

Abstract
Thalamic relay neurons have well-characterized dual firing modes: bursting and tonic spiking. Studies in brain slices have led to a model in which rhythmic synchronized spiking (phasic firing) in a population of relay neurons leads to hyper-synchronous oscillatory cortico-thalamo-cortical rhythms that result in absence seizures. This model suggests that blocking thalamocortical phasic firing would treat absence seizures. However, recent in vivo studies in anesthetized animals have questioned this simple model. Here we resolve this issue by developing a real-time mode-switching approach to drive thalamocortical neurons into or out of a phasic firing mode in two freely-behaving genetic rodent models of absence epilepsy. Toggling between phasic and tonic firing in thalamocortical neurons launched and aborted absence seizures, respectively. Thus a synchronous thalamocortical phasic firing state is required for absence seizures and switching to tonic firing rapidly halts absences. This approach should be useful for modulating other networks that have mode-dependent behaviors.
eTOC blurb
Here Sorokin, J., et al. investigate the roles of phasic and tonic firing modes of thalamocortical relay cells in absence epilepsy, and discover that unilaterally toggling TC phasic firing initiates bilateral absence seizures, while switching to tonic aborts seizures in real-time.
Reciprocal communication between thalamus and cortex is critical to normal sensory experience (Steriade 2000; Steriade et al. 1993a; Contreras & Steriade 1995), and disorders in these circuits are associated with seizures (Huguenard 1999; Beenhakker & Huguenard 2009; Kostopoulos 2000) and altered cognitive processing (Buzsáki & Watson 2012). The firing modes of individual thalamocortical (TC) and corticothalamic (CT) neurons, which can fluctuate between burst and tonic firing, can alter cortico-thalamo-cortical (CTC) function (Steriade et al. 1993a; Steriade 2000). One form of epilepsy associated with changes in firing mode is generalized absence epilepsy. Genetic animal models of this disorder display stereotyped bilaterally synchronous spike-wave discharge (SWD) electrographic seizure activity and episodes of behavioral arrest (Danober et al. 1998; Coenen et al. 1992; Marescaux et al. 1992). SWDs, in both human patients and animal models, result from abnormally synchronous oscillations in CTC networks (for review, see (Danober et al. 1998; Beenhakker & Huguenard 2009)). Within the CTC circuit, the necessity of TC versus CT output and firing mode in generating and/or maintaining global epileptic activity has been debated (Meeren, Hanneke; Lopes da Silva 2005; Polack & Charpier 2006; Huguenard 1999). One current working model is that CT output is sufficient to initiate and maintain SWDs, and that active TC phasic output (see experimental procedures) is not necessary (Polack et al. 2007; Polack & Charpier 2006; Pinault 2003). This conclusion, obtained by multiple research groups, is based in part on the findings that (1) intra-cortical infusions of tetrodotoxin (TTX), lidocaine or ethosuximide can effectively suppress SWDs in epileptic rats (Polack et al. 2009; Manning et al. 2004; Sitnikova & Van Luijtelaar 2004), while intra-thalamic delivery of these compounds is less effective(Richards et al. 2003; Polack et al. 2009), and (2) TC neurons in epileptic rats and cats are silent or only fire sparse single spikes during SWDs due to the shunting output of GABAergic reticular thalamic (RT) neurons (Timofeev et al. 1998; Timofeev & Steriade 2004; Steriade & Contreras 1995; Pinault et al. 1998; Pinault 2003; Polack & Charpier 2006).
An alternate view is that phasic TC firing promoted by T-current dependent post-inhibitory rebound (PIR) bursts (Jahnsen & Llinás 1984; Coulter et al. 1989) is required for SWDs. This view is supported by the observation that lesions of the RT, which normally drives TC PIR bursts (for review see (Huguenard & McCormick 2007)), reduce both SWDs and high-voltage neocortical spindles (Avanzini et al. 1993; Buzsaki et al. 1988). Further, many in vitro studies using rodent thalamic slices (Huguenard & Prince 1994; Huguenard & McCormick 1992; Sohal et al. 2000) strongly argue that the intra-thalamic RT-TC loop undergoes prominent seizure related oscillatory activity with burst firing in both RT and TC cells. In contrast with these in vitro studies, researchers have surprisingly found in vivo that RT output tends to shunt TC activity rather than promote oscillatory PIR bursts (Steriade & Contreras 1995). However, these in vivo findings were obtained in animals under anesthesia, which can alter CTC firing dynamics.
While the specific contribution of TC phasic firing has not been shown, previous studies have shown that the thalamus at large plays a role in SWDs. For instance, pharmacological manipulations such as intra-thalamic infusion of GABA agonists into Genetic Absence Epilepsy Rats from Strasbourg (GAERS) promoted SWD occurrence, while KCl, GABA and glutamate antagonists prevented SWDs (Paz et al. 2007; Cope et al. 2009; Danober et al. 1998; Marescaux et al. 1992). Further, lesions to certain thalamic nuclei affect SWDs of the same hemisphere (Avanzini et al. 1993; Vergnes & Marescaux 1992), and lesions and pharmacological manipulations of the basal ganglia and neuromodulator pathways affect SWDs (Danober et al. 1998; Danober et al. 1993). Nonetheless, these results stem from non-specific manipulations that confirm only (1) the necessity of a fully intact CTC network for SWD expression, and (2) that modulatory pathways, which affect diffuse brain regions and cognitive states, influence absence seizures. Thus, these studies do not address the specific roles of various pathways and firing states in the CTC network for SWDs. These controversies lead to three questions to be addressed in this study, as follows:
Why is it important to determine whether the thalamus is actively involved in SWDs? If the thalamus simply follows cortically generated SWDs, then there is little point in further studying thalamic channelopathies or developing novel antiepileptic therapies that target TC firing. On the other hand, if phasic firing in TC cells does contribute to SWD generation, then therapies that target phasic firing may have powerful anti-absence effects. Given that phasic TC output is mediated by Cav3.1 T-channels, and that novel, selective T-type channel blockers (compared to the non-selective blocker ethosuximide (Gomora et al. 2001)) are under development (Dreyfus et al. 2010), then those that specifically target Cav3.1 may be legitimate treatments for absence epilepsy and potentially other disorders involving aberrant TC phasic firing, perhaps with fewer side effects.
What are potential explanations for opposing findings? Previous studies that were performed under anesthesia, neuroleptanalgesia, or in brain slices, which allow for stable high-quality intracellular recordings, have yielded important insights on intrinsic membrane properties and subthreshold synaptic activities from epileptic cortical and thalamic (TC and RT) neurons that can not be obtained with extracellular recordings. However, such preparations affect CTC network dynamics. Moreover, given the adjacency and reciprocity of TC and CT projection pathways (Jones 2002; Adams 1997), it is challenging to specifically isolate the contributions of the thalamus and the cortex via techniques used in other previous studies (lesions, pharmacology, electrical stimulations), as these can indirectly affect neighboring networks.
How can the opposition be resolved? Recent advances in optogenetic tools now enable cell-type-specific manipulations of firing mode in real-time, positioning us to resolve these long-standing issues regarding the necessity of active TC phasic output for the maintenance of naturally occurring absence seizures during free behavior.
Here we demonstrate that TC neurons rhythmically fire synchronized, phasic clusters of action potentials that are time-locked with natural SWDs in freely behaving mice and rats, and that such TC phasic output is necessary to maintain SWDs and behavioral absences. We first used electrophysiology to reveal the endogenous properties of TC neurons in behaving rodents. Then we applied novel optogenetic approaches to selectively manipulate the unilateral TC component of the CTC circuit to determine if TC phasic firing alone is sufficient to initiate and necessary to maintain generalized SWDs. We manipulated the firing modes of TC neurons in the ventrobasal (VB) complex—the primary somatosensory relay center (Chmielowska et al. 1989)—in healthy mice and rats and in two genetic models of absence epilepsy: Stargazer (STG) mice (Lacey et al. 2012; Noebels et al. 1990) and Wistar Albino Glaxo rats from Rijswijk (WAGRij) (Coenen et al. 1992; Sarkisova & van Luijtelaar 2011).
RESULTS
Unilaterally manipulating TC output is sufficient to alter bilateral cortical states in non-epileptic rats
Normal oscillatory output from the thalamus, which is believed to recruit ipsilateral cortical activity (Steriade et al. 1993a), is primarily driven by RT-mediated PIR bursts in TC neurons (Steriade et al. 1993a; Cox et al. 1997). To test whether unilateral TC oscillatory output can drive bilateral cortical rhythms, we expressed an inhibitory opsin, halorhodopsin (eNpHR3.0) (Gradinaru et al. 2008) unilaterally in excitatory VB TC neurons in non-epileptic rats. In vitro photoinhibition of eNpHR–expressing TC neurons in acute brain slices with brief 594nm pulses at various frequencies (see experimental procedures) reliably drove robust PIR bursts (Jahnsen & Llinás 1984) with ~25ms latencies (Fig. 1a,c) mediated by T-type calcium channels (Coulter et al. 1989). Our in vitro experiments documented unambiguous single-cell, multi-spike PIR bursts after each and every 594nm pulse within the pulse trains (Fig. 1a).
Figure 1. 12 Hz train of 594nm light drives sleep-like spindles in normal rats.
