart and lung diseases such as endocarditis and necrotizing pneumonia, which are now being diagnosed in the younger community populations, rather than remaining solely a hospital acquired (HA) infection. Deaths have been reported in relation to these heart and lung infections [2].
Methicillin-resistant Staphylococcus aureus (MRSA) bacteria are resistant to all beta-lactam antibiotics such as penicillin, methicillin, amoxicillin, and oxacillin. In 2011, the Center for Disease Control estimate 80,000 invasive MRSA infections and 11,285 related deaths in the United States annually [3]. Most of these are nosocomial infections, though there are increases in community acquired (CA) MRSA infections, particularly among immunocompromised patients. Others in the community setting that have shown a tendency to acquire MRSA are those of younger age rather than older. According to Casey, the median age for CA-MRSA in 2010 was 24 years versus a median age of 61 years for nosocomial MRSA infections [4]. Another predictor of CA-MRSA infections was an increased number of antibiotics prescribed in the year before infection. There is no significant data showing a link between race and CA-MRSA infection rates, but obesity was determined as a risk factor.
CA-MRSA in the United States is of particular concern due to the USA300 strain gaining momentum globally, relative to its excessive production of exotoxins and its genetics of polyamine-resistance [5]. One of the most prominent mechanisms responsible for the virulence of USA300 is its Arginine Catabolic Mobile Element (ACME), which ultimately inhibits polyamines that perpetuate wound healing in patients [6]. The ACME of this CA-MRSA allows it to survive acidic environments that normally limit its colonization.
Due to documented increases of a global spread of CA-MRSA in just the past 20 years, a worldwide need for innovative therapies that target these divergent strains in new ways is of ultimate concern [2]. This directs attention to prediction work with hypothetical proteins in silico, which allows for further investigation into the S. aureus genome. Upon analyzing the phylogeny of its protein sequencing, prediction of the bacteria’s next mutation is possible, thus enhancing knowledge of its mechanisms of action. By this, science gains insight into receptor targets that inhibits reproduction of such a resistant and virulent species.
Approximately 50% of the S. aureus NCTC 8325 genome is comprised of hypothetical proteins. Hypothetical proteins are protein sequences by nucleic acid sequence only with unknown function [7]. These sequences have little to no experimental evidence for their function’s existence, characterized by a low identity to proteins with known function. Frequently, these nonconserved proteins do not follow established phylogenetic lineage. There are two groups of hypothetical proteins: uncharacterized protein families and domains of unknown function. The latter are experimentally identified proteins with no known structural domains related to function.
Several studies have characterized hypothetical proteins. Mohan and Venugopal examined ten hypothetical plasmid proteins in S. aureus in 2012 [8]. They characterized an ABC transporter ATPbinding protein, export proteins, and a protein related to the multiple antibiotic resistance family among others. In 2015, Varma and colleagues examined one hypothetical protein from S. aureus, selected for its size and Basic Local Alignment Search Tool (BLAST) result, which appears to bind to ribosomal subunits [7]. Shahbaaz and researchers predicted the function of 83 hypothetical proteins in Mycoplasma pneumoniae type 2a strain 309, several of which appear virulent [9]. Islam, et al., characterized six hypothetical proteins in Vibrio cholerae O139 predicting the function of an antibiotic resistance protein, an integrase enzyme, and a restriction endonuclease [10]. All used similar methods to those presented in this study.
With approximately half of all genomic protein sequences currently annotated as hypothetical, great potential exists for the discovery of new drug targets [10]. The pharmaceutical industry is struggling to discover and develop new drugs quickly and cheaply. Increasing the number of available targets that pharmaceutical agents could act on by characterizing hypothetical proteins may alleviate some of the pharmaceutical industry’s pressure. This could lead to novel and improved therapeutic agents for better patient care, increased corporate and hospital profits, and decreased drug prices for consumers.
Methodology
This study randomly selected 35 proteins from the Staphylococcus aureus NCTC 8325 chromosomal protein table that the National Center for Biotechnology Information (NCBI) classified as hypothetical. The protein loci were SAOUHSC_00010, SAOUHSC_00077, SAOUHSC_00082, SAOUHSC_00085, SAOUHSC_00091, SAOUHSC_00136, SAOUHSC_00145, SAOUHSC_00156, SAOUHSC_00219, SAOUHSC_00238, SAOUHSC_00303, SAOUHSC_00307, SAOUHSC_00308, SAOUHSC_00328, SAOUHSC_00423, SAOUHSC_00455, SAOUHSC_00548, SAOUHSC_00751, SAOUHSC_00766, SAOUHSC_00837, SAOUHSC_00972, SAOUHSC_01024, SAOUHSC_01291, SAOUHSC_01306, SAOUHSC_01402, SAOUHSC_01851, SAOUHSC_01931, SAOUHSC_01937, SAOUHSC_02471, SAOUHSC_02570, SAOUHSC_02770, SAOUHSC_02889, SAOUHSC_02901, SAOUHSC_02911, and SAOUHSC_02934.
Several algorithms characterized these hypothetical proteins. Position-Specific Iterative BLAST (PSI-BLAST) at NCBI identified potential homologs through secondary protein structure alignments. ExPASy’s Protparam server computed the number of amino acids, amino acid composition and frequencies, molecular weight, the total number of charged residues (aspartic acid plus glutamic acid for positively charged and the sum of arginine and lysine for negatively charged), theoretical isoelectric point (pI), extinction coefficient, instability index (II), aliphatic index (AI), and grand average hydropathy (GRAVY) [11].
Both Pfam and the conserved domain database BLAST (CDDBLAST) from NCBI, performed protein domain identification. Pfam is a comprehensive collection of multiple sequence alignments and Hidden Markov Models that represent protein domains and families [12]. The CDD-BLAST algorithm uses a PSI-BLAST variant to establish position-specific scoring matrices with the protein sequence [13]. Researchers frequently use Pfam and CDD-BLAST together to characterize parts of the protein involved in binding capability [8,9,10].
