

Abstract
A highly chemo-, diastereo- and enantioselective catalytic method that efficiently combines a silyl hydride, vinyl–B(pin) (pin = pinacolato) and (E)-1,2-disubstituted allylic phosphates is introduced. Reactions, best promoted by a Cu-based complex with a chiral sulfonate-containing N-heterocyclic carbene, are broadly applicable. Aryl-, heteroaryl-, alkenyl, alkynyl and alkyl-substituted allylic phosphates may thus be converted to the corresponding homoallylic boronates and then alcohols (after C–B bond oxidation) in 46–91% yield and in up to >98% SN2’:SN2 ratio, 96:4 diastereomeric ratio and 98:2 enantiomeric ratio. The reasons why an NHC–Cu catalyst is uniquely effective (vs. the corresponding phosphine systems) and the basis for different trends in stereoselectivity are provided with the aid of DFT calculations.
Keywords: boron, copper, enantioselective catalysis, enantioselective synthesis, multicomponent reactions
Graphical abstract

A desirable combination: A silyl hydride and vinyl–B(pin) (pin = pinacolato), both commercially available, may be merged with readily accessible (E)-1,2-disubstituted allylic phosphates to afford an assortment of homoallylic boronates efficiently and with selectivity. A sulfonate containing N-heterocyclic carbene ligand is found to be optimal.
Catalytic strategies for enantioselective preparation of organic molecules with a boron-substituted stereogenic carbon center are much sought after in organic chemistry.[1] Notable examples are enantioselective boron–hydride additions to alkenes with Rh- or Ir-based complexes[2] and related diboryl additions with Pt-, Pd- or carbohydrate-derived catalysts[3] (Scheme 1a). An alternative approach entails site- and enantioselective Cu–B(pin) (pin = pinacolato) addition to an olefin followed by in situ protonation (proto-boryl addition)[4] or allylic substitution (boron–allyl addition)[5] of the Cu–C bond; these transformations are typically promoted by a chiral Cu-based complex (Scheme 1a).
Scheme 1.

Related previous work, the key objectives of this study and the associated challenges. Abbreviations: pin = pinacolato; G, R = various functional groups; L = ligand.
One other disconnection would entail enantioselective Cu–H addition[6] to commercially available vinyl–B(pin) and an ensuing SN2’-selective allylic substitution involving a 1,2-disubstituted allylic phosphate (Scheme 1b). Products bearing an allylic C-substituted and a homoallylic (pin)B-substituted carbon stereogenic center would be obtained. With G = Me simple oxidation to a secondary homoallylic alcohol could result in diastereo- and enantiomerically enrich product expected from crotyl addition to acetaldehyde, a transformation that, to the best of our knowledge, remains without a catalytic enantioselective variant.[7] Nonetheless, the above plan might present several complications. A chiral catalyst must promote efficient, diastereo- and enantioselective Cu–H addition followed by allylic substitution in preference to two potentially competing routes. One pathway could involve reaction of Cu–alkoxide with vinyl–B(pin) (vs. a hydride reagent) to yield a vinyl–Cu complex, which might then react with an allylic phosphate;[8] alternatively, the Cu–H might react directly with the allyl electrophile.[9]
Here, we demonstrate that a sulfonate-containing chiral NHC–Cu complex can efficiently promote the general transformation in Scheme 1b with high chemo-, SN2’-, diastereo- and enantioselectivity.[10] While this manuscript was being prepared, reactions involving (E)-1,2-disubstituted alkenyl–B(pin) substrates and allylphosphate with bis-phosphine–Cu catalysts were disclosed (to generate products with a single stereogenic center);[11] processes were highly enantioselective but the only reported case with an (E)-1,2-disubstituted allylic phosphate furnished the desired product in 78:22 diastereomeric ratio (d.r.) and 73:27 enantiomeric ratio (e.r.), hinting at the difficulty of the proposed class of reactions.