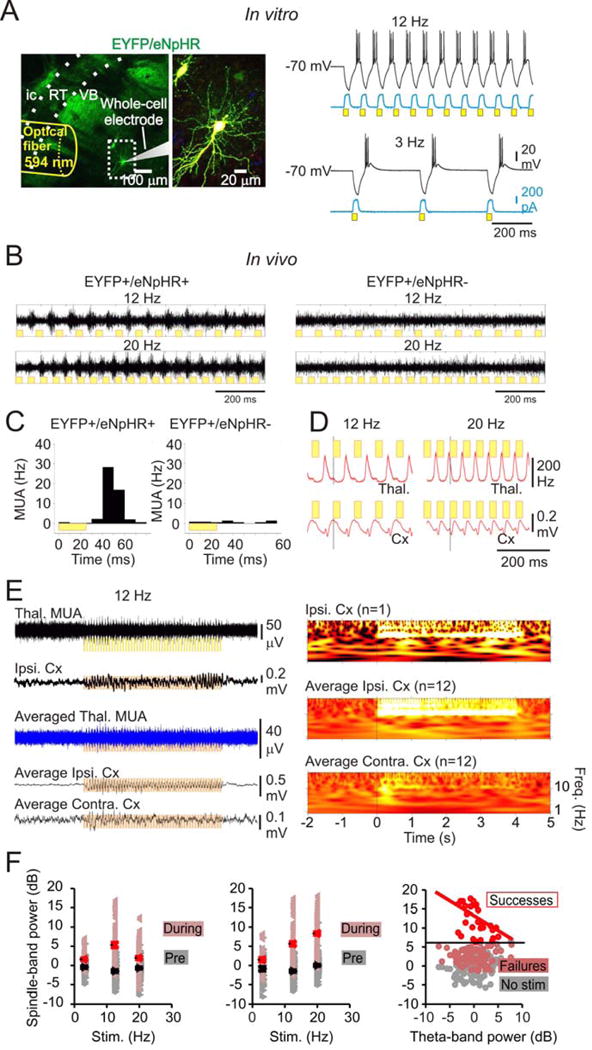
(a) In vitro 594nm trains induced eNpHR currents (blue) and rhythmic hyperpolarizations (black) followed by PIR bursts in TC neurons (right) expressing eNpHR-EYFP (left, image of recorded slice showing eYFP fluorescence in VB). (b) in vivo 12 Hz 594nm trains delivered via optrodes induced synchronized oscillatory TC firing in animals expressing EYFP-eNpHR (left) but not in controls (right). (c) Quantification of multi-unit activity (MUA) following the light pulse in eNpHR+ and eNpHR- TC neurons in vivo. (d) Superimposed average thalamic MUA rate and averaged cortical ECoG during 594nm pulses. (e) Representative single and average traces (left) and spectrograms (right) showing in vivo 594nm-induced TC clusters drive 12 Hz cortical oscillations. (f) 594nm trains fail to produce cortical oscillations during theta rhythm. Left: Ipsilateral cortical spindle power (dark red triangles) induced by thalamic optical stimulation. Bright red triangles: mean + SE. Grey: pre-stim cortical spindle power. Black triangles: mean + SE. Middle: Contralateral cortical spindle power (dark red triangles) induced by thalamic optical stimulation. Right: 12 Hz light train is more likely to evoke strong cortical spindles (bright red circles) in the absence of cortical theta activity. After eliminating stimuli not inducing spindle power > 2 S.D above the mean power (dark red circles), the evoked spindle power and cortical theta power are negatively correlated (bright red line). Grey: power in the spindle band immediately prior to the stimulus. S.D. ~3dB. Pearson’s R2 = 0.22 for the linear fit. p = 0.008, 10 trials, 3 animals.
Our in vivo results were consistent with this finding, where we recorded multi-unit (MU) activity with chronic optrode devices (Yizhar et al. 2011) implanted in the right VB of adult non-epileptic rats and mice along with electrocorticogram (ECoG) electrodes in ipsilateral and contralateral cortical hemispheres, as described in (Paz et al. 2013) (see experimental procedures). We found that unilaterally-delivered 594nm pulse trains at various frequencies (see Fig. 1f) robustly drove phasic action potentials in a local population of TC cells (that we will refer to as clustered spiking for simplicity, see experimental procedures). Strikingly, these pulses drove 12 Hz ECoG oscillations in both hemispheres (Fig. 1b–e), suggesting that unilateral TC clustered spiking can evoke bilateral thalamo-cortical network responses independent of the stimulation frequency (Fig. 1f). Although we did not isolate individual single units from our recordings, our results clearly show that 594nm pulses organize MU TC activity into high-frequency clusters with similar post-light latencies to in vitro T-current-mediated PIR bursts (~25ms; see Fig. 1a,b, Fig. S1a). In some cases just a few light pulses (<10) initiated ECoG oscillations that outlasted the optical train, indicating that the oscillation, once initiated in thalamus, became fully established in the CTC circuit. The effect of evoking phasic TC output on cortical oscillations depended on the behavioral state of the animal: in awake animals during pronounced cortical theta rhythm (5–12 Hz), which is associated with attention, exploration, and learning (Buzsáki & Moser 2013; Hasselmo et al. 2002), photoinhibition of TC cells still produced oscillatory TC clustered spiking, yet failed to induce cortical oscillations (Fig. 1f).
These results establish that coordinated rhythmic firing of a group of TC cells, even unilaterally, can entrain a global network CTC oscillator.
TC firing mode switches from tonic to phasic during spontaneous SWDs
To quantify innate TC firing behavior during spontaneous SWDs, we recorded ECoG (Fig. 2a, top) and MU activity from the VB (Fig. 2a, bottom) in epileptic WAGRij rats. All in vivo recordings were obtained from animals in states of quiet wakefulness. These rats displayed highly stereotyped seizures, with prominent SWDs in the ECoG and robust, rhythmic TC clustered spikes time-locked to the ECoG (Fig. 2a,b), which were infrequent and disorganized during inter-ictal periods. Interestingly, the overall rate of TC firing and the occurrence of clusters increased from pre-ictal to ictal periods in all rats (Fig. 2c,d left), with low variance among SWDs (Fig. 2c,d middle). Additionally, the inter-spike interval (ISI) decreased (Fig. 2c, right) due to the organization of MU spikes into clusters. Most notably, the inter-cluster interval (ICI) became very regular (~100–120ms, Fig. 2d, right), matching the principle oscillatory frequency of rodent SWDs (Danober et al. 1998; Coenen et al. 1992).
Figure 2. related to Fig. S5, Table S1: VB displays rhythmic, high firing-rate output throughout the duration of naturally occurring seizures.
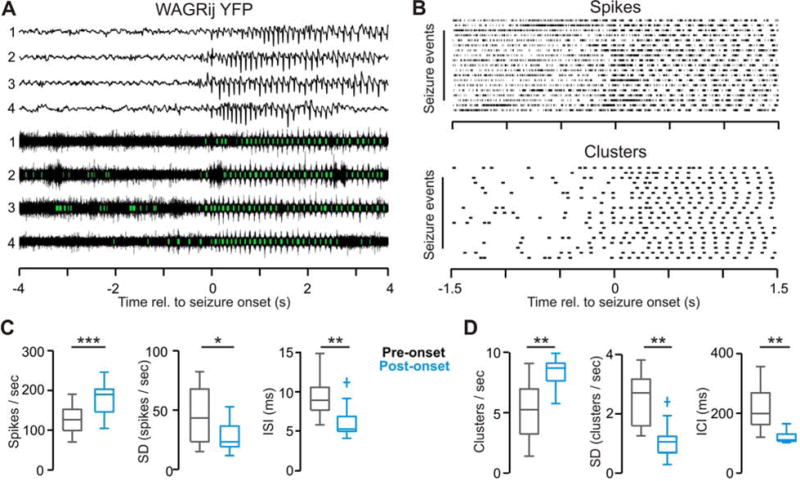
(a) Example ipsilateral ECoG (top) and thalamic multi-unit activity (bottom) with detected clusters (green) from four consecutive spontaneous seizures in a WAGRij-YFP rat. (b) Rasters of detected spikes (top) and spike clusters (bottom) from the same animals/trial as in (a). (c) The population spike rate (left), intra-trial s.d. of the spike rate (middle), and the inter-spike interval (right) averaged over 2-second windows prior to the seizure onset (gray) and following the seizure onset (blue) across all WAGRij-YFP animals. Note that the population spike rate actually increases during seizures, though becomes highly organized and phasic. (d) The cluster rate (left), intra-trial s.d. of the cluster rate (middle), and the inter-cluster interval (right) across all WAGRij-YFP animals. n = 13 trials, three animals; Wilcoxon Signed-Rank test; * p < 0.05, ** p < 0.01, *** p < 0.001
Unilaterally driving TC phasic firing induces generalized absence seizures and behavioral arrest in epileptic rodents
Given that absence seizures appear to emerge from similar elements of the thalamocortical network that drive normal oscillations as in sleep spindles (Kostopoulos 2000; Beenhakker & Huguenard 2009), we next investigated whether selectively driving TC phasic firing in epileptic animals could initiate SWDs. We expressed eNpHR-eYFP unilaterally in TC neurons of STG mice and WAGRij rats and delivered 8 Hz pulses of 594nm light to TC neurons to mimic the fundamental frequency of rodent absence seizures (van Luijtelaar & Coenen 1986; Noebels et al. 1990). Repetitive 594nm (but not sham) pulses produced phasic clustered spikes in TC neurons, with similar post-light latencies (~28ms) to our in vitro results (Fig. 3g, Fig. S1a). Robust CTC oscillations were detected in the thalamic local field potential (LFP) and cortical ECoG in both mouse and rat models of epilepsy (Fig. 3a, Fig. S1b (bottom)), which were not seen in eYFP-controls (Table S1). We discovered that unilateral photoinhibition initiated bilateral cortical SWDs with classic features of absence epilepsy: synchronous 8 Hz rhythmic activity and behavioral absences identical to spontaneously occurring seizures (Fig. 3c, Video S1) (van Luijtelaar & Coenen 1986).
Figure 3. related to Fig. S1–3, Table S1: Rhythmic activation of eNpHR in TC drives SWD seizures in STG mice and WAGRij rats.