Tertiary structure predictions were completed by (PS)2, which is an automatic homology modeling server uses a protein sequence in pair-wise and multiple alignments though unions of PSI-BLAST, integrated molecular pathway level analysis, and multiple sequence alignment methods [14]. This approach combines information on sequence and secondary structure to detect homologous proteins with remote similarity and the target-template alignment. MODELLER software builds the protein’s three-dimensional structure containing all non-hydrogen atoms from homology or comparative modeling. The product is a Protein Data Bank (PDB) file used by 3DLigandSite for identifying potential active sites when possible otherwise 3DLigandSite used Pyre2 to attempt to model proteins from sequence [15].
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) is a database of known and predicted protein interactions. It draws from genomic context, high-throughput experiments, conserved co-expression, and previous PubMed literature [16]. STRING integrates interaction information on functional and physical relationships. Search Tool for Interactions of Chemicals (STITCH) is a database equal to STRING in its capacity to predict a hypothetical protein’s functional associates in a biological network [17]. STITCH integrates information from scientific literature with several databases to focus on drug-target interactions, binding affinities, and biological pathways to predict chemical and protein associates to the protein query.
Two programs analyzed protein location within the cell. PSortB predicts the location of each protein [18]. The SOSUI server characterized a protein’s solubility and identified potential transmembrane regions [19]. Examining how cysteine forms disulfide bonds to stabilize the protein may be helpful. The DISULFIND predicted disulfide bridges and examined structural and functional properties of hypothetical proteins [20]. Default program settings were used for all analyses except for STITCH where the required confidence (score) was set to highest confidence (0.900).
Discussion:
Thirty-five chromosomal hypothetical proteins from S. aureus NCTC 8325 were randomly selected from 1509 possible hypothetical proteins. Characterization included homolog identification, physiochemical measurements, domain identification, active site description, binding partners, cellular location, and solubility calculations.
Sequence Similarity
PSI-BLAST compares protein secondary structures among proteins. Top PSI-BLAST result for each hypothetical protein is listed in Table 1. All hypothetical proteins matched proteins in S. aureus with 100% query coverage, except for SAOUHSC_01937, as PSIBLAST could not match SAOUHSC_01937. SAOUHSC_00010 fit a protein in S. aureus MRSA131. SAOUHSC_00328 matched a protein in S. aureus A5948. SAOUHSC_001024 hit a protein in S. aureus VRS1. Percent identity ranged from 97% to 100% with e-values of 0.0 to 4e-11, indicating strong matches between hypothetical proteins and their homologs.
Table 1. Top PSI-BLAST result for hypothetical proteins.
| Locus Tag | PSI-BLAST Match | Identity | E-value |
| SAOUHSC_00010 | azaleucine resistance protein | 100% | 8.00E-163 |
| SAOUHSC_00077 | siderophore biosynthesis protein | 99% | 0 |
| SAOUHSC_00082 | diaminopimelate decarboxylase | 99% | 0 |
| SAOUHSC_00085 | membrane protein | 99% | 6.00E-146 |
| SAOUHSC_00091 | ligase | 99% | 0 |
| SAOUHSC_00136 | sulfonate ABC transporter ATP-binding protein | 99% | 6.00E-179 |
| SAOUHSC_00145 | 4'-phosphopantetheinyl transferase | 99% | 8.00E-157 |
| SAOUHSC_00156 | outer surface protein 99% | 0 | |
| SAOUHSC_00219 | galactitol-1-phosphate 5-dehydrogenase | 99% | 0 |
| SAOUHSC_00238 | hypothetical protein | 98% | 3.00E-20 |
| SAOUHSC_00303 | hypothetical protein | 97% | 6.00E-25 |
| SAOUHSC_00307 | deacetylase SIR2 | 99% | 0 |
| SAOUHSC_00308 | lipoate-protein ligase A | 99% | 0 |
| SAOUHSC_00328 | twin arginine-targeting protein translocase TatC | 99% | 2.00E-151 |
| SAOUHSC_00423 | methionine ABC transporter ATP-binding protein | 99% | 0 |
| SAOUHSC_00455 | signal peptidase II | 99% | 0 |
| SAOUHSC_00548 | glycosyl transferase family 1 | 99% | 0 |
| SAOUHSC_00751 | hypothetical protein | 99% | 2.00E-69 |
| SAOUHSC_00766 | competence protein ComF | 100% | 1.00E-163 |
| SAOUHSC_00837 | hypothetical protein | 100% | 7.00E+00 |
| SAOUHSC_00972 | hypothetical protein | 99% | 4.00E-57 |
| SAOUHSC_01024 | hypothetical protein MQA_00274 | 100% | 1.00E+00 |
| SAOUHSC_01291 | hypothetical protein | 97% | 2.00E+00 |
| SAOUHSC_01306 | LSM domain protein | 98% | 8.00E-34 |
| SAOUHSC_01402 | MSA protein | 99% | 4.00E-83 |
| SAOUHSC_01851 | hypothetical protein | 97% | 4.00E+00 |
| SAOUHSC_01931 | NTPase | 99% | 0 |
| SAOUHSC_02471 | hypothetical protein | 99% | 0 |
| SAOUHSC_02570 | AraC family transcriptional regulator | 99% | 0 |
| SAOUHSC_02770 | diaminopimelate epimerase | 99% | 0 |
| SAOUHSC_02889 | hypothetical protein | 100% | 4.00E-20 |
| SAOUHSC_02901 | GTPase | 99% | 0 |
| SAOUHSC_02911 | ATPase or DNA integration/ recombination1 | 99% | 0 |
| SAOUHSC_02934 | hypothetical protein | 97% | 4.00E+00 |
| 1Ranked equal in top hit | |||
Physiochemical Characterization
ExPASy calculated the physiochemical parameters listed in Table 2. Number of amino acids ranged from 30 to 1370 with molecular weights from 3544.3 to 163266.7. The theoretical isoelectric point, the pI where the protein would be most stable, was calculated from the number of negative and positive residues (Asp and Glu, Arg and Lys, respectively). The extinction coefficient values are for 280nm because that is the wavelength where proteins absorb light strongly while other substances common to protein solutions do not. The extinction coefficient for two smaller hypothetical proteins, SAOUHSC_01024 and SAOUHSC_01291, could not be determined because there were no Trp, Tyr, or Cys in the protein, so the protein should not be visible by UV spectrophotometry. The instability index (II) predicts if a protein would be stable in a test tube under normal conditions. Proteins with II values over 40 considered unstable. The aliphatic index (AI) represents the protein’s volume taken up by aliphatic side chains (Ala, Val, Leu, and Ile). The higher the AI, the wider the temperature range at which the protein will be stable. GRAVY measures the protein’s hydrophobicity. Values spanned -1.984 to 1.096 with higher scores meaning increased hydrophobicity for the protein.