We started by gauging the effectiveness of various chiral bis-phosphine ligands to promote the reaction involving vinyl–B(pin), (E)-1,2-disubstiuted allylic phosphate 1a with polymethylhydrosiloxane (PMHS).[12] None afforded 2a as the major product however (Scheme 2). In three cases, the product from SN2 mode of addition (3a) was the major component with moderate selectivity (phos-1, phos-2 and phos-5), and in two instances it was formed almost exclusively (phos-4 and phos-6). With every bis-phosphine ligand, 2a and 3a were generated in low d.r. and e.r. This was somewhat surprising since chiral phosphines – and some of these exact ligands – have been shown to be optimal in several transformations that begin with enantioselective Cu–H addition to an alkene (phos-2,[13] phos-3,[11] and phos-6[6f–h]). We took these findings as an indication that the desired sequence of reactions demands a distinct set of catalysts.
Scheme 2.

Identification of an effective chiral catalyst. Conv. and d.r. determined by analysis of unpurified product mixtures by 1H NMR analysis. Enantioselectivity determined by HPLC analysis. Yields are for purified products. Abbreviations: phos = phosphine ligand; imid = imidazolinium salt; nd = not determined. See the Supporting Information for details.
Results were more encouraging with N-heterocyclic carbene (NHC) systems (Scheme 2). With imid-1[14] as NHC precursor, 2a was the major component of the product mixture (2a:3a, 69:31) but stereoselectivity remained low. There was further improvement with the NHC–Cu complex derived from sulfonate-containing imid-2,[15] which afforded 2a exclusively and in appreciable d.r. and e.r. [13:87 and 80:20 (for the major diastereomer), respectively]. Another unexpected observation was that the catalyst derived from imid-3,[16] where the Mes unit (2,4,6-trimethylphenyl) is replaced by a 3,5-(2,4,6-triisopropoylphenyl) group, high 2a:3a ratio persisted (94:6) with stereoselectivity improving greatly as well (94:6 d.r. and e.r. for major diastereomer of 2a). Hence, there was a mechanistically suggestive reversal in diastereoselectivity along with substantially lower enantioselectivity with imid-2 (vs. imid-3; see below for further discussion).
Many aryl-substituted allylic phosphates could be converted to isolable homoallylic boronates, which were converted to the corresponding alcohols after C–B bond oxidation (Scheme 3). Reactions were performed at ambient temperature with 5.5 mol % imid–3 and 5.0 mol % CuCl along with three equivalents of inexpensive PMHS and with LiOtBu as base (identified as optimal based with further screening);[17] only a small excess of vinyl–B(pin) sufficed (1.1 equiv.). γ-Addition products (SN2’ pathway) were uniformly high (2:3, 86:14 to >98:2) as was the diastereo- and enantioselectivity with which 2a-2l were formed (90:10–96:4 d.r., 94:6–98:2 e.r.). Pure 2a-2l were obtained in 60–84% yield after silica gel chromatography. Transformations proceeded with similarly high efficiency and selectivity regardless of whether the aryl group within the allylic phosphate was sterically hindered (cf., 2c-f), electron withdrawing (cf. 2k-l) or electron donating (cf. 2d, 2h). The lower SN2’ selectivity with 2l may be attributed to direct alkylation of the exceptionally electrophilic p-nitrophenyl-substituted allylphosphate (vs. Cu–alkene complexation and allyl transfer; see below for further analysis).
Scheme 3.

Reactions with aryl-substituted allylic phosphates. Same conditions as in Scheme 2, except that LiOtBu was used as base. Conv. and d.r. determined by analysis of unpurified product mixtures by 1H NMR analysis. Enantioselectivity determined by HPLC analysis. Yields are for purified products. See the Supporting Information for details.