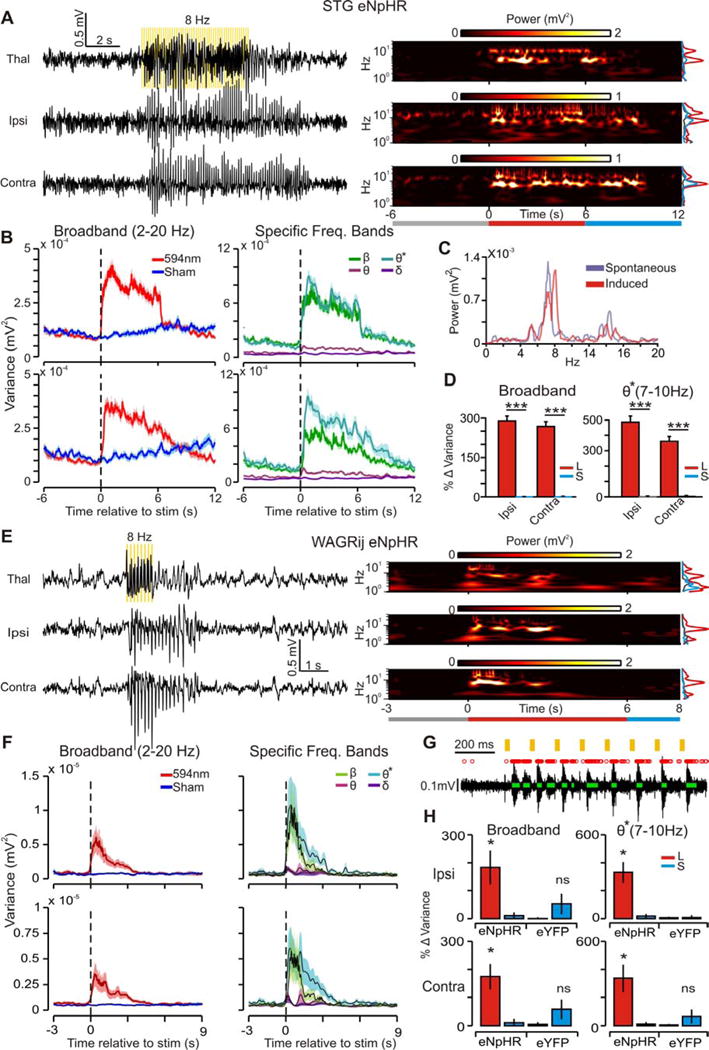
(a) Left: example induced seizure from STG-eNpHR mouse with thalamic LFP (top), ipsilateral (middle), and contralateral (bottom) ECoG. Yellow bars indicate 8 Hz pulses of 594nm light, delivered to the right VB. Right: time-frequency wavelet decomposition of traces. Right Inset: Average prestim, stim, and postsim power (times indicated by the colored bars below). (b) Left: trial-averaged broadband (BB, 2–20 Hz) power (variance in this and all subsequent figures) over time for all laser (red) and sham (blue) trials. Right: trial-averaged power for laser-induced seizures, decomposed into different frequency bands (ß=10–20 Hz; Ø*=7–10 Hz; Ø=4–7 Hz; ∂=2–4 Hz). (c) Contralateral ECoG from spontaneous (purple) and induced (red) seizures; note the peak power at ~7 Hz and harmonics at ~15 Hz in both conditions. (d) Left: median BB power percent change + s.e.m.) from prestim → stim periods for induced (red) and sham (blue) trials across STG mice. Right: median Ø* power percent change. (107/195 laser/sham events, seven trials, three animals; p < 0.001, Wilcoxon-ranksum). (e–f) Same as a–b, but for 1s 8 Hz pulse in WAGRij-eNpHR. (g) Response of thalamic multi-unit (MU) activity in vivo to 594nm pulses. Red X’s = detected spikes, green bars = detected clusters (see online methods). Note that clusters time-lock to 594nm pulses. (h) Median % change in BB (left) and Ø* (right) power +.s.e.m) for WAGRij-eNpHR and WAGRij-eYFP from prestim → stim for ipsilateral (top) and contralateral (bottom) ECoG. Red = laser, blue = sham (p < 0.05, Wilcoxon-ranksum; eNpHR: seven trials, four animals; eYFP: four trials, four animals; see also Video S1).
To compare the optically induced seizures to spontaneously occurring events, we used wavelet decomposition (Torrence & Compo 1998) to analyze changes in ECoG power centered around five frequency bands (see experimental procedures). We found significant changes in BB, ß, and Ø* power for both WAGRij and STG expressing eNpHR-eYFP, but not eYFP (Fig. 3b,d, Table S1). The Ø* and ß bands reflect the fundamental and harmonic frequencies, respectively, of SWDs (see Fig. 3a,c,e). Notably, wavelet power in each ECoG increased relatively more for ß and Ø* bands than it did for Ø, ∂, or BB (Fig. 3b right, Table S1), which was not evident in low-power 8 Hz controls, nor with 488nm light (which does not strongly activate eNpHR (Zhang et al. 2006); Fig. S2b,c,e,f, Video S2).
Although long (~6s) unilateral 8 Hz trains could drive bilateral SWDs, we wondered whether brief (1s) 8 Hz trains could reproduce this result. Indeed, 1s unilateral 8 Hz trains reliably drove bilateral SWDs with elevated Ø*, ß, and BB power that outlasted the pulse train (Fig. 3e,f, Table S1). Seizures were induced on average in 40–50% of stimulations, but only occurred by chance in ~0–5% of cases following sham pulses and in eYFP animals (Fig. S3b,c). Further, BB and Ø* ECoG power was elevated in both hemispheres after photoinhibition relative to sham and eYFP controls (Fig. 3h). This is a striking result given that TC neurons of primary thalamic nuclei project exclusively to ipsilateral cortex (Jones 2007).
Thalamic induction of SWD is pulse-frequency invariant, and even a single cluster of TC spikes can initiate an SWD
We next asked whether various pulse frequencies could induce SWDs. We found that pulsed 594nm light at 3, 12, and 20 Hz drove phasic clustered spikes in TC neurons that followed the pulse frequency (Fig. 4a–e), and increased the incidence of spike clusters compared to pre-stim periods and eYFP controls (Fig. 4f,g). Although TC spike clusters followed the stimuli, we found that the different pulse frequencies consistently induced bilateral ECoG SWDs with maximal power in ß and Ø* bands (Fig. 4h, Table S1, Video S3). These findings argue that the induced cortical oscillations do not simply reflect an extrinsic thalamic drive, but rather arise from intrinsic epileptic CTC network activity that is rapidly activated by a synchronous switch of TC neurons into a rhythmic, phasic state.
Figure 4. related to Fig. S1–3, Table S1: In vivo clusters from eNpHR rodents follow various pulse-train frequencies, yet seizure-specific ECoG power is maximally elevated.
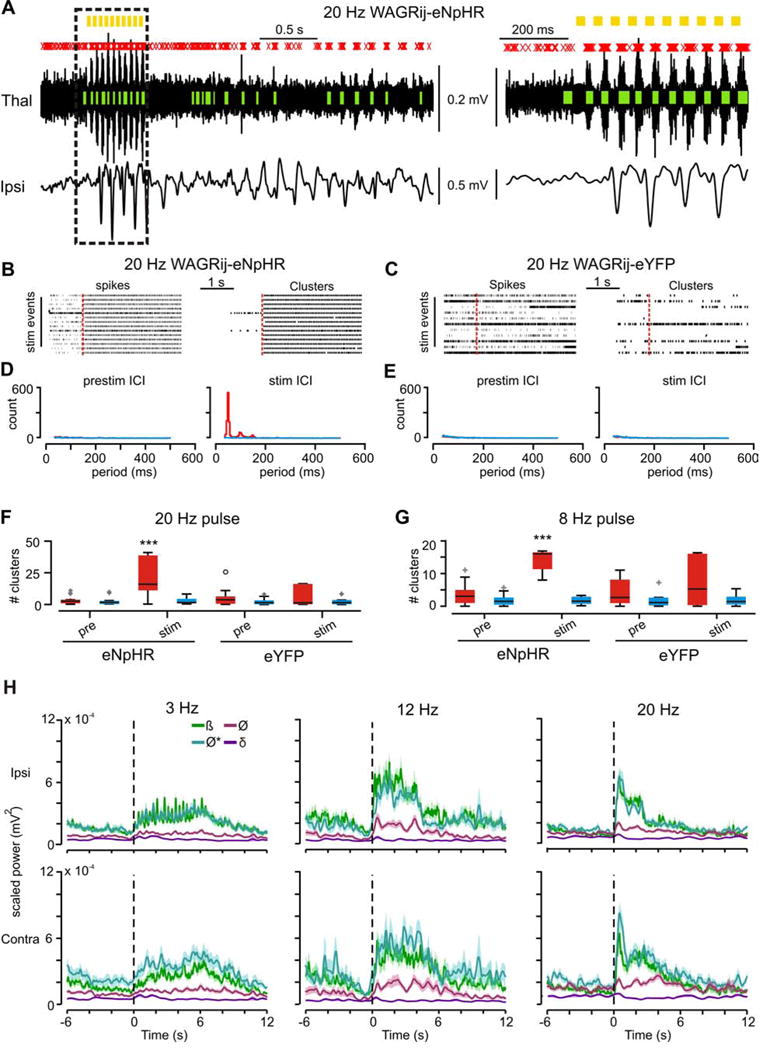
(a) TC Multi-unit (MU) activity (top) and contralateral ECoG (bottom) from a WAGRij-eNpHR animal. MU spikes (red x’s) cluster into phasic bouts (green bars) that consistently follow the 20 Hz pulses (yellow bars). Right: expanded trace from the highlighted region. Note that ECoG spike-wave dischages rapidly converge to ~ 8 Hz despite the 20 Hz pulse frequency. (b) In vivo spike (left) and cluster (right) rasters from WAGRij-eNpHR before and during a 20 Hz train of 594nm light (red bar = 594nm onset). (c) Same as b, but for a WAGRij-eYFP control. (d) Prestim (left) and during-stim (right) inter-cluster interval (ICI) across all laser (red) and sham (blue) pulses for same animal as in b, showing that bursts organize around ~50 ms. (e) Same as d, but for WAGRij-eYFP, demonstrating lack of cluster organization during the stim. (f) Boxplots of the # of prestim (left) and stim (right) clusters during a 20 Hz pulse train across all recording trials for all animals, averaged on a per-trial basis for laser (red) and sham (blue) conditions. Only WAGRij-eNpHR show an elevated # of clusters during the laser stim period (p < .001, ANOVA, eNpHR: 10 trials, 4 animals; eYFP: 7 trials, 4 animals). (g) Same as f, but for 8 Hz (p < .001, ANOVA, eNpHR: 21 trials, four animals; eYFP: 11 trials, 5 animals). (h) Average power across different frequency bands (ß=10–20Hz; Ø*=7–10 Hz; Ø=4–7 Hz; ∂=2–4 Hz) separated by pulse-frequency (columns) for ipsilateral (top row) and contralateral (bottom row) ECoG. Note the preferential increase in Ø* and ß ECoG power regardless of the pulse frequency. Shaded error represents + s.e.m. (n=76/77, (3Hz), 107/195, (8Hz), 33/38, (12Hz), and 78/55 (20Hz) laser/sham events, from 5 trials (3Hz, 12Hz, 20Hz), or 7 trials (8Hz), 3 STG animals. See also Video S3).
This raised the question: could a single, unilateral TC population action potential cluster also trigger a seizure? Surprisingly, one 50ms pulse of 594nm light delivered to WAGRij-eNpHR rats was sufficient to initiate sustained phasic TC firing at ~ 8 Hz (Fig. 5c,d,g), along with synchronous oscillations with elevated Ø* power in thalamic LFP and bilateral ECoG (Fig. 5b,c,h (left),i). These were accompanied by behavioral absences, which were not evoked in eYFP controls (Fig. 5a,h right) nor with sham pulses (Fig. 5e–g). Induced SWDs showed a mean onset latency of 850ms following the stimulus and duration of 3.24s, as well as pronounced changes in Ø* and ß power relative to Ø and ∂ (Fig. 5h bottom, Table S1). Seizures were induced in ~50% of stimulations on average versus ~0–5% of cases with sham pulses and in eYFP controls (Fig. S3a,d).
