

Abstract
Objective
To examine the effects of apoB100 structure, specifically a mutation in the LDLr binding region, on the production of LDL and development of atherosclerosis in vivo.
Methods and Results
Ldlr−/− Apobec1−/− mice lacking the LDLR and apoB editing enzyme accumulated LDL in plasma and developed severe atherosclerosis when they had wild-type apoB100. In marked contrast, in Ldlr−/− Apobec1−/− mice carrying the Apob100-β mutation, in the 2 putative LDLR-binding domains of apoB prevented both LDL accumulation and atherosclerosis. Intestinal absorption of lipids and triglyceride secretion from the liver were not affected. However, the VLDL particles with apoB100-β were larger in volume by about 70%, and carried approximately four times as much apoE per particle. ApoB100-β synthesis rate in the primary hepatocytes was normal, but its intracellular degradation was enhanced. Additionally, mutant apoB100 VLDL cleared from the circulation more quickly in vivo through apoE-LRP–mediated mechanism than VLDL with wild-type apoB100. In contrast, uptake of the 2 VLDL by macrophages were not different.
Conclusion
While conformational change to apoB100 during conversion of VLDL to LDL exposes LDLR binding domains and facilitates LDLR-mediated lipoprotein clearance, it may also inhibit LRP-mediated VLDL uptake and contribute to LDL accumulation in familial hypercholesterolemia.
Keywords: lipoprotein clearance, atherosclerosis, apolipoprotein B100, familial hypercholesterolemia, animal models
Apolipoprotein (apo) B is an essential component of VLDL, LDL, and chylomicrons. ApoB normally exists in 2 forms, apoB100 and apoB48; both are the products of the same gene. ApoB100 comprises 4536 amino acids, synthesized in the liver, and secreted into the circulation as a structural component of VLDL. ApoB48 is 48% of full-length apoB, and is formed as a result of posttranslational editing of ApoB mRNA by the apoB editing complex (apo-BEC), which changes Gln at codon 2153 to a stop codon.1 ApoB48 is synthesized in the small intestine and is required for the packaging of lipids into chylomicrons. Whereas human liver makes exclusively apoB100, a large proportion of message in the mouse liver is edited and consequently mice produce both apoB48 and apoB100 from the liver.2
In addition to maintaining the structural integrity of li-poprotein particles, apoB100 also functions as a ligand for the LDLR and is therefore a primary determinant of circulating LDL cholesterol levels. The LDLR-binding domain of apoB100 has not been fully defined; however, biochemical, immunochemical, and genetic evidence suggests that it is a region of net positive change located in the carboxyl-terminal portion of apoB100. Two sequences, residues 3147 to 3157 and 3359 to 3367, are enriched in basic amino acid residues and have been proposed as putative LDLR-binding domains in both species.3 The sequence at 3359 to 3367 is highly conserved among mammalian species and is also similar to the LDLR-binding site of apoE. Also, Boren et al showed that the removal of positive charges from residues 3359 to 3367 by site-directed mutagenesis renders the LDL containing the modified apoB defective in LDLR binding.3
To define the regions of apoB that bind the LDLR, we previously introduced mutations into the mouse Apob gene.4 The apoB100-β protein is the same length as apoB100 but contains 2 peptide sequences for human β-globin in place of the residues 3147 to 3157 and 3359 to 3367. The modification also drastically reduced the net positive charges and amphipathic helicies of the 2 domains. We expected that the mice producing apoB100-β would model defective apolipoprotein B100 in humans by accumulating binding-defective LDL in plasma.5 However, we found that the apoB100-β/B100-β mice have slightly, but not significantly lower than normal, total plasma cholesterol and HDL cholesterol, and the amount of plasma LDL was not different from that in wild-type mice.4 One explanation is that these 2 regions are not essential for apoB100 binding to the LDLR in vivo. The interpretation, however, is complicated because mice normally have very little apoB100-containing LDL particles in circulation. In addition, the production of apoB48 from the liver and the efficient clearance of apoB48-containing remnants mediated by apoE make the metabolism of apoB100 difficult to study in vivo in mice.