Table 2. Physiochemical properties of hypothetical proteins.
| Protein | # AA | MW | pI | # neg | # pos | EC | II | AI | GRAVY |
| SAOUHSC_00010 | 231 | 25147 | 6.06 | 10 | 9 | 28670 | 28.84 | 125.45 | 1.048 |
| SAOUHSC_00077 | 584 | 66433.1 | 5.1 | 73 | 50 | 75540 | 37.33 | 98.18 | -0.152 |
| SAOUHSC_00082 | 400 | 45759.8 | 5.86 | 52 | 37 | 53080 | 41.31 | 88.45 | -0.3 |
| SAOUHSC_00085 | 208 | 22980.1 | 6.9 | 22 | 22 | 13200 | 23.01 | 123.7 | 0.455 |
| SAOUHSC_00091 | 412 | 47053.3 | 8.66 | 23 | 27 | 51020 | 28.94 | 138.37 | 0.966 |
| SAOUHSC_00136 | 246 | 28095.3 | 6.76 | 28 | 26 | 13200 | 44.53 | 105 | -0.296 |
| SAOUHSC_00145 | 214 | 25261.9 | 8.26 | 21 | 23 | 47120 | 37.21 | 86.87 | -0.335 |
| SAOUHSC_00156 | 346 | 39868.6 | 6.05 | 42 | 35 | 21360 | 46.22 | 91.82 | -0.323 |
| SAOUHSC_00219 | 347 | 38401.3 | 5.84 | 44 | 37 | 39350 | 31.27 | 93.54 | -0.069 |
| SAOUHSC_00238 | 33 | 5199.3 | 9.6 | 2 | 5 | 9970 | 34.69 | 150.68 | 0.766 |
| SAOUHSC_00303 | 30 | 3544.3 | 9.9 | 3 | 8 | 0 | 15.53 | 65 | -0.8 |
| SAOUHSC_00307 | 314 | 36397.1 | 5.29 | 44 | 31 | 44600 | 40.63 | 74.9 | -0.505 |
| SAOUHSC_00308 | 340 | 38847 | 5.1 | 53 | 42 | 48040 | 22.53 | 91.18 | -0.394 |
| SAOUHSC_00328 | 218 | 25382.7 | 9.32 | 6 | 13 | 38640 | 41.54 | 121.65 | 1.096 |
| SAOUHSC_00423 | 341 | 38675.7 | 8.2 | 41 | 43 | 13200 | 22.69 | 91.96 | -0.199 |
| SAOUHSC_00455 | 267 | 30232.8 | 4.84 | 47 | 34 | 16430 | 33.71 | 102.92 | -0.244 |
| SAOUHSC_00548 | 496 | 58418.3 | 6.41 | 61 | 57 | 77380 | 38.68 | 87.22 | -0.422 |
| SAOUHSC_00751 | 104 | 12723.7 | 8.31 | 13 | 16 | 19535 | 70.95 | 56.25 | -0.645 |
| SAOUHSC_00766 | 224 | 26347.7 | 8.86 | 27 | 35 | 24005 | 37.86 | 90.62 | -0.373 |
| SAOUHSC_00837 | 37 | 4265.2 | 9.7 | 2 | 5 | 8480 | 29.62 | 121.35 | 0.584 |
| SAOUHSC_00972 | 95 | 11193.6 | 4.59 | 16 | 9 | 10430 | 26.75 | 102.53 | -0.5 |
| SAOUHSC_01024 | 44 | 5162.6 | 5.05 | 12 | 10 | None1 | 50.22 | 48.86 | -1.984 |
| SAOUHSC_01291 | 36 | 4267.4 | 10.47 | 2 | 9 | None1 | 30.75 | 135.28 | 0.578 |
| SAOUHSC_01306 | 63 | 7193.2 | 5.08 | 9 | 8 | 4470 | 29.79 | 117.46 | -0.168 |
| SAOUHSC_01402 | 133 | 15657.1 | 6.71 | 10 | 10 | 16390 | 36.75 | 152.41 | 1.021 |
| SAOUHSC_01851 | 37 | 4504.4 | 10.17 | 0 | 7 | 5960 | 2.76 | 105.14 | 0.273 |
| SAOUHSC_01931 | 1370 | 163266.7 | 5.95 | 201 | 185 | 241090 | 39.01 | 92.5 | -0.481 |
| SAOUHSC_01937 | 35 | 4041.9 | 9.4 | 1 | 3 | 8480 | 20.97 | 147.71 | 0.843 |
| SAOUHSC_02471 | 468 | 56151.2 | 5.91 | 73 | 68 | 64765 | 33.09 | 90.51 | -0.494 |
| SAOUHSC_02570 | 651 | 76005.7 | 8.3 | 71 | 75 | 66170 | 40.78 | 104.62 | -0.225 |
| SAOUHSC_02770 | 273 | 31004 | 6.08 | 26 | 19 | 36370 | 43.01 | 78.13 | -0.301 |
| SAOUHSC_02889 | 42 | 5160 | 6.06 | 6 | 6 | 4595 | 9.37 | 88.1 | -0.06 |
| SAOUHSC_02901 | 296 | 32835.3 | 5.69 | 34 | 26 | 13910 | 43.56 | 101.08 | 0.118 |
| SAOUHSC_02911 | 240 | 27789 | 8.25 | 32 | 35 | 28350 | 36.67 | 76.46 | -0.464 |
| SAOUHSC_02934 | 31 | 3652.3 | 8.16 | 2 | 3 | 2980 | 27.75 | 106.77 | 0.245 |
| # AA, number of amino acids; MW, molecular weight; pI, theoretical isoelectric point; # neg, total number of negatively charged residues (Asp + Glu); # pos, total number of positively charged residues (Arg + Lys); EC, extinction coefficient assuming all pairs of Cys residues form cystines; II, instability index; AI, aliphatic index; GRAVY, grand average hydropathy. 1As there are no Trp, Tyr, or Cys in the region considered, protein should not be visible by UV spectrophotometry. | |||||||||
Domain Identification
CDD-BLAST and Pfam identified domains for hypothetical proteins. CDD-BLAST and Pfam results and domain descriptions in Table 3,Table 4, Table 5,Table 6, respectively. The programs could not find domains within proteins not listed. If both programs identified a domain, the CDD-BLAST tables identified and defined it (Table 3 and Table 4) and not repeated in the Pfam tables.