Figure 5. related to Fig. S3, Table S1: Single 50ms 594nm pulse induces 8 Hz TC clusters and bilateral seizures.
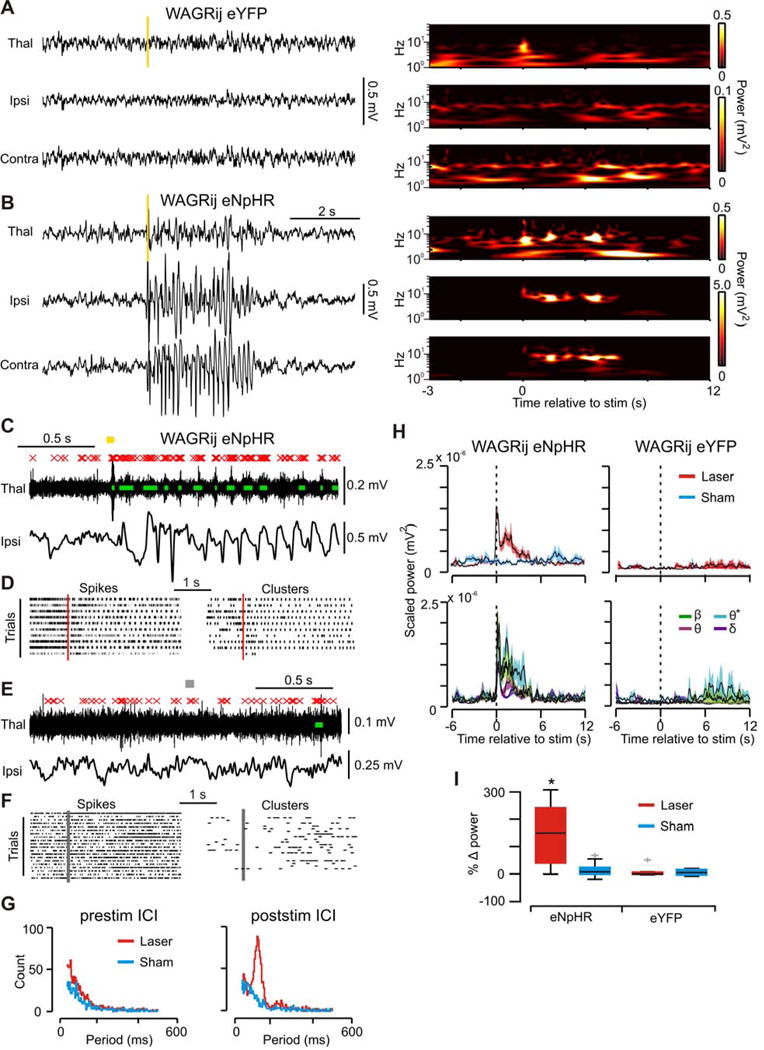
(a) Left: Thalamic LFP (top) and cortical ECoG (middle, bottom) from WAGRij-eYFP demonstrating no effect of 50ms 594nm pulse (yellow bar) on cortical ECoG. Right: wavelet transformations of traces. Notice the transient increase in power in the thalamic LFP due to the light artifact. (b) Same as a, but for WAGRij-eNpHR. The 50ms light pulse induces a seizure that almost instantly generalizes bilaterally. (c) In vivo thalamic MU activity during the same event as in b (red X’s = detected spikes, green bars = detected clusters, yellow bar = 594nm light). Bottom: ipsilateral ECoG from b. Note that ECoG spikes phase and time-lock to clusters. (d) Spike (left) and cluster (right) rasters of all 594nm events from same animal/trial as in b (red bar = 594nm onset). (e,f) Same as c,d, but for sham pulses. (g) Inter-cluster interval (ICI) histograms across all pulses/animals for laser/sham pulses 2 seconds before (left) or after (right) the pulse onset. Poststim ICI peaks at ~125ms, or 8 Hz. (h) Top: mean + s.e.m. BB power across laser/sham pulses for WAGRij-eNpHR (left) and WAGRij-eYFP (right). Bottom: mean + s.e.m. band-specific power across laser pulses. Note that Ø* power remains elevated longer compared to other bands. (i) Percent change in BB power from prestim → stim periods for all WAGRij-eNpHR and WAGRij-eYFP (red = laser, blue = sham). 594nm pulses produced a change in BB power, but only in eNpHR rats (p < .05, ANOVA; eNpHR: 15 trials, four animals; eYFP: 13 trials, four animals).
These results indicate that toggling phasic firing in TC neurons – even via a single unilateral cluster of action potentials – can initiate self-sustained bilateral epileptic activity that evolves irrespective of the frequency of direct, light-induced TC responses, likely by recruiting epileptic CTC networks (van Luijtelaar & Sitnikova 2006).
Depolarizing TC neurons switches TC firing mode from a phasic to tonic and eliminates clustered spiking
Having shown that eNpHR photoinhibition reliably drives phasic firing of TC cells and CTC rhythms, we next asked: can the same TC pathway be targeted to destabilize ongoing rhythms? Indeed, closed-loop control of thalamus and hippocampus is sufficient to interrupt spontaneously occurring seizures in animal models of acquired epilepsy (Paz et al. 2013; Armstrong et al. 2013), and closed-loop cerebellar stimulation (Kros et al. 2015) and transcranial stimulation (Berenyi et al. 2012) can disrupt SWDs. Despite these studies, the necessity of active TC phasic firing for SWD maintenance has never been studied in freely behaving rodents with absence epilepsy. To address this question, we developed our own closed-loop stimulation protocol (see experimental procedures) in order to unilaterally manipulate the TC firing mode during spontaneous seizures in both mice and rats.
We hypothesized that subthreshold depolarization of TC cells would enhance tonic firing associated with behavioral arousal (McCormick et al. 2015; McCormick 1992; Steriade et al. 1993b; Hirata & Castro-Alamancos 2010; Poulet et al. 2012) and reduce the probability of phasic firing in response to SWD-related inhibitory RT output (Coulter et al. 1989; Huguenard 2002; Huguenard & McCormick 2007), and thus the ability to sustain an ongoing CTC oscillation. For this purpose, we expressed Stable Step Function Opsin (SSFO), a bistable depolarizing opsin (Yizhar et al. 2011), unilaterally in VB neurons in both STG mice and WAGRij rats.
We describe first the intracellular recordings from SSFO-expressing VB neurons in vitro from rats that had been used for successful in vivo recordings (see below and experimental procedures). SSFO activation and subsequent deactivation increased and decreased tonic firing in TC cells, respectively (Fig. 6a,b). Notably, the SSFO activation strongly reduced the likelihood of eliciting TC PIR bursts, increased the likelihood of tonic spiking (Fig. 6c,d), and reduced the burst index, a measure of burst vs. tonic firing propensity (BI, Fig. 6d,e).
Figure 6. related to Fig. S4–5: SSFO activation in TC neurons increases tonic firing, reduces phasic clusters.
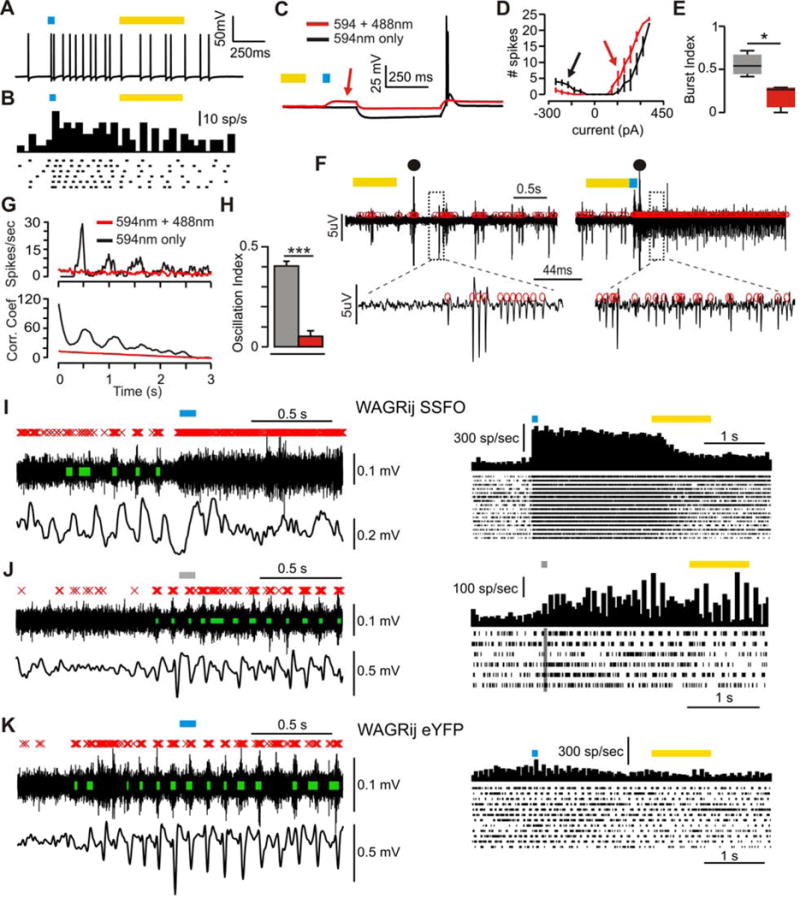
(a) Example in vitro whole-cell patch recording from an SSFO-expressing TC neuron demonstrating increased firing rate with SSFO activation (blue bar). (b) Raster and PSTH of same experiment as a over many sweeps. (c) −150pA current pulse in SSFO-inactive (594nm light, black) or SSFO-active (594nm + 488nm light, red) sweeps. SSFO activation depolarizes the cell (red arrow) and abolishes the rebound burst. (d) Total # spikes vs. injected current for SSFOs and SSFOa sweeps. SSFO activation increases the rebound-spike threshold and reduces # of rebound spikes (black arrow), but inversely affects tonic spiking (red arrow). (e) Burst index (BI) for SSFOs and SSFOa cells (p < .05, Wilcoxon-ranksum, n = 3 cells). (f) Extracellular multi-unit activity (red circles = spikes) from thalamic slice showing oscillatory multi-unit clusters following electrical stimulation (●) without SSFO activation (left), and non-oscillatory, desynchronized multi-unit tonic firing with SSFO activation (right). (g) Top: average PSTHs of extracellular multi-unit activity across SSFO active and inactive sweeps. Bottom: autocorrelations of the PSTHs. (h) Average oscillatory index (OI), for each condition (p < .001, T-test, n = 17 slices). (i) Left: in vivo thalamic multi-unit activity (top; red X’s = spikes, green bars = clusters), and cortical ECoG (bottom) during a closed-loop experiment in WAGRij-SSFO. Clusters and ECoG oscillations at the seizure start are abolished with SSFO activation. Right: PSTH and raster of spikes across all detected seizures with laser activation from same trial. (j) Same as i, but for sham pulses. (k) Same as i, but for WAGRij-eYFP control.