Allyl electrophiles containing a heterocyclic substituent such as a pyridyl (4, Scheme 4) or a benzothiophene group can be used (5). However, SN2’:SN2 and diastereoselectivities were somewhat lower in such instances and the final products at times contained a small amount of impurity from the SN2 addition (cf. 4, Scheme 4). Similar results were obtained with a dienylphosphate (cf. 6, Scheme 4). The transformation with the corresponding enynylphosphate (cf. 7) was more SN2’- (>98% vs. 87% for 6) and enantioselective (97:3 vs. 92:8 e.r. for 6). In the case of 6 none of the product from SN2” mode of reaction was detected, and the lower yield for 7 (46%) might be due to competitive Cu–H addition to the alkynyl group.[18]
Scheme 4.

Reactions with heteroaryl-, alkenyl- and alkynyl-substituted allylic phosphates. Same conditions as in Scheme 3, except 7.5 mol % imid-3 and 7.0 mol % CuCl were used for 4 and 6. Conv. and d.r. determined by analysis of unpurified product mixtures by 1H NMR analysis. Enantioselectivity determined by HPLC analysis. Yields are for purified products. See the Supporting Information for details.
Alkyl-substituted, and particularly Me-substituted allylic phosphates, are suitable substrates (Scheme 5). As highlighted by synthesis of 8 and 9 (Scheme 5), while somewhat less enantioselective compared to when aryl-substituted allylic phosphates are utilized (Scheme 2), reaction with the larger cyclohexyl-substituted allylic phosphate was efficient with 7.0 mol % catalyst loading. In both cases SN2’selectivity was exceptional (>98%), diastereoselectivities were high 92:8–93:7 d.r. and pure products were obtained in 91% and 79% yield, respectively.
Scheme 5.

Reactions with alkyl-substituted allylic phosphates. Same conditions as in Scheme 3, except 7.5 mol % imid-3 and 7.0 mol % CuCl was used for 9. Conv. and d.r. determined by analysis of unpurified product mixtures by 1H NMR analysis. Enantioselectivity determined by HPLC analysis. Yields are for purified products. See the Supporting Information for details.
Of special value are the transformations involving Me-substituted allylic phosphate 1b, which, when performed on 5 mmol scale and with 2.0 mol % catalyst loading, afforded 10 in 55% yield (volatile compound), >98:2 SN2’:SN2, 92:8 d.r. and 93:7 e.r. after purification (Scheme 5). This is a valuable fragment that has been used in a total synthesis of a biologically active analog of natural product chondramide C.[19] Previously, however, preparation of enantiomerically pure 10 entailed the use of Brown’s chiral auxiliary,[20] necessitating somewhat forcing conditions along with the use of stoichiometric amounts of an exceptionally strong base (nBuLi/KOtBu to give nBuK); there is also the need for an equivalent of an organoboron reagent. Another functionalization procedure entails conversion of the homoallylic boronate formed by the reaction of allylic phosphate 1c to the corresponding 2-furyl product [Eq. (1)].[21]
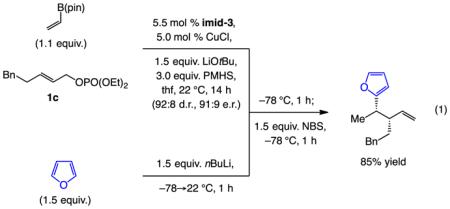 |
For insight regarding the unique effectiveness of imid-3-derived catalyst and the selectivity differences with imid-2, a series of DFT calculations were carried out. These studies indicate[17] that the most favored mode of Cu−H addition to vinyl–B(pin) with imid-3 probably arises from coordination of a pinacolato oxygen atom to the alkali metal counter-ion (I, Figure 1a).[22] The corresponding mode of reaction with the NHC–Cu complex derived from imid-2 (Scheme 2) suffers from steric repulsion between an o-methyl substituent of the ligand (II, Figure 1a), resulting in diminished e.r.
Figure 1.

Regarding the origin of diastereo- and enantioselectivity. Computations have been performed at the MN12SX/Def2TZVPPthf(SMD) level after geometry optimization performed with the ONIOM method M06L/Def2SVP:UFFthf(PCM); Ar = 2,6-(iPr)2C6H3. See the Supporting Information for details.