Table 3. CDD-BLAST domain data for hypothetical proteins.
| Locus Tag | Domains | E-value |
| SAOUHSC_00010 | AzlC | 3.45E-67 |
| SAOUHSC_00077 | lucA_lucC, FhuF, RhbC | 6.96e-63, 4.42e-15, 7.18e-180 |
| SAOUHSC_00082 | PLPDE_III_PvsE_like, LysA | 0e00, 1.42e-118 |
| SAOUHSC_00085 | MFS | 1.49E-04 |
| SAOUHSC_00091 | O-antigen_lig | 1.24E-05 |
| SAOUHSC_00136 | ABC_NrtD_SsuB_transporters, TauB | 2.95e-106, 7.68e-115 |
| SAOUHSC_00145 | ACPS, Sfp | 1.85e-10, 2.37e-63 |
| SAOUHSC_00156 | COG3589 | 9.06E-161 |
| SAOUHSC_00219 | sugar_DH, Tdh | 0e00, 7.15e-97 |
| SAOUHSC_00307 | SIR2 | 1.32E-67 |
| SAOUHSC_00308 | LplA, Lip_prot_lig_C, lipoyltrans | 4.74e-82, 2.33e-30, and 1.09e-69 |
| SAOUHSC_00328 | TatC | 1.55E-50 |
| SAOUHSC_00423 | ABC_MetN_methionine_transporter, NIL, AbcC | 3.27e-138, 6.03e-11, 7.16e-176 |
| SAOUHSC_00455 | YaaT | 5.26E-102 |
| SAOUHSC_00548 | GT1_gtfA_like, DUF1975, TIGR02918 | 6.56e-146, 6.42e-56, 1.53e-31 |
| SAOUHSC_00751 | COG4357 | 3.47E-50 |
| SAOUHSC_00766 | PRTases_typeI, ComFC | 2.38e-10, 4.47e-52 |
| SAOUHSC_01931 | AAA_16, AAA | 5.36e-05, 5.87e-03 |
| SAOUHSC_02570 | HTH_AraC, HTH_ARAC | 4.36e-04, 6.20e-14 |
| SAOUHSC_02770 | DapF | 7.31E-74 |
| SAOUHSC_02901 | cobW, CobW_C, YejR | 1.81e-57, 9.02e-10, 1.06e-77 |
| SAOUHSC_02911 | COG1636 | 3.25E-101 |
Table 4. Description of CDD-BLAST domains.
| Superfamily | Description |
| AzlC | Predicted branched-chain amino acid permease (azaleucine resistance) |
| IucA_IucC | IucA / IucC family |
| FhuF | Bacterial ferric iron reductase protein |
| RhbC | Siderophore synthetase component |
| PLPDE_III_PvsE_like | Type III Pyridoxal 5-phosphate (PLP)-Dependent Enzyme PvsE |
| LysA | Diaminopimelate decarboxylase |
| O-antigen_lig | O-antigen ligase like membrane protein |
| MFS | Major Facilitator Superfamily |
| ABC_NrtD_SsuB_transporters | ATP-binding cassette domain of the nitrate and sulfonate transporters |
| TauB | ABC-type nitrate/sulfonate/bicarbonate transport system, ATPase component |
| ACPS | 4'-phosphopantetheinyl transferase superfamily |
| Sfp | Phosphopantetheinyl transferase |
| COG3589 | Uncharacterized protein [Function unknown] |
| sugar_DH | NAD(P)-dependent sugar dehydrogenases |
| Tdh | Threonine dehydrogenase or related Zn-dependent dehydrogenase |
| SIR2 | NAD-dependent protein deacetylase |
| Lip_prot_lig_C | Bacterial lipoate protein ligase C-terminus |
| lipoyltrans | Lipoyltransferase and lipoate-protein ligase |
| LplA | Lipoate-protein ligase A |
| TatC | Sec-independent protein secretion pathway component |
| ABC_MetN_methionine_transporter | ATP-binding cassette domain of methionine transporter |
| NIL | NIL domain |
| AbcC | ABC-type methionine transport system, ATPase component |
| YaaT | Cell fate regulator YaaT, PSP1 superfamily |
| GT1_gtfA_like | GT1 family of glycosyltransferases |
| DUF1975 | Domain of unknown function |
| TIGR02918 | Accessory Sec system glycosylation protein GtfA |
| COG4357 | Uncharacterized protein, contains Zn-finger domain of CHY type |
| PRTases_typeI | Phosphoribosyl transferase (PRT)-type I domain |
| ComFC | Predicted amidophosphoribosyltransferases |
| AAA_16 | AAA ATPase domain |
| AAA | ATPases associated with a variety of cellular activities |
| HTH_AraC | Bacterial regulatory helix-turn-helix proteins, AraC family |
| HTH_ARAC | Helix_turn_helix, arabinose operon control protein |
| DapF | Diaminopimelate epimerase |
| cobW | CobW/HypB/UreG, nucleotide-binding domain |
| CobW_C | Cobalamin synthesis protein cobW C-terminal domain |
| YejR | GTPase, G3E family |
| COG1636 | Predicted ATPase, Adenine nucleotide alpha hydrolases (AANH) superfamily |
Table 5. Pfam domain data for hypothetical proteins.