Next, we determined the in vitro effects of SSFO on isolated thalamic network activity by recording extracellular MU activity in the VB of WAGRij rats from brain slices that preserved intra-thalamic connectivity (Huguenard & Prince 1994; Paz et al. 2011; Paz et al. 2013) (see experimental procedures). In control conditions (594nm light only), internal capsule stimulation evoked intra-thalamic oscillations with bouts of clustered spikes that lasted for many seconds (Fig. 6f left, Supplementary Fig. 4a,b). However, SSFO activation in TC cells by 488nm light eliminated the evoked oscillations, increased desynchronized MU tonic firing (Fig. 6f right, Fig. S4a,b), and reduced the oscillatory index (OI)—a measurement of the regularity and robustness of oscillations (Fig. 6g,e), as well as the number of clusters per oscillation and the oscillation duration (Fig. S4c,d). These data validate SSFO as a robust tool to toggle TC tonic firing and effectively desynchronize thalamic network oscillations.
Disrupting TC phasic firing in vivo desynchronizes cortical oscillations
We next asked if toggling TC tonic firing in vivo could disrupt CTC rhythms. We developed an algorithm to unilaterally activate SSFO in the VB following SWD-detection (see experimental procedures). Closed-loop SSFO activation reliably and rapidly eliminated TC clusters and toggled tonic firing, which was not observed with sham pulses (Fig. 6j, Fig. S4e–g, S5a–c), nor in eYFP controls (Fig. 6k, S5d). The effect of unilaterally switching the firing mode from phasic to tonic on bilateral SWD was rapid and robust. SSFO activation bilaterally shortened SWDs in STG and WAGRij rats vs. sham pulses, but not in eYFP controls (Fig. 7e–g, Fig. S5a, Table S2, Video S4), and heightened arousal and exploratory behavior and reduced Ø* (Fig. 7a–d) ECoG power; this did not occur with 594nm light alone (Video S5).
Figure 7. related to Fig. S4–5, Table S2: SSFO activation in TC cells aborts automatically detected SWD seizures in STG and WAGRij.
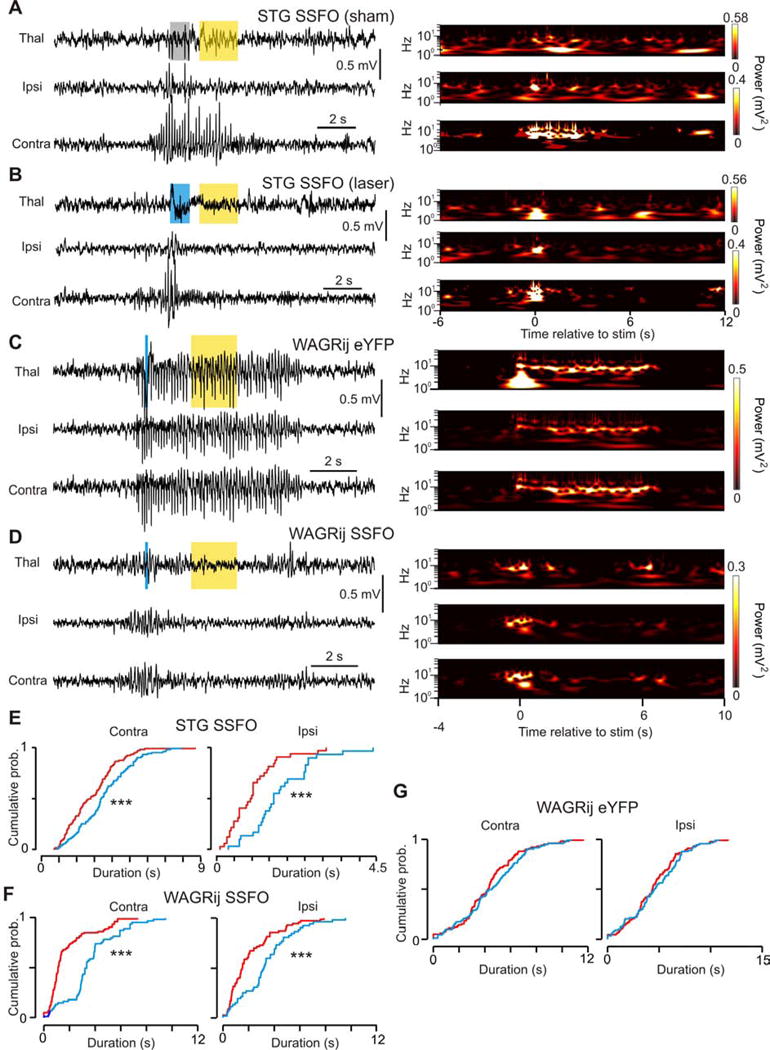
(a) Left: example seizure from an SSFO-expressing STG mouse showing thalamic LFP (top), ipsilateral (middle) and contralateral (bottom) ECoG. Gray/yellow bars: sham/594nm light. Right: wavelet transformations of the channels. (b) Same as a, but with a 488nm pulse that interrupts the seizure. Blue/yellow bars: 488nm/594nm light. Right: wavelet transformations; note the silence following the blue pulse. (c) Detected seizure from WAGRij-eYFP control + 488nm/594nm light. Blue light has no effect on the seizure. Note the light artifact in the thalamic wavelet power. (d) Detected seizure from WAGRij SSFO + 488nm/594nm light. Like b, the wavelet power is abolished following blue light. (e) Contralateral (left) and ipsilateral (right) empirical cumulative distributions of ECoG total seizure durations across all laser (red) and sham (blue) pulses for STG-SSFO. Laser seizure durations for ipsi- and contralateral cortices were significantly shortened (p < 0.001, Kolmogorov-Smirnov test; 110/112 laser/sham seizures, four trials, four animals). (f–g) Same as e, but for WAGRij-SSFO (f, p < 0.001, 155/111 laser/sham seizures, 10 trials, three animals) and WAGRij-eYFP (g, ns, 96/98 laser/sham seizures, 13 trials, three animals. See also Vid. S4–5).
Thus, closed-loop toggling of TC tonic firing mode via SSFO-mediated depolarization is a novel and reliable mechanism to abolish synchronized TC phasic firing in vivo and desynchronize bilateral SWDs and behavioral absences. Moreover, our experiments show that phasic TC output is a consistent feature of spontaneous absence seizures (Fig. 2, 5i–k, Fig. S5a left), and that such phasic firing is required for SWD maintenance.
Given the robustness of TC phasic firing during SWDs and the strong effects of SSFO activation, we wondered whether the TC output is necessary for SWD expression. To test this, we used a similar closed-loop protocol for our STG-eNpHR mice, but delivered 594nm light (see experimental methods). eNpHR activation strongly reduced MU TC firing compared to sham (Fig. S6a,b), and bilaterally shortened SWDs in all three STG-eNpHR mice (Fig. S6c–f). In combination with our SSFO results, this experiment argues that the continued phasic TC output is necessary for SWD maintenance during free behavior.
DISCUSSION
We investigated the role of TC neurons in generating cortical rhythms relevant to non-epileptic and absence behavior. For this purpose, we developed two novel optogenetic strategies to drive or disrupt phasic and tonic firing states unilaterally in VB neurons, with high temporal resolution. We demonstrate in non-anesthetized, freely behaving mice and rats that TC activity can bi-directionally control SWDs and behavioral absences in real-time.
Specifically, we demonstrated that (1) TC firing is heightened and organized into phasic clusters during seizures, (2) unilaterally driving TC phasic firing can initiate bilateral CTC oscillations and SWDs, (3) switching TC firing mode from phasic to tonic is sufficient to globally terminate SWDs, and 4) reducing TC firing shortens SWDs. Finally, we demonstrated that TC clustered spiking is robust throughout spontaneous and evoked SWDs. These results were supported by in vivo and in vitro experiments, and were validated in vitro in tissue from animals used for in vivo recordings.
The striking finding that a single TC spike cluster can initiate an SWD (Fig. 5) suggests that the TC pathway can rapidly recruit global CTC oscillators. These results are in agreement with our previous studies using the GluA4 knockout mouse in which “normal” excitatory inputs to the thalamus cause an abnormally large population of VB cells to fire, which promotes SWDs (Paz et al. 2011).
Origins of the phasic firing during SWDs
The in vivo TC clustered spiking following photoinhibition is likely due to a combination of single action potentials and PIR bursts. Although intracellular recordings are required to isolate the relative contribution of single cell bursts to population clusters, the latency between the end of the 594nm pulse and the start of the spike cluster was similar to the latency of PIR bursts obtained in slices (Fig. 1a,c, 2g, Fig. S1a). Therefore, the TC clustered spiking we observe during SWDs could be due in part to T-channel mediated PIR bursts.
Nonetheless, independent of the origin of the clustered spiking, the major point of this study is that at the population level, TC activity is heightened and organized into phasic, rhythmic bouts during seizures, and that this phasic firing is necessary for seizure maintenance. This last point is supported by our SSFO results, which demonstrate that even unilaterally disrupting phasic firing halts SWDs. While the effects of SSFO may be partially attributed to an increase in TC firing rate, we observed a significant increase in TC firing rate during spontaneous seizures (Fig. 2) compared to pre-seizure periods, which corroborates our hypothesis that it is the phasic firing mode of the TC network that is relevant for seizure expression, and that seizures depend on more than just reciprocal excitatory feedback between cortex and thalamus. Moreover, our finding that closed-loop eNpHR-mediated reduction of TC output shortens SWDs (Fig. S6) further supports our hypothesis.