Allylic substitution with imid-3 is most favorable with the allyl electrophile approaching such that chelation with the more Lewis acidic Li cation is most effective and there is less steric repulsion between its substituent (Ph) and the NHC’s sizeable N-aryl moieties (III, Figure 1b).[23] Another consequence of the sulfonate/Li/phosphate chelation is the exceptional SN2’-selectivity; otherwise, as is the case with the transformations involving phosphine ligands, the linear products are generated preferentially (see 3a, Scheme 2). In IV and V, arising from the imid-2-based Cu complex (Figure 1c), the allyl electrophile is forced to coordinate with its CH2OPO(OEt)2 moiety pointing away from the large mesityl group.[23] A different diastereomer is preferred compared to when imid-3 is used because the reaction proceeds via IV with the minor isomer being formed with the opposite sense of enantioselectivity (via V).
These investigations put forth a highly diastereo- and enantioselective protocol for direct catalytic access to an assortment of valuable homoallylic alcohols typically viewed as products of crotyl-type additions to acetaldehyde. By providing an efficient, chemo-, SN2’-, diastereo- and enantioselective method for accessing otherwise difficult-to-prepare and readily alterable homoallylic boronate compounds, we provide further evidence regarding the importance of sulfonate-containing chiral NHC ligands. These Cu-based complexes have formerly proven optimal in catalyzing enantioselective allylic substitution reactions[24] and conjugate addition processes[15,25] with C-based nucleophiles as well as Cu–B(pin) additions to alkenes[26] and allenes;[27] this is however the first time that a member of this catalyst class has emerged as the most effective for enantioselective Cu–H additions to an alkene.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (GM-47480) and the National Science Foundation (CHE-1362763).
Footnotes
Dedicated to Professor Samuel J. Danishefsky
References & Footnotes
- [1].a) Hartmann E, Vyas DJ, Oestreich M. Chem. Commun. 2011;47:7917–7932. doi: 10.1039/c1cc10528k. [DOI] [PubMed] [Google Scholar]; b) Takaya J, Iwasawa N. ACS Cat. 2012;2:1993–2006. [Google Scholar]
- [2].Carroll A-M, O’Sullivan TP, Guiry PJ. Adv. Synth. Catal. 2005;347:609–631. [Google Scholar]
- [3].a) Burks HE, Morken JP. Chem. Commun. 2007:4717–4725. doi: 10.1039/b707779c. [DOI] [PubMed] [Google Scholar]; b) Ref. 1b. [Google Scholar]; c) Coombs JR, Morken JP. Angew. Chem. Int. Ed. 2016;55:2636–2649. doi: 10.1002/anie.201507151. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Fang L, Yuan L, Haeffner F, Morken JP. J. Am. Chem. Soc. 2016;138:2508–2511. doi: 10.1021/jacs.5b13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234–18235. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Corberán R, Mszar NW, Hoveyda AH. Angew. Chem. Int. Ed. 2011;50:7079–7082. doi: 10.1002/anie.201102398. [DOI] [PubMed] [Google Scholar]; d) Meng F, Jang H, Hoveyda AH. Chem. Eur. J. 2013;19:3204–3214. doi: 10.1002/chem.201203803. [DOI] [PubMed] [Google Scholar]
- [5].Jia T, Cao TP, Wang B, Lou Y, Yin X, Wang M, Liao J. J. J. Am. Chem. Soc. 2015;137:13760–13763. doi: 10.1021/jacs.5b09146. [DOI] [PubMed] [Google Scholar]