The present study examined the effect of apoB100-β-containing LDL by introducing the mutation onto a background of Ldlr−/− Apobec1−/− double mutants; a model of human familial hypercholesterolemia with severe atherosclerosis. Apobec1−/− mice that lack the mRNA editing enzyme produce only apoB100,7 whereas Ldlr−/− mice that lack LDLR accumulate LDL cholesterol in plasma.6,7 Surprisingly, when these mice also carry the apoB100-β mutation, they are completely protected from hypercholesterolemia and atherosclerosis that normally occurs in Ldlr−/− Apobec1−/− mice.
Methods
Please see supplemental methods (available online at http://atvb.ahajournals.org) for details on plasma lipid, lipoprotein, tissue weight, triglyceride secretion, lipolysis, uptake of DiI and 125I-labeled lipoproteins, LRP inhibition by adenoRAP, gene expression, and apoB synthesis, secretion, and degradation.
Animals and Diets
The apoB100-β allele codes for “VHLTPVEKSAVT” and “KEFT-PPVQAAYQ” instead of “LSVKAQYKKNSD” and “GTSRLM-RKRGLK” of the wild-type apoB100 allele at residues 3143 to 3154 and 3356 to 3366, respectively.4 Ldlr−/− mice (B6;129S7-Ldlrtm1Her/J) were obtained from the Jackson Laboratory. Apobec1−/− mice were obtained from Dr Eddy Rubin at the Lawrence Berkeley National Laboratory.7 Three strains of mutants were crossed to generate mice that are heterozygous for the Apob locus and doubly homozygous for the Apobec1 and the Ldlr loci. These mice were then crossbred, and Ldlr−/− Apobec1−/− mice with Apob genotypes of 100/100 (wild type), 100/100-β (heterozygous), and100-β/100-β (homozygous) were generated for experiments. Their genetic backgrounds were complex mixes between C57BL/6J, 129/SvEv, and129/Ola. Animals were maintained on normal chow (NC; 4.5% fat, 0.022% cholesterol; Prolab Isopro 3000; Agway Inc), or were fed a high-fat Western-type diet (HFW; 21% fat, 0.2% cholesterol; TD 88137; Harlan Teklad). Mice in all experiments were age-matched within 3 weeks. All procedures for the handling of mice were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Biochemical Analyses and Atherosclerosis Evaluation
Mice were fasted 4 hours before analysis. Liver and fecal lipids were extracted with chloroform/methanol.8 Plasma lipids, lipoprotein distribution, and triglyceride secretion rate, were determined as described.9 Lipoprotein particle diameters were determined by dynamic light scattering analysis using a Microtrac 250.10 Peritoneal macrophages and hepatocytes were isolated as described.11,12 The VLDL (d <1.006 g/mL) and LDL (d=1.06 to 1.10 g/mL) fraction was isolated from pooled plasma by ultracentrifugation and labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI C18; Molecular Probes Inc)13 or with 125I (Iodine-125 Radionuclide, Perkin Elmer)14 for clearance assays. Fibroblasts were kindly provided by Dr J. Herz at the University of Texas Southwestern Medical Center. Cellular lipids were extracted with isopropanol and measured with a microscope fluorometer.13 Gene expression in the liver was analyzed by real-time polymerase chain reaction (PCR), and quantification of atherosclerosis was carried out as described.11
Data Analysis
Values are reported as mean±SEM unless otherwise stated. Data were analyzed by ANOVA using JMP software (SAS Inc).
Results
ApoB100-β Causes Marked Reduction of LDL in Ldlr−/− Apobec1−/− Mice
Ldlr−/− Apobec1−/− mice with wild-type apoB100 had high levels of plasma cholesterol and triglycerides on NC, and further increased plasma lipids on a HFW diet (Table). In contrast, both plasma cholesterol levels in the Ldlr−/− Apobec1−/− mice that are heterozygous and homozygous for the apoB100-β mutation were reduced in an allele dose-dependent manner. The protective effect of the apoB100-β mutation compared to controls was retained when mice were fed a HFW diet, although plasma cholesterol levels increased about 3-fold in all mice. Plasma levels of triglyceride, free cholesterol, and phospholipids in mice with apoB100-β were also significantly lower than those with apoB100 mice.