Our results in light of the controversy
Dissecting the causal involvement of the TC versus CT pathways in rhythmic oscillations has been challenging given their highly reciprocal connections (Jones 2002; Adams 1997), and traditional, non-specific approaches such as lesions or electrical stimulation have yielded conflicting results. Thus, the active role of TC output—in particular TC cell bursts—in SWDs has been debated (Avanzini et al. 1996; De Curtis & Avanzini 1994; Tsakiridou et al. 1995; Polack et al. 2007; Polack et al. 2009; Meeren et al. 2002; Danober et al. 1998; Manning et al. 2004; Pinault et al. 1998; Pinault et al. 2006).
Many studies have shown that TC neurons are essentially silent or only fire sparse single action potentials throughout SWDs in vivo (Timofeev & Steriade 2004; Steriade & Contreras 1995; Polack & Charpier 2006; Polack et al. 2007; Pinault 2003; Pinault et al. 1998). These studies, corroborated by findings that pharmacological manipulations of the cortex but not the thalamus block SWDs (Polack et al. 2009; Manning et al. 2004; Sitnikova & Van Luijtelaar 2004; Leresche et al. 1998), have resulted in the hypothesis that TC output is not required for SWD initiation or maintenance. Our results refute this theory, as we have shown that TC neurons are highly active and organized into rhythmic, phasic clusters during SWDs, and that even unilaterally disrupting TC phasic firing bilaterally alters seizure dynamics. One potential reason our results contrast with those of previous in vivo studies is that stable and high-quality intracellular recordings of TC neurons has required anesthesia or neuroleptanalgesia, which may affect CTC dynamics.
Our results in light of the cortical-focus theory
A popular hypothesis that has emerged over the last 10–15 years is that absence seizures are initiated in the cortex – specifically in somatosensory cortex (S1) in rodents (Meeren et al. 2002; Meeren, Hanneke; Lopes da Silva 2005; Polack et al. 2009; Lüttjohann et al. 2013) – known as the cortical focus theory. Our results do not argue against the cortical focus theory but instead emphasize that TC neurons themselves are able to rapidly recruit epileptic cortical networks. Our results tie the previous controversies regarding which cellular substrates are required for an SWD, by suggesting that either CT or TC neurons can be causally involved in SWD initiation by recruiting inhibitory RT neurons that toggle TC phasic firing, a feature that we have shown to be necessary for the maintenance of cortical SWDs and associated absence behavior.
Translational relevance and opportunities
We propose that TC phasic firing is a potential therapeutic target to abort or possibly prevent SWDs. Given that T-type calcium channels are mainly responsible for TC burst behavior (Beenhakker & Huguenard 2009), we propose that designing drugs to specifically target these channels (primarily Cav3.1, the major T-channel in TC neurons(Talley et al. 1999)) would lead to an effective anti-absence therapy, and may reduce side effects associated with non-specific anti-absence medication such as ethosuximide, which also acts on Na+ channels (Leresche et al. 1998).
Our results are in agreement with complementary findings that systemic or intrathalamic administration of agents that increase GABAergic inhibition and hyperpolarize neurons (e.g., vigabatrin, tiagabin) promote burst firing and initiate or worsen SWDs in humans and rodents with epilepsy(Cope et al. 2009; Danober et al. 1998; Hosford & Wang 1997). Thus an optimal therapy would consist of an online switch from a phasic to tonic TC firing state at the onset of or just prior to the SWD. Until such closed-loop therapies are available, systemic administration of drugs that specifically target phasic TC firing could be used to prevent seizures.
Altering firing states as a tool for controlling global networks
We have shown that the firing mode of TC neurons can control global cortical rhythms, and that such firing modes can be manipulated in real-time for large-scale control of brain state. With tools such as SSFO, we are able to rapidly alter the firing mode of specific neurons, which may not necessarily be replicated by other optogenetic constructs with fast kinetics such as ChR2. SSFO in particular is an extremely sensitive and powerful tool to investigate the causal sources of other healthy and pathological brain states in real-time, and to assess the effects of manipulating brain regions that have multi-modal firing states. For instance, one could investigate the role of burst versus tonic output of cerebellar purkinje neurons on motor control, or the effects of disrupting hippocampal or cortical bursts during sleep.
Remaining challenges: finding key cells within brain wide circuits
The ability to selectively target specific cells in complex networks with powerful tools such as optogenetics is crucial for understanding and modifying pathologies, including epilepsy. Although seizures can initiate focally, they rapidly engage widespread circuits; thus disrupting this spread requires identifying “choke points” that may not necessarily be the seizure foci. Although there might be other targets, we propose that the TC pathway is a choke point for SWDs, similar to the subthalamus for Parkinson’s disease (for review see (Paz & Huguenard 2015)).
EXPERIMENTAL PROCEDURES
We performed all experiments according to protocols approved by the Institutional Animal Care and Use Committee, precautions were taken to minimize stress and the number of animals used in each set of experiments. Animals were separately housed after viral injections and surgical implants.
Viral injections
Stereotactic viral injections were carried out as described(Paz et al. 2011; Paz et al. 2013). 500nL of concentrated virus (2×1012 genome copies per milliliter) carrying genes for eNpHR3.0 (rAAV5/CaMKIIα-eNpHR-EYFP), SSFO (rAAV5/CaMKIIα-SSFO-EYFP), or eYFP alone (rAAV5/CaMKIIα-EYFP) was injected stereotaxically into the right somatosensory thalamus (VB) of P30–90 mice and rats with a 34-gauge beveled needle at 120nL/min. Injection of viral DNA under CaMKIIα promoter results in expression only in excitatory TC neurons (Gradinaru et al. 2010; Paz et al. 2011; Paz et al. 2013). The stereotaxic coordinates of the injections were 2.6–2.7mm posterior, 2.8mm lateral, and 5.8–6.0 mm ventral (rats) or 1.7mm posterior, 1.5mm lateral, and 3.5mm ventral (mice) relative to Bregma.
Slice preparation
Mice and rats were anesthetized with pentobarbital (100 mg per kg, intraperitoneal) and decapitated. The thalamic slice preparation was performed as described(Paz et al. 2011; Paz et al. 2013). For in vitro recordings from SSFO-expressing animals containing chronic implants, the animals were anesthetized and perfused with ice-cold oxygenated sucrose solution. Implants were then pulled off orthogonal to the skull prior to decapitation. All in vitro SSFO experiments were performed in the dark to avoid chronic activation of SSFO.
Extracellular thalamic oscillations
400-μm horizontal slices containing TC and RT were placed in a humidified, oxygenated interface chamber and perfused at a rate of 2 mL/min at 34°C with oxygenated ACSF supplemented with 300 μM glutamine. Oscillations were evoked by a square current pulse (240–260μA, 30μs duration) delivered to the internal capsule every 35s through two parallel tungsten electrodes (50–100Ω, FHC) 50–100μm apart. Extracellular potentials were recorded with a tungsten electrode placed in the VB. Signals were amplified at 10,000× and band-pass filtered between 10 Hz and 10 kHz. Photostimulation of thalamocortical neurons in TC consisted a pulse of 594nm light (1000ms, 2.6mW), immediately followed by a pulse of 488nm light (100ms, 1.2mW) on every other sweep. Sweeps with current stimulation were immediately followed by sweeps that consisted of photostimulation, but no current stimulation, to determine if photostimulation alone could sufficiently elicit thalamo-cortical oscillations. Alternating sweeps were repeated 10–20 times in a single recording.
Patch-clamp electrophysiology from thalamic slices
Recordings were performed as described(Paz et al. 2011; Paz et al. 2013). Recording electrodes made of borosilicate glass had a resistance of 2.5–4 MΩ when filled with intracellular solution. Potentials were corrected for −15mV liquid junction potential. Access resistance was monitored in all the recordings, and cells were included for analysis only if the access resistance was < 18 MΩ and resistivity change was < 20% over the course of the experiment. Cells were filled with 0.2–0.5% biocytin (Sigma-Aldrich), and whole slices were fixed and processed using standard avidin–biotin peroxidase (Horikawa & Armstrong 1988). Immunofluorescence was assessed with a laser confocal microscope (Zeiss LSM 510). Numerical values are given as means ± s.e.m., unless stated otherwise. Data analysis was performed with MATLAB 2013a and Origin 8.0 (Microcal Software).
To quantify changes in burst probability from neurons expressing SSFO by activation with 488nm light, we injected DC currents ranging from −250pA to 350 pA and developed a burst index (BI, see Fig. 6e) defined as:
where IR is the negative current threshold for eliciting a rebound burst, IT is the positive current threshold for eliciting tonic firing, SRR is burst spike count divided by the current pulse length, and SRT is the tonic spike count divided by the current pulse length.
In vivo data acquisition
ECoG and LFP signals were recorded using XLTek (Natus Medical Incorporated, Pleasanton, CA) or RZ5 (TDT) and sampled at 500 and 2441 Hz, respectively, while thalamic extracellular multi-unit signals were recorded using Axoscope (Molecular Devices) or RZ5 at 10 and 24 kHz. Two contralateral and two ipsilateral ECoG channels and four thalamic MU/LFP channels were obtained via an implanted optrode(Yizhar et al. 2011; Anikeeva et al. 2011) (see supplemental procedures). A small video camera mounted on a flexible arm was used to continuously monitor the animals. Each recording trial lasted 20–60 min. To control for circadian rhythms, we housed our animals using a regular light/dark cycle, and performed recordings from roughly 9:00AM – 4:00PM.
Spike detection and oscillation analysis
We first differentiated and smoothed raw MU data, then calculated baseline RMS values to detect spikes via threshold crossing. Spikes were excluded if their waveforms lasted longer than 2ms (in vitro) or 1ms (in vivo), and artifact rejection was implemented to eliminate detected signals that were 10-fold larger in amplitude than the mean spike amplitude. We define the various firing modes of the TC network that were detected as follows: 1) burst firing – the intrinsic generation of bursts of action potentials driven by T-type calcium channels (Coulter et al. 1989), 2) clustered spiking – refers to the coordinated and time delimited phasic action potential firing of a population of TC neurons, likely but not necessarily composed of individual TC burst responses, 3) tonic firing - unclustered action potential firing, and 4) phasic firing - the rhythmic occurrence of well-defined MU clusters in a sustained oscillation.