| Locus Tag | Pfam Domain | E-value |
| SAOUHSC_00010 | AzlC | 1.20E-37 |
| SAOUHSC_00077 | lucA_lucC, FhuF | 2.1e-54, 5e-15 |
| SAOUHSC_00082 | Orn_Arg_deC_N,Orn_DAP_Arg_deC | 9.7e-37, 2e-10 |
| SAOUHSC_00091 | Wzy_C | 2.20E-13 |
| SAOUHSC_00136 | ABC_tran | 3.90E-30 |
| SAOUHSC_00145 | ACPS | 9.10E-08 |
| SAOUHSC_00156 | DUF871 | 3.50E-86 |
| SAOUHSC_00219 | ADH_N, ADH_zinc_N | 8.8e-30, 2.8e-22 |
| SAOUHSC_00308 | BPL_LplA_LipB, Lip_prot_lig_C | 4.6e-09,4.6e-26 |
| SAOUHSC_00328 | TatC | 4.20E-41 |
| SAOUHSC_00423 | ABC_tran, NIL | 2.8e-35, 4.3e-10 |
| SAOUHSC_00455 | PSP1 | 4.10E-26 |
| SAOUHSC_00548 | Glycos_transf_1 | 3.30E-32 |
| SAOUHSC_00751 | Zf-CHY | 8.40E-12 |
| SAOUHSC_02570 | HTH_18 | 1.90E-14 |
| SAOUHSC_02901 | cobW, CobW_C | 1.4e-45, 4.4e-09 |
| SAOUHSC_02911 | DUF208 | 8.90E-61 |
Table 6. Description of Pfam domains.
| Superfamily | Description |
| Orn_Arg_deC_N | Pyridoxal-dependent decarboxylase, pyridoxal binding domain |
| Orn_DAP_Arg_deC | Pyridoxal-dependent decarboxylase, Cterminal sheet domain |
| Wzy_C | O-Antigen ligase |
| ABC_tran | ABC transporter |
| DUF871 | Bacterial protein of unknown function |
| ADH_N | Alcohol dehydrogenase GroES-like domain |
| ADH_zinc_N | Zinc-binding dehydrogenase |
| BPL_LplA_LipB | Biotin/lipoate A/B protein ligase family |
| PSP1 | PSP1 C-terminal conserved region |
| Glycos_transf_1 | Glycosyl transferases group 1 |
| Zf-CHY | CHY zinc finger |
| HTH_18 | Helix-turn-helix domain |
| DUF208 | Uncharacterized BCR, COG1636 |
Active Site and Substrate Characterization
The (PS)2 server attempted to model each hypothetical protein. Template information including percent identity and e-value is in Table 7. Several proteins could not be modeled by (PS)2. Hypothetical proteins, SAOUHSC_01931 and SAOUHSC_02570, yielded an error message of computer language when (PS)2 attempted to model them. Attempted to report the problem to (PS)2 at chieh.bi91g@nctu.edu.tw, but no correction was made. The program could not find significant templates for other hypothetical proteins not listed.
Table 7. (PS)2 model data for hypothetical proteins.
| Protein | Template Structure | Template %ID | E-value |
| SAOUHSC_00010 | Rh50 in NH3 transport | 3B9W | 14.46 0.053 |
| SAOUHSC_00077 | NRPS Condensation Enzyme | 1L5A | 11.36 3 |
| SAOUHSC_00082 | BTRK decarboxylase | 2J66 | 28.79 2.20E-17 |
| SAOUHSC_00085 | Aquaporin-4 | 2D57 | 14.89 3.5 |
| SAOUHSC_00091 | GltPh transport protein | 2NWL | 13.46 0.024 |
| SAOUHSC_00136 | Multiple sugar binding transport ATP-binding protein | 2D62 | 31.12 4.70E-29 |
| SAOUHSC_00145 | 4'-phosphopantetheinyl transferase SFP-coenzyme A | 1QR0 | 21.12 4.60E-18 |
| SAOUHSC_00156 | Conserved Protein of Unknown Function | 2P0O | 27.53 5.50E-24 |
| SAOUHSC_00219 | NAD+-dependent alcohol dehydrogenase | 1RJW | 28.73 2.50E-11 |
| SAOUHSC_00307 | Sir2 homologue F159A mutant-ADP ribose complex | 1M2K | 16.55 9.80E-10 |
| SAOUHSC_00308 | Lipoate-protein ligase a | 1VQZ | 35.99 1.20E-28 |
| SAOUHSC_00328 | Complex III with bound cytochrome C | 3CX5 | 15.9 0.0051 |
| SAOUHSC_00548 | Family GT4 glycosyltransferase | 2JJM | 16.67 1.60E-06 |
| SAOUHSC_00751 | CHY zinc finger domain-containing protein 1 RING finger | 2DKT | 25.77 6.90E-06 |
| SAOUHSC_00766 | Glutamine PRPP amidotransferase | 1ECF | 15.64 0.024 |
| SAOUHSC_00972 | Methane monooxygenase regulatory protein | 1CKV | 23.33 8.4 |
| SAOUHSC_01291 | Conserved Protein of Unknown Function | 1RLK | 34.29 4.8 |
| SAOUHSC_01402 | Acetylcholine receptor pore | 1OED | 11.85 0.99 |
| SAOUHSC_01937 | S-adenosylmethionine decarboxylase | 1VR7 | 31.25 2.5 |
| SAOUHSC_02471 | Site-specific DNA nickase | 2EWF | 12.18 2.7 |
| SAOUHSC_02770 | Diaminopimelate epimerase | 2OTN | 15.18 5.90E-09 |
| SAOUHSC_02889 | GTPase | 1YRB | 28.57 3.5 |
| SAOUHSC_02901 | Yija protein | 1NIJ | 24.61 2.50E-10 |
| SAOUHSC_02911 | Isoluecyl-tRNA lysidine synthetase | 1WY5 | 12.24 0.81 |
| SAOUHSC_02934 | Human TPP1 | 2I46 | 17.61 1.1 |
3DLigandSite characterized the active site for hypothetical proteins. Figure 1 depicts the predicted active site with binding heterogens for the 12 of 22 proteins with the largest active sites. Table 8 lists predicted residues responsible for forming active sites and heterogens. There were insufficient homologous structures with ligands bound for other hypothetical proteins not listed in the table.
Figure 1.