- [6].a) Noh D, Chea H, Ju J, Yun J. Angew. Chem. Int. Ed. 2009;48:6062–6064. doi: 10.1002/anie.200902015. For representative studies regarding catalytic processes that commence with an enantioselective Cu–H addition to an alkene, see: [DOI] [PubMed] [Google Scholar]; b) Miki Y, Hirano K, Satoh T, Miura M. Angew. Chem. Int. Ed. 2013;52:10830–10834. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]; c) Miki Y, Hirano K, Satoh T, Miura M. Org. Lett. 2014;16:1498–1501. doi: 10.1021/ol5003219. [DOI] [PubMed] [Google Scholar]; d) Nishikawa D, Hirano K, Miura M. J. Am. Chem. Soc. 2015;137:15620–15623. doi: 10.1021/jacs.5b09773. [DOI] [PubMed] [Google Scholar]; e) Pirnot MT, Wang Y-M, Buchwald SL. Angew. Chem. Int. Ed. 2016;55:48–57. doi: 10.1002/anie.201507594. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang Y-M, Buchwald SL. J. Am. Chem. Soc. 2016;138:5024–5027. doi: 10.1021/jacs.6b02527. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Bandar JS, Ascic E, Buchwald SL. J. Am. Chem. Soc. 2016;138:5821–5824. doi: 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Yang Y, Perry IB, Lu G, Liu P, Buchwald SL. Science. 2016;353:144–150. doi: 10.1126/science.aaf7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Kim IS, Han SB, Krische MJ. J. Am. Chem. Soc. 2009;131:2514–2520. doi: 10.1021/ja808857w. For reports regarding related types of catalytic enantioselective additions to other types of aldehydes, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao X, Townsend IA, Krische MJ. J. Org. Chem. 2011;76:2350–2354. doi: 10.1021/jo200068q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McIntruff EL, Yamaguchi E, Krische MJ. J. Am. Chem. Soc. 2012;134:20628–20631. doi: 10.1021/ja311208a. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zbeig JR, Yamaguchi E, McIntruff EL, Krische MJ. Science. 2012;336:324–327. doi: 10.1126/science.1219274. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Liang T, Zhang W, Chen T-Y, Nguyen KD, Krische MJ. J. Am. Chem. Soc. 2015;137:13066–13071. doi: 10.1021/jacs.5b08019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Gao F, Carr JL, Hoveyda AH. Angew. Chem. Int. Ed. 2012;51:6613–6617. doi: 10.1002/anie.201202856. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao F, Carr JL, Hoveyda AH. J. Am. Chem. Soc. 2014;136:2149–2161. doi: 10.1021/ja4126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nguyen TNT, Thiel NO, Pape F, Teichert JF. Org. Lett. 2016;18:2455–2458. doi: 10.1021/acs.orglett.6b00941. [DOI] [PubMed] [Google Scholar]
- [10].a) Kim J, Park S, Park J, Cho SH. Angew. Chem. Int. Ed. 2016;55:1498–1501. doi: 10.1002/anie.201509840. For non-diastereo- and non-enantioselective catalytic methods for synthesis of similar types of products through reaction of 1,1-diborylalkanes and allylic chlorides, see: [DOI] [PubMed] [Google Scholar]; b) Zhang Z-Q, Zhang B, Lu X, Liu J-H, Liu X-Y, Xiao B, Fu Y. Org. Lett. 2016;18:952–955. doi: 10.1021/acs.orglett.5b03692. [DOI] [PubMed] [Google Scholar]
- [11].Han JT, Jang WJ, Kim N, Yun J. J. Am. Chem. Soc. DOI: 10.1021/jacs.6b11229. [Google Scholar]
- [12].Senapati KK. Synlett. 2005:1960–1961. [Google Scholar]
- [13].a) Zhu S, Niljianskul N, Buchwald SL. J. Am. Chem. Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu S, Buchwald SL. J. Am. Chem. Soc. 2014;136:15913–15916. doi: 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ref. 6d. [Google Scholar]
- [14].Clavier H, Coutable L, Toupet L, Guillemin J-C, Mauduit M. J. Organomet. Chem. 2005;690:5237–5254. [Google Scholar]
- [15].Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem. Int. Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]