In vitro and in vivo clustered spikes were determined as periods containing at least four spikes within 10ms with spike rates greater than 2 times the spike rates of adjacent 40ms periods of MU activity, as typical rhythmic activity occurs in isolated bouts of firing (see Fig 2a, 6i–k for examples). In vitro oscillations were determined as periods containing at least two clusters within 600ms. For each sweep, spikes were aligned to the stimulus onset, a peri-stimulus time histograms (PSTH) with a bin width of 10ms was calculated, and the PSTH auto-correlation was used to calculate the oscillatory index (OI). OI was defined as:
where ti = the ith local minimum of the auto-correlation (trough) and pi = the ith local maximum of the auto-correlation (peak). See Fig. 6c,d and Fig. S5 for details.
For in vivo recordings, we also rejected high-frequency movement artifacts by following previous methods (Paralikar et al. 2009). We then organized spikes into sweeps of 18s surrounding each seizure and calculated PSTHs using a bin width of 40ms as well as cluster statistics for laser/sham groups (see Fig. 2, 4, 5, Fig. S4–6).
In vivo optogenetics
We simultaneously passed a fiber optic with an inline rotating joint (Doric) through a concentric channel in the electrical commutator, and connected it to the 200um-core fiber optic in each animal’s headpiece while recording ECoG/MU. The fiber optic was connected to a 488nm/594nm dual-wavelength laser control box, which was triggered externally using the RZ5 and custom software. The tip of the fiber rested 200 um from the most dorsal tungsten electrode on each optrode to allow maximal activation of the TC without physically obstructing the electrodes. We used 8mW (mice) and 15mW (rats) of 594nm/488nm power, measured at the end of the optical fiber prior to connecting to the animals.
During induced-seizure experiments, our software periodically triggered either 594nm light or sham pulses (pulses that did not open our laser shutter), selected at random from a uniform distribution, while recording the digital-out from the TDT system. We used different protocols to deliver either a single 50ms pulse, or 25ms pulses at 3, 8, 12, or 20 Hz, each composed of 50 pulses per train except for the 3-Hz train which was composed of 20 pulses. A 30 or 60 second time-out was implemented after each trigger.
For closed-loop experiments, we wrote custom software to calculate line-length(Paz et al. 2013) from an ECoG channel to trigger either a laser or sham pulse based on threshold crossing. Briefly, line-length was calculated by splitting signal into two streams, bandpass filtering between 7–9 Hz or 14–18 Hz, summed, and finally applying a 1s moving average window. Upon user-defined threshold crossing, a 1s 594nm/sham pulse (eNpHR trials) or 100ms 488nm/sham pulse and then a 2sec 594nm pulse (SSFO trials) was delivered, followed by a 15s time-out. We detected seizures in ~585msec on average.
ECoG spectral analysis
We used the continuous wavelet transformation for spectral analysis (Torrence & Compo 1998) to decompose signals into both time and frequency. We used a basis of Morlet wavelets from 1–128 Hz with seven octaves and 10 wavelets per octave. We analyzed individual frequency bands defined by the equation:
where BV is the band-specific variance (or power), l and u are the lower and upper boundaries of the bandwidth of choice, ∂j is a scalar for the number of sub-octaves used, ∂t is our sampling interval, C∂ is a scalar for the Morlet wavelet, P is total wavelet power, and S is an array containing the scales from the wavelet transform(Torrence & Compo 1998). Although we did not z-score normalize raw signals, seizure power is relative to the overall power of each recording session. Further, the equation for calculating BV normalizes wavelet power for each scale by the scale itself, and our statistics were based on relative changes in BV on a per-seizure basis, not on absolute BV or wavelet power.
To compare the effects of light on seizure power in both induced and interrupted trials, we calculated relative changes in seizure-averaged BV between pre-stimulus (PreStim), stimulus (Stim), and post-stimulus (PostStim) periods across recording trials (Fig. 3d,h). We focused on five bandwidths for our analyses: broadband (BB) (2–20 Hz), ß (10–20 Hz), Ø* (7–10 Hz), Ø (4–6 Hz), and ∂ (2–4 Hz), as these bands have been studied extensively in previous epilepsy literature. For induced trials, PreStim was defined as 6s before Stim onset, Stim periods were defined by the start and end of the pulse train, which were either 6.67 sec (3 Hz), 6.25 sec (8 Hz), 6 sec (single pulse), 4.17 sec (12 Hz), or 2.5 sec (20 Hz) long, and PostStim was defined as 6 sec after the end of the Stim period. We calculated relative changes in BV from PreStim → Stim for laser and sham, and repeated this analysis for each pulse condition to statistically evaluate the effect of various pulse frequencies (Fig. 4k).
Seizure length statistics
Seizure lengths were compared between laser and sham pulses during closed-loop experiments to characterize the effect of SSFO activation. Seizure durations were semi-automatically computed as periods in which BB power crossed and remained above a significance threshold (0.99, calculated from the wavelet transformation) for longer than 1.5s. Seizures were considered to end if BB power returned to and stayed at baseline levels for longer than 1s. Post-stimulus seizure lengths were calculated by subtracting the time of the stimulus onset relative to the start of the seizure from the total seizure length.
Seizure lengths were separated between laser and sham seizures and were averaged for each recording session. Summary statistics (Table S2) and cumulative distributions of these statistics (Fig. 7) for laser/sham groups were calculated and statistically compared via the Kolmogorov-Smirnov test.
Seizure probability statistics
For short 8 Hz (Fig. 3e–h) and single pulse (Fig. 5) eNpHR activation experiments, we calculated the probability of inducing a seizure by counting the number of instances in which a seizure (measured via ECoG recordings) occurred within 2s following a sham or laser trigger, and assessed the differences using the Kruskal-Wallis test (see Fig. S3).
Code availability
Please contact Jordan Sorokin (Jorsor@stanford.edu) or John Huguenard (huguenar@stanford.edu).
Supplementary Material
Highlights.
TC output is synchronized, phasic, and rhythmic during spontaneous SWDs
Unilaterally toggling TC phasic spiking via eNpHR induces bilateral SWDs
Unilaterally toggling TC tonic spiking via SSFO bilaterally aborts SWDs
Unilaterally suppressing TC output via eNpHR bilaterally shortens SWDs
Acknowledgments
J.T.P. is supported by NIH-NINDS R00NS078118-01 and Gladstone Institutes. J.R.H. is supported by NIH-NINDS 5R01NS034774. J.S. is supported by the Stanford Neuroscience Graduate program. K.D. is supported by Howard Hughes Medical Institute, CIRM, NIH and DARPA REPAIR Program. We thank Carl Pisaturo for designing and fabricating custom electronics, Jessica Wong for her help with animal husbandry, and Stefanie Ritter-Makinson for critical feedback on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
JMS and JTP performed in vivo experiments related to seizures; JTP and TJD performed the in vivo experiments in non-epileptic animals; JTP and JMS performed surgeries, JTP and AMA performed the in vitro experiments, and JMS performed in vitro experiments in Fig. 4a–e; TJD helped implement in vivo acquisition. JMS analyzed all seizure data, while EF analyzed non-epileptic in vivo data. JMS, JRH and JTP wrote the manuscript. KD provided reagents/tools.
References
- Adams NC. Complexities in the thalamocortical and corticothalamic pathways. European Journal of Neuroscience. 1997;9(2):204–209. doi: 10.1111/j.1460-9568.1997.tb01391.x. [DOI] [PubMed] [Google Scholar]
- Anikeeva P, et al. Optetrode: a multichannel readout for optogenetic control in freely moving mice. Nature Neuroscience. 2011;15(1):163–170. doi: 10.1038/nn.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, et al. Closed-loop optogenetic intervention in mice. Nature protocols. 2013;8(8):1475–93. doi: 10.1038/nprot.2013.080. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3988315&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzini G, et al. Calcium-dependent regulation of genetically determined spike and waves by the reticular thalamic nucleus of rats. Epilepsia. 1993;34(1):1–7. doi: 10.1111/j.1528-1157.1993.tb02369.x. [DOI] [PubMed] [Google Scholar]
- Avanzini G, et al. Cortical versus thalamic mechanisms underlying spike and wave discharges in GAERS. Epilepsy Research. 1996:37–44. doi: 10.1016/s0920-1211(96)00037-x. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that Fire Together Also Conspire Together: Is Normal Sleep Circuitry Hijacked to Generate Epilepsy? Neuron. 2009;62(5):612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenyi A, et al. Closed-Loop Control of Epilepsy by Transcranial Electrical Stimulation. Science. 2012;337(6095):735–737. doi: 10.1126/science.1223154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, et al. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci. 1988;8(11):4007–4026. doi: 10.1523/JNEUROSCI.08-11-04007.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Moser EI. Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nature neuroscience. 2013;16(2):130–8. doi: 10.1038/nn.3304. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4079500&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Watson BO. Brain rhythms and neural syntax: Implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues in Clinical Neuroscience. 2012;14(4):345–367. doi: 10.31887/DCNS.2012.14.4/gbuzsaki. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos Ma. Dynamics of sensory thalamocortical synaptic networks during information processing states. Progress i1. M. a. Castro-Alamancos, Dynamics of sensory thalamocortical synaptic networks during information processing states Prog Neurobiol 74, 213–247 (2004) .n Neurobiology. 2004;74(4):213–247. doi: 10.1016/j.pneurobio.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Chmielowska J, Carvell GE, Simons DJ. Spatial organization of thalamocortical and corticothalamic projection systems in the rat SmI barrel cortex. The Journal of comparative neurology. 1989;285(3):325–338. doi: 10.1002/cne.902850304. [DOI] [PubMed] [Google Scholar]
- Coenen AML, et al. Genetic models of absence epilepsy, with emphasis on the WAG/Rij strain of rats. Epilepsy Research. 1992;12(2):75–86. doi: 10.1016/0920-1211(92)90029-s. [DOI] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1995;15(1 Pt 2):604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nature medicine. 2009;15(12):1392–1398. doi: 10.1038/nm.2058. Available at: http://www.nature.com/doifinder/10.1038/nm.2058%5Cnpapers3://publication/doi/10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Calcium currents in rat thalamocortical relay neurones: kinetic properties of the transient, low-threshold current. The Journal of physiology. 1989;414:587–604. doi: 10.1113/jphysiol.1989.sp017705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Nucleus reticularis neurons mediate diverse inhibitory effects in thalamus. Neurobiology. 1997;94:8854–8859. doi: 10.1073/pnas.94.16.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Curtis M, Avanzini G. Thalamic regulation of epileptic spike and wave discharges. Functional Neurology. 1994;9(6):307–326. [PubMed] [Google Scholar]
- Danober L, et al. Effects of cholinergic drugs on genetic absence seizures in rats. European journal of pharmacology. 1993;234(2–3):263–268. doi: 10.1016/0014-2999(93)90962-h. [DOI] [PubMed] [Google Scholar]
- Danober L, et al. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Progress in Neurobiology. 1998;55(1):27–57. doi: 10.1016/s0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, et al. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(1):99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomora JC, et al. Block of cloned human T-type calcium channels by succinimide antiepileptic drugs. Molecular pharmacology. 2001;60(5):1121–1132. [PubMed] [Google Scholar]