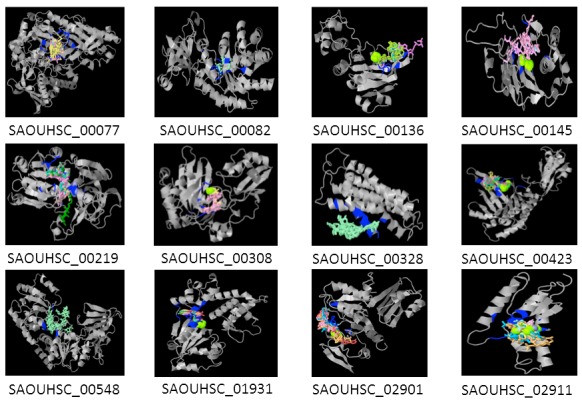
3DLigandSite Models for Proteins with the largest active site. Hypothetical proteins shown in grey with potential metallic heterogens shown as space fill and non-metallic heterogens as wireframe. Residues involved in bindings are blue.
Table 8. 3DLigandsite active site predictions.
| Protein | Predicted Binding Site | Heterogens |
| SAOUHSC_00077 | GLN140, SER263, SER264, SER265, ILE270, HIS278, LYS280, CYS326, ARG354,LYS355, PRO357, SER370, HIS424, GLN426, ASN427, LEU429, ARG443, ASP444 | STU, ADP, MG, AMP, FMM, ATP |
| SAOUHSC_00082 | MET49, GLU69, ARG138, HIS185, HIS187, SER190, GLY226, GLY227, GLY228,ILE229, GLU266, CYS267, GLY268, ARG269, PHE270, TYR373 | PLP |
| SAOUHSC_00091 | PHE340, PHE344, MET371, MET374 | HEM, MG, FAD, FE, CA, FE2, ZN |
| SAOUHSC_00136 | PHE11, VAL16, LYS35, SER36, GLY37, CYS38, GLY39, LYS40, SER41, THR42 | ADP, MG, CA, ATP |
| SAOUHSC_00145 | HIS43, ARG72, LYS74, LEU91, SER92, TYR93, ASP110, GLU149, LYS153 | COA, MG |
| SAOUHSC_00156 | PHE24, TYR133, PHE165 | FMN, MG, CU, ZN |
| SAOUHSC_00219 | CYS38, GLY39, SER40, HIS59, GLU60, GLU144, GLY168, CYS169, GLY170,SER171, ILE172, ASP192, ILE193, LYS197, SER212, SER235, SER236, THR241,ASN287 | CHD, NDP, NAP, NAD, ZN |
| SAOUHSC_00307 | CYS154, ARG184, CYS185, PRO186, LYS187, CYS188, ASP189, ALA190 | ZN |
| SAOUHSC_00308 | VAL80, ARG127, ASP129, LYS136, SER154, LEU156, VAL187 | ADP, MG, AMP, ATP |
| SAOUHSC_00328 | SER2, VAL4, ILE5, THR6, VAL7, ILE8, VAL9, VAL10, VAL12, GLN42, MET46,PHE49, VAL59 | HEA |
| SAOUHSC_00423 | PHE11, TYR39, GLY41, ALA42, GLY43, LYS44, SER45, THR46, LEU47, HIS199 | ADP, MG, CA, ATP |
| SAOUHSC_00455 | THR108, LYS111, LYS113 | GTP, ADP, MG, FAS, CA, ATP, ZN |
| SAOUHSC_00548 | LYS18, HIS246, ARG329, GLY406, PHE408, LEU410, ALA411 | F6P, ADP, G6P, G1P, PLP, GLC |
| SAOUHSC_00751 | ASN91, CYS94 | ZN |
| SAOUHSC_00766 | SER126, ASP193, ASP194 | ADP, MG, AMP, CA |
| SAOUHSC_01931 | GLY359, ILE360, GLY361, LYS362, SER363, HIS364, HIS489, PHE492, GLU496,PRO525, LEU526, LYS529 | ADP, MG, ATP |
| SAOUHSC_02471 | GLU1 | MG |
| SAOUHSC_02570 | MET15, VAL18, GLU20, ILE26, ILE52 | NI, ARA, ZN |
| SAOUHSC_02770 | GLU52 | CA |
| SAOUHSC_02901 | LEU10, GLY11, GLY12, GLY13, LYS14, THR15, THR16, GLU90, SER92, ASN154 | ADP, MG, ADX, ATP |
| SAOUHSC_02911 | HIS41, CYS43, CYS44, ALA45, PRO46, CYS47, SER48, TYR64, ALA66, SER68,ASN69, ARG79, MET135, ARG136, SER154 | GTP, MG, AMP, NAP, ATP, ZN,SAM, G6P, CA, NAD |
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) and Search Tool for Interactions of Chemicals (STITCH) predicted interactions with hypothetical proteins. Table 9 shows the top non-hypothetical protein interactions with the highest confidence from STRING. If a protein is not listed in the table, it did not have predicted functional partners or all predicted partners were other hypothetical proteins. Figure 2 illustrates the highest confidence interactions STITCH predicted with multiple proteins. Since the findings between STITCH and STRING were similar, if one non-hypothetical protein was predicted with highest confidence, it is listed in Table 9 but not shown in Figure 2.
Table 9. Top STRING predicted substrates.