- [16].Jung B, Hoveyda AH. J. Am. Chem. Soc. 2012;134:1490–1493. doi: 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. See the Supporting Information for details. [Google Scholar]
- [18].a) Semba K, Fujihara T, Xu T, Terao J, Tsuji Y. Adv. Synth. Catal. 2012;354:1542–1550. For examples of catalytic processes involving Cu–H addition to an alkyne, see: [Google Scholar]; b) Shi S-L, Buchwald SL. Nature Chem. 2015;7:38–44. doi: 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Uehling MR, Suess AM, Lalic G. J. Am. Chem. Soc. 2015;137:1424–1427. doi: 10.1021/ja5124368. [DOI] [PubMed] [Google Scholar]
- [19].a) Tannert R, Milroy L-G, Ellinger B, Hu T-S, Arndt H-D, Waldmann H. J. Am. Chem. Soc. 2010;132:3063–3077. doi: 10.1021/ja9095126. [DOI] [PubMed] [Google Scholar]; b) Hassfeld J, Eggert U, Kalesse M. Synthesis. 2005:1183–1199. For another application to complex molecule synthesis involving fragment 10, see: [Google Scholar]
- [20].a) Brown HC, Bhat K. J. Am. Chem. Soc. 1986;108:5919–5923. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]; b) Chen JL–Y, Scott HK, Hesse MJ, Willis CL, Aggarwal VK. J. Am. Chem. Soc. 2013;135:5316–5319. doi: 10.1021/ja401564z. For a more recent chiral auxiliary based approach, see: [DOI] [PubMed] [Google Scholar]
- [21].Bonet A, Odachowski M, Leonori D, Essafi S, Aggarwal VK. Nature Chem. 2014;6:584–589. doi: 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]
- [22].a) Yamamoto H, Futatsugi K. Angew. Chem. Int. Ed. 2005;44:1924–1942. doi: 10.1002/anie.200460394. For representative reports where coordination of a Lewis acid to a B(pin) moiety has been suggested to play a key role, see: [DOI] [PubMed] [Google Scholar]; b) Rauniyar V, Zhai H, Hall DG. J. Am. Chem. Soc. 2008;130:8481–8490. doi: 10.1021/ja8016076. [DOI] [PubMed] [Google Scholar]; c) Barnett DS, Moquist PN, Schaus SE. Angew. Chem. Int. Ed. 2009;48:8679–8682. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang H, Jain P, Antilla JC, Houk KN. J. Org. Chem. 2013;78:1208–1215. doi: 10.1021/jo302787m. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) van der Mei FW, Miyamoto H, Silverio DL, Hoveyda AH. Angew. Chem. Int. Ed. 2016;55:4701–4706. doi: 10.1002/anie.201600546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shi Y, Jung B, Torker S, Hoveyda AH. J. Am. Chem. Soc. 2015;137:8948–8964. doi: 10.1021/jacs.5b05805. A similar mode of reaction was recently proposed (based on DFT calculations) for enantioselective allylic substitutions reactions involving propargylcopper intermediates and the same class of allylic phosphates. See: [DOI] [PubMed] [Google Scholar]
- [24].Shi Y, Hoveyda AH. Angew. Chem. Int. Ed. 2016;55:3455–3458. doi: 10.1002/anie.201600309. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Peese KM, Gin DY. Chem. Eur. J. 2008;14:1654–1665. doi: 10.1002/chem.200701290. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Slutskyy Y, Jamison CR, Lackner GL, Müller DS, Dieskau AP, Untiedt NL, Overman LE. J. Org. Chem. 2016;81:7029–7035. doi: 10.1021/acs.joc.6b00697. [DOI] [PubMed] [Google Scholar]
- [26].Meng F, Jang H, Hoveyda AH. Chem. Eur. J. 2013;19:3204–3214. doi: 10.1002/chem.201203803. and references cited therein. [DOI] [PubMed] [Google Scholar]
- [27].Jang H, Jung B, Hoveyda AH. Org. Lett. 2014;16:4658–4661. doi: 10.1021/ol5022417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.