- Gradinaru V, et al. Molecular and Cellular Approaches for Diversifying and Extending Optogenetics. Cell. 2010;141(1):154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Thompson KR, Deisseroth K. eNpHR: A Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biology. 2008;36(1–4):129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Bodelón C, Wyble BP. A proposed function for hippocampal theta rhythm: separate phases of encoding and retrieval enhance reversal of prior learning. Neural computation. 2002;14(4):793–817. doi: 10.1162/089976602317318965. [DOI] [PubMed] [Google Scholar]
- Hirata A, Castro-Alamancos MA. Neocortex network activation and deactivation states controlled by the thalamus. Journal of neurophysiology. 2010;103(3):1147–1157. doi: 10.1152/jn.00955.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa K, Armstrong WE. A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. Journal of neuroscience methods. 1988;25(1):1–11. doi: 10.1016/0165-0270(88)90114-8. [DOI] [PubMed] [Google Scholar]
- Hosford DA, Wang Y. Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia. 1997;38(4):408–414. doi: 10.1111/j.1528-1157.1997.tb01729.x. [DOI] [PubMed] [Google Scholar]
- Huguenard JR. Block of T -Type Ca2+ Channels Is an Important Action of Succinimide Antiabsence Drugs. Epilepsy Currents. 2002;2(2):49–52. doi: 10.1046/j.1535-7597.2002.00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. Neuronal circuitry of thalamocortical epilepsy and mechanisms of antiabsence drug action. Advances in neurology. 1999;79:991–999. [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends in Neurosciences. 2007;30(7):350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. Journal of neurophysiology. 1992;68(4):1373–83. doi: 10.1152/jn.1992.68.4.1373. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1279135. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1994;14(9):5485–5502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. The Journal of physiology. 1984;349:205–26. doi: 10.1113/jphysiol.1984.sp015153. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1199334&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. Thalamic circuitry and thalamocortical synchrony. Philosophical Transactions of the Royal Society B: Biological Sciences. 2002;357(1428):1659–1673. doi: 10.1098/rstb.2002.1168. Available at: http://rstb.royalsocietypublishing.org/cgi/doi/10.1098/rstb.2002.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The Thalamus. History. 2007;326(2):419–442. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2669290&tool=pmcentrez&rendertype=abstract. [Google Scholar]
- Kostopoulos GK. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: The continuing development of a hypothesis. Clinical Neurophysiology. 2000;111(SUPPL 2) doi: 10.1016/s1388-2457(00)00399-0. [DOI] [PubMed] [Google Scholar]
- Kros L, et al. Controlling Cerebellar Output to Treat Refractory Epilepsy. Trends in Neurosciences. 2015 doi: 10.1016/j.tins.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Lacey CJ, et al. Enhanced NMDA Receptor-Dependent Thalamic Excitation and Network Oscillations in Stargazer Mice. Journal of Neuroscience. 2012;32(32):11067–11081. doi: 10.1523/JNEUROSCI.5604-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche N, et al. On the action of the anti-absence drug ethosuximide in the rat and cat thalamus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18(13):4842–4853. doi: 10.1523/JNEUROSCI.18-13-04842.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Luijtelaar ELJM, Coenen AML. Two types of electrocortical paroxysms in an inbred strain of rats. Neuroscience Letters. 1986;70(3):393–397. doi: 10.1016/0304-3940(86)90586-0. Available at: http://linkinghub.elsevier.com/retrieve/pii/0304394086905860. [DOI] [PubMed] [Google Scholar]
- van Luijtelaar G, Sitnikova E. Global and focal aspects of absence epilepsy: The contribution of genetic models. Neuroscience and Biobehavioral Reviews. 2006;30(7):983–1003. doi: 10.1016/j.neubiorev.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Lüttjohann A, Schoffelen JM, van Luijtelaar G. Peri-ictal network dynamics of spike-wave discharges: Phase and spectral characteristics. Experimental Neurology. 2013;239(1):235–247. doi: 10.1016/j.expneurol.2012.10.021. Available at: http://dx.doi.org/10.1016/j.expneurol.2012.10.021. [DOI] [PubMed] [Google Scholar]
- Manning JPA, et al. Cortical-area specific block of genetically determined absence seizures by ethosuximide. Neuroscience. 2004;123(1):5–9. doi: 10.1016/j.neuroscience.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Marescaux C, Vergnes M, Depaulis A. Genetic absence epilepsy in rats from Strasbourg–a review. Journal of neural transmission Supplementum. 1992;35:37–69. doi: 10.1007/978-3-7091-9206-1_4. FEBRUARY 1992. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Progress in Neurobiology. 1992;39(4):337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- McCormick DA, McGinley MJ, Salkoff DB. Brain state dependent activity in the cortex and thalamus. Current Opinion in Neurobiology. 2015;31:133–140. doi: 10.1016/j.conb.2014.10.003. Available at: http://dx.doi.org/10.1016/j.conb.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren Hanneke, Lopes da Silva F. Evolving Concepts on the Pathophysiology of Absence Seizures. Archives of neurology. 2005;62:371–376. doi: 10.1001/archneur.62.3.371. [DOI] [PubMed] [Google Scholar]
- Meeren HKM, et al. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22(4):1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, et al. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy research. 1990;7(2):129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- Paralikar KJ, Rao CR, Clement RS. New approaches to eliminating common-noise artifacts in recordings from intracortical microelectrode arrays: Inter-electrode correlation and virtual referencing. Journal of Neuroscience Methods. 2009;181(1):27–35. doi: 10.1016/j.jneumeth.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, et al. A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy. Nature neuroscience. 2011;14(9):1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, et al. Activity of ventral medial thalamic neurons during absence seizures and modulation of cortical paroxysms by the nigrothalamic pathway. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(4):929–941. doi: 10.1523/JNEUROSCI.4677-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, et al. Closed-loop optogenetic control of thalamus as a new tool to interrupt seizures after cortical injury. Nature neuroscience. 2013;16(1):64–70. doi: 10.1038/nn.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Huguenard JR. Microcircuits and their interactions in epilepsy: is the focus out of focus? Nature neuroscience. 2015;18(3):351–9. doi: 10.1038/nn.3950. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25710837 [Accessed July 4, 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. Cellular interactions in the rat somatosensory thalamocortical system during normal and epileptic 5–9 Hz oscillations. The Journal of physiology. 2003;552(Pt 3):881–905. doi: 10.1113/jphysiol.2003.046573. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2343451&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, et al. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. Journal of Physiology. 1998;509(2):449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Slézia A, Acsády L. Corticothalamic 5–9 Hz oscillations are more pro-epileptogenic than sleep spindles in rats. The Journal of physiology. 2006;574(Pt 1):209–27. doi: 10.1113/jphysiol.2006.108498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack P-O, et al. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(24):6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack P-O, Charpier S. Intracellular activity of cortical and thalamic neurones during high-voltage rhythmic spike discharge in Long-Evans rats in vivo. J Physiol Lond. 2006;571(Pt 2):461–76. doi: 10.1113/jphysiol.2005.100925. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1796797&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, et al. Inactivation of the somatosensory cortex prevents paroxysmal oscillations in cortical and related thalamic neurons in a genetic model of absence epilepsy. Cerebral Cortex. 2009;19(9):2078–2091. doi: 10.1093/cercor/bhn237. [DOI] [PubMed] [Google Scholar]
- Poulet JFA, et al. Thalamic control of cortical states. Nature Neuroscience. 2012;15(3):370–372. doi: 10.1038/nn.3035. [DOI] [PubMed] [Google Scholar]
- Richards DA, et al. Targeting thalamic nuclei is not sufficient for the full anti-absence action of ethosuximide in a rat model of absence epilepsy. Epilepsy research. 2003;54(2–3):97–107. doi: 10.1016/s0920-1211(03)00060-3. [DOI] [PubMed] [Google Scholar]
- Sarkisova K, van Luijtelaar G. The WAG/Rij strain: A genetic animal model of absence epilepsy with comorbidity of depressiony. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(4):854–876. doi: 10.1016/j.pnpbp.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Sitnikova E, Van Luijtelaar G. Cortical control of generalized absence seizures: Effect of lidocaine applied to the somatosensory cortex in WAG/Rij rats. Brain Research. 2004;1012(1–2):127–137. doi: 10.1016/j.brainres.2004.03.041. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Huntsman MM, Huguenard JR. Reciprocal inhibitory connections regulate the spatiotemporal properties of intrathalamic oscillations. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20(5):1735–45. doi: 10.1523/JNEUROSCI.20-05-01735.2000. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10684875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101(2):243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. Journal of neuroscience. 1995;15(1 Pt 2):623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science (New York, N.Y.) 1993a;262(5134):679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science (New York, N.Y.) 1993b doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Talley EM, et al. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19(6):1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. Available at: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10066243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Spike-Wave Complexes and Fast Components of Cortically Generated Seizures. IV. Paroxysmal Fast Runs in Cortical and Thalamic Neurons. J Neurophysiol. 1998;80(3):1495–1513. doi: 10.1152/jn.1998.80.3.1495. Available at: http://jn.physiology.org/content/80/3/1495.long [Accessed August 28, 2015] [DOI] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Neocortical seizures: Initiation, development and cessation. Neuroscience. 2004;123(2):299–336. doi: 10.1016/j.neuroscience.2003.08.051. [DOI] [PubMed] [Google Scholar]
- Torrence C, Compo GP. A Practical Guide to Wavelet Analysis. Bulletin of the American Meteorological Society. 1998;79(1):61–78. [Google Scholar]
- Tsakiridou E, et al. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1995;15(4):3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7722649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C. Cortical and thalamic lesions in rats with genetic absence epilepsy. Journal of neural transmission Supplementum. 1992;35:71–83. doi: 10.1007/978-3-7091-9206-1_5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1512595. [DOI] [PubMed] [Google Scholar]
- Yizhar O, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477(7363):171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, et al. Channelrhodopsin-2 and optical control of excitable cells. Nature methods. 2006;3(10):785–92. doi: 10.1038/nmeth936. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16990810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.