| Protein | Substrate | Score |
| SAOUHSC_00077 | Ornithine cyclodeaminase | 0.842 |
| SAOUHSC_00082 | Ornithine cyclodeaminase | 0.876 |
| SAOUHSC_00085 | Acetoin reductase | 0.488 |
| SAOUHSC_00091 | Superoxide dismutase | 0.461 |
| SAOUHSC_00136 | Putative DNA-binding/iron metalloprotein/AP endonuclease | 0.488 |
| SAOUHSC_00145 | D-alanine-poly(phosphoribitol) ligase subunit 1 | 0.933 |
| SAOUHSC_00156 | N-acetylmuramic acid-6-phosphate etherase | 0.974 |
| SAOUHSC_00219 | PTS system protein | 0.968 |
| SAOUHSC_00307 | DNA-directed RNA polymerase subunit beta | 0.541 |
| SAOUHSC_00308 | Lipoyl synthase | 0.934 |
| SAOUHSC_00328 | mttA/Hcf106 family protein | 0.919 |
| SAOUHSC_00423 | ABC transporter permease | 0.999 |
| SAOUHSC_00455 | DNA polymerase III subunit delta | 0.939 |
| SAOUHSC_00548 | Capsular polysaccharide biosynthesis protein | 0.785 |
| SAOUHSC_00751 | UDP-N-acetylenolpyruvoylglucosamine reductase | 0.805 |
| SAOUHSC_00766 | Biotin synthase | 0.65 |
| SAOUHSC_00837 | Glycine cleavage system protein H | 0.657 |
| SAOUHSC_00972 | Glycosyl transferase | 0.859 |
| SAOUHSC_01402 | Cold shock protein | 0.498 |
| SAOUHSC_01851 | Catabolite control protein | 0.638 |
| SAOUHSC_01937 | Serine protease | 0.636 |
| SAOUHSC_02570 | Protein A (spA) | 0.762 |
| SAOUHSC_02770 | Peptide ABC transporter peptide-binding protein | 0.782 |
| SAOUHSC_02901 | Imidazole glycerol phosphate synthase subunit hisF | 0.441 |
| SAOUHSC_02911 | Ribonuclease HII (rnhB) | 0.762 |
| SAOUHSC_02934 | Betaine aldehyde dehydrogenase | 0.648 |
Figure 2.
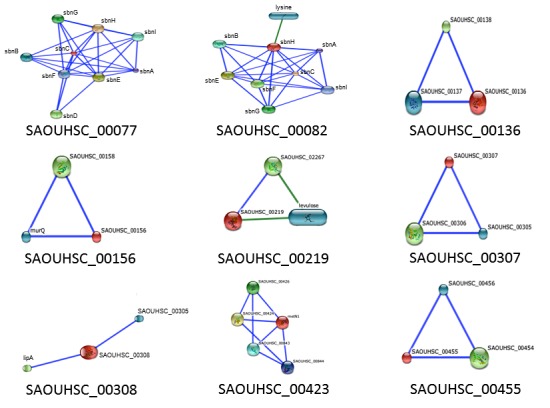
STITCH Chemical-Protein Interactions. Hypothetical proteins with more than one highest confidence binding partner (0.900) shown.
Cellular Location, Solubility, and Stability
PSortB predicted the cellular location of hypothetical proteins with results summarized in Table 10. PSortB was unable to determine cellular location for unlisted proteins.
Table 10. SOSUI results for transmembrane hypothetical proteins.
| Protein | Location | Localization Scores |
| SAOUHSC_00010 | cytoplasmic membrane | 10 |
| SAOUHSC_00077 | cytoplasmic membrane | 8.16 |
| SAOUHSC_00082 | cytoplasmic | 7.5 |
| SAOUHSC_00085 | cytoplasmic membrane | 10 |
| SAOUHSC_00091 | cytoplasmic membrane | 10 |
| SAOUHSC_00136 | cytoplasmic membrane | 8.78 |
| SAOUHSC_00145 | cytoplasmic | 7.5 |
| SAOUHSC_00156 | cytoplasmic | 7.5 |
| SAOUHSC_00219 | cytoplasmic | 9.67 |
| SAOUHSC_00238 | cytoplasmic membrane | 9.55 |
| SAOUHSC_00303 | extracellular | 8.91 |
| SAOUHSC_00307 | cytoplasmic | 7.5 |
| SAOUHSC_00308 | cytoplasmic | 9.97 |
| SAOUHSC_00328 | cytoplasmic membrane | 10 |
| SAOUHSC_00423 | cytoplasmic membrane | 8.78 |
| SAOUHSC_00455 | cytoplasmic | 7.5 |
| SAOUHSC_00548 | cytoplasmic | 7.5 |
| SAOUHSC_00751 | cytoplasmic | 7.5 |
| SAOUHSC_00766 | cytoplasmic | 7.5 |
| SAOUHSC_00837 | cytoplasmic membrane | 9.55 |
| SAOUHSC_00972 | cytoplasmic | 7.5 |
| SAOUHSC_01291 | cytoplasmic membrane | 9.55 |
| SAOUHSC_01402 | cytoplasmic membrane | 10 |
| SAOUHSC_01931 | cytoplasmic | 7.5 |
| SAOUHSC_01937 | cytoplasmic membrane | 9.55 |
| SAOUHSC_02471 | cytoplasmic | 7.5 |
| SAOUHSC_02889 | cytoplasmic | 7.5 |
| SAOUHSC_02901 | cytoplasmic | 7.5 |
| SAOUHSC_02911 | cytoplasmic | 7.5 |
| SAOUHSC_02934 | extracellular | 8.91 |
SOSUI calculates the average hydrophobicity and determines if the protein is soluble from it. If hydrophobicity exists, that portion of the protein is labeled as a transmembrane region. Table 11 shows the transmembrane regions of the eight proteins. SOSUI deemed all other proteins soluble.
Table 11. PSortB cellular location of hypothetical proteins.
| Locus Tag | N terminal | Transmembrane Region | C terminal | Type | Length | |
| SAOUHSC_00010 | 12 | QECIPTLLGYAGVGISFGIVASS | 34 | SECONDARY | 23 | |
| 40 | LEIVLLCLVIYAGAAQFIMCALF | 62 | PRIMARY | 23 | ||
| 70 | AIVLTVFIVNSRMFLLSMSLAPN | 92 | PRIMARY | 23 | ||
| 132 | HGLNITAYLFWAISCVAGALFGE | 154 | PRIMARY | 23 | ||
| 161 | TLGLDFAITAMFIFLAIAQFESI | 183 | SECONDARY | 23 | ||
| 197 | AVIVMMLSLSMFMPSYLAILIAA | 219 | PRIMARY | 23 | ||
| SAOUHSC_00085 | 15 | YFQIAYIVLMAITLCGFVICYGL | 37 | PRIMARY | 23 | |
| 56 | TIVISAIISIFVIILSIVPVIVL | 78 | PRIMARY | 23 | ||
| 93 | LIVLAIIALVLCNFVSAILWFVS | 115 | PRIMARY | 23 | ||
| 8 | KLLTLLLIGLAVFIQQSSVIAGV | 30 | PRIMARY | 23 | ||
| 33 | SIADFITLLILVYLLFFANHLLK | 55 | PRIMARY | 23 | ||
| SAOUHSC_00091 | 63 | FIILYTYRMIITLCLLFFDDLIF | 85 | PRIMARY | 23 | |
| 94 | STVKYAFVVIYFYLGMIIFKLGN | 116 | PRIMARY | 23 | ||
| 120 | VIVTSYIISSVTIGLFCIIAGLN | 142 | PRIMARY | 23 | ||
| 167 | YFAMTQIITLVLAYKYIHNYIFK | 189 | SECONDARY | 23 | ||
| 197 | LWSLTTTGSKTAFIILIVLAIYF | 219 | PRIMARY | 23 | ||
| 228 | NAVSVVSMSVIMLILLCFTFYNI | 250 | PRIMARY | 23 | ||
| 288 | SVVWINAISVIKYTLGFGVGLVD | 310 | SECONDARY | 23 | ||
| 333 | FAEWGILFGALFIIFMLYLLFEL | 355 | PRIMARY | 23 | ||
| 358 | FNISGKNVTAIVVMLTMLIYFLT | 380 | PRIMARY | 23 | ||
| 382 | SFNNSRYVAFILGIIVFIVQY | 402 | SECONDARY | 21 | ||
| SAOUHSC_00238 | 5 | IINIAYLYAIIWKLKRLQKIVTS | 27 | PRIMARY | 23 | |
| 2 | SFVITVIVVYVSSFWWMTPFITY | 24 | SECONDARY | 23 | ||
| 42 | QIYVMIIFFIAFCFISPVMFYQL | 64 | PRIMARY | 23 | ||
| 85 | FFSVLLFCAGVAFAFYVGFPMII | 107 | PRIMARY | 23 | ||
| 126 | KAYLIELIRWLFTFGLLFQLPIL | 148 | PRIMARY | 23 | ||
| SAOUHSC_00328 | 180 | IIAPPDLTLNILLTLPLILLFEF | 202 | PRIMARY | 23 | |
| SAOUHSC_01291 | 15 | KIEFLIGTFIIILVILGFKIMK | 36 | PRIMARY | 22 | |
| 24 | NINILAAMMIVLVIPIMISGILF | 46 | PRIMARY | 23 | ||
| SAOUHSC_01402 | 50 | NIDKTYIFFNIIFIDFYYYIYNV | 72 | SECONDARY | 23 | |
| 103 | FGFDEILFYTLYLLLILIVLYYL | 125 | PRIMARY | 23 | ||
| SAOUHSC_01851 | 9 | KYIVRYHLAFVFISSFSLNFS | 29 | PRIMARY | 21 |
Despite 24 proteins having cysteine resides that could form disulfide bonds; DISULFIND was unable to find potential disulfide bonding in any hypothetical protein evaluated here.
Conclusion:
Annotation of a genome does not stop after the sequence is published. We must update genomic annotations as new information on protein homology and structures are discovered. Since the annotation of over half of the S. aureus NCTC 8325 genome is as hypothetical, this study characterized 35 hypothetical proteins using bioinformatics tools and various databases for homology similarity comparisons, physiochemical characterization, domain identification, active site characterization, predicted protein-protein interactions, cellular location and stability. The examination revealed some hypothetical proteins with potentially virulent domains and protein-protein interactions including Oantigen, superoxide dismutase, siderophore synthesis, and bacterial ferric iron reductase. Other hypothetical proteins appear to metabolic or transport proteins including major facilitator superfamily, ABC transporters, GTPases, and Sadenosylmethionine decarboxylase. While this contributes to the current understanding of S. aureus, there is more work to do. More homology and structural information is needed in public repositories to be able to fully evaluate some hypothetical proteins, especially the smaller ones. This process will have to be repeated at regular intervals until the entire genome is properly annotated and should be done with all genomes as part of regular maintenance. Automation of this process would help ensure up-to-date databases. Until then, these data describing what is currently available for these 35 hypothetical proteins will contribute to the scientific understanding of S. aureus, aiding in the discovery of therapeutic targets.
Edited by P Kangueane
Citation: School et al. Bioinformation 12(3): 209-220(2016)
References
- 1. http://www.ncbinlmnihgov/books/NBK8448/
- 2.Stinear TP,, et al. Genome Biol Evol. 2014;6(2):366. doi: 10.1093/gbe/evu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. http://www.cdcgov/drugresistance/pdf/carb_national_strategypdf.
- 4.Casey JA, et al. Epidemiology and infection. 2013;141(06):1166. doi: 10.1017/S0950268812001872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee SS, Otto M. Clinical epidemiology. 2013;5:205. doi: 10.2147/CLEP.S37071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thurlow LR,, et al. Cell host & microbe. 2013;13(1):100. doi: 10.1016/j.chom.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bharat Siva Varma P,, et al. J Infect Public Health. 2015;8(6):526. doi: 10.1016/j.jiph.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Mohan R, Venugopal S. Bioinformation. 2012;8(15):722. doi: 10.6026/97320630008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shahbaaz M,, et al. Computational Biology and Chemistry. 2015;59:67. doi: 10.1016/j.compbiolchem.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Islam MS,, et al. Genomics Inform. 2015;13(2):53. doi: 10.5808/GI.2015.13.2.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gasteiger E,, et al. Methods Mol Biol. 1999;112:531. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 12.Sonnhammer EL,, et al. Proteins. 1997;28:405. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 13.Marchler-Bauer A,, et al. Nucleic Acids Res. 2015;43:D222. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen CC,, et al. BMC Bioinformatics. 2009;10:366. doi: 10.1186/1471-2105-10-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wass MN,, et al. NAR. 2010;38:W469. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szklarczyk D,, et al. Nucleic Acids Res. 2015;43:D447. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhn M,, et al. Nucleic Acids Res. 2014;42(D1):D401. [Google Scholar]
- 18.Yu NY,, et al. Bioinformatics. 2010;26(13):1608. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirokawa T, et al. Bioinformatics. 1998;14:378. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 20.Ceroni A, et al. Nucleic Acids Res. 2006;34:W177. doi: 10.1093/nar/gkl266. [DOI] [PMC free article] [PubMed] [Google Scholar]