Table.
Plasma Lipid Levels in Ldlr−/− Apobec1−/− Mice With Wild-Type ApoB100 or ApoB100-β Mutation
| Apob Genotype | Normal Chow Diet
|
High-Fat Western Diet
|
|||||
|---|---|---|---|---|---|---|---|
| TC (mg/dl) | TG (mg/dl) | TC (mg/dl) | TG (mg/dl) | FC (mg/dl) | PL (mg/dl) | ||
| F | 100/100 | 380±31(15) | 78±6 (15) | 1005±35 (24) | 134±14 (20) | 253±37 (15) | 627±23 (10) |
| 100/100-β | 196±7 (10) | 36±3 (10) | 476±82 (6) | 124±56 (6) | 143±36 (10) | 342±21 (10) | |
| 100-β/100-β | 93±6 (15) | 23±3 (15) | 292±24 (19) | 60±5 (15) | 84±14 (8) | 236±8 (8) | |
| M | 100/100 | 478±23 (7) | 99±7 (7) | 1161±26 (11) | 599±22 (11) | n.d. | n.d. |
| 100/100-β | 256±10 (8) | 60±9 (8) | n.d. | n.d. | n.d. | n.d. | |
| 100-β/100-β | 126±7 (8) | 39±4 (8) | 329±20 (7) | 149±29 (7) | n.d. | n.d. | |
Data are mean±SE in mg/dl. Plasma samples were collected from mice after a 4-hr fast. The numbers in parentheses are the No. of animals. F indicates females; M, males; TC, total cholesterol; TG, triglycerides; FC, free cholesterol; PL, phospholipids; n.d., not determined. Effects of Apob genotypes are highly significant in all categories (P<0.0001).
When plasma lipoproteins from male mice on normal chow diet were analyzed by fast protein liquid (FPLC), more than 60% of the plasma cholesterol was in the LDL fraction in Ldlr−/− Apobec1−/− mice with wild-type apoB100. In contrast, a striking absence of LDL-cholesterol was noted in the plasma of Ldlr−/− Apobec1−/− mice homozygous for apoB100-β (Figure 1A). Mice with one copy of apoB100-β had approximately half the amount of LDL as those with wild-type apoB100. All mice had very low levels of VLDL cholesterol, and there was no difference in the amount of HDL. The possession of apoB100-β resulted in a similar reduction of triglycerides in the LDL fraction (Figure 1B). SDS gel electrophoresis of lipoprotein fractions from plasma of mice fed a HFW diet showed that the distribution of apoB100-β among various classes of lipoproteins was similar to that of normal apoB100 with the highest concentration in the LDL range (1.02 g/mL>d >1.04 g/mL, Figure 1C). However, total apoB100-β in these mice was much less than wild-type apoB100, because samples of 3 times of apoB100-β plasma volume was loaded compared to apoB100 plasma. Total plasma apoE was also less in mice with apoB100-β than in mice with apoB100, but the ratio of apoE/apoB on the lipoprotein particles in the apoB100-β mice was about 4 times higher than in apoB100 mice.
Figure 1.

Plasma lipoprotein distribution by FPLC (A) cholesterol, (B) triglycerides. C, SDS-PAGE of apoB100, apoB-100β, and apoE in VLDL to LDL (1.006 to 1.06 g/mL) density fractions. Samples equivalent to 5 μL of apoB100 plasma and 15 μL of apoB100-β plasma were loaded. M, weight markers. D, Tissue weight normalized to body weight, (E) liver lipid contents, and (F) fecal lipids. Number of animals is in each bar. Error represents SEM. *P<0.005, **P<0.001.
Although there was no difference in adipose tissue weight, the liver weight per body weight was slightly but significantly smaller in mice with apoB100-β (P<0.005, Figure 1D). Hepatic intracellular cholesterol pools in the 2 groups of mice were not significantly different after 2 months on HFW diet. In contrast, the liver triglyceride content of Ldlr−/− Apo-bec1−/− males with apoB100-β was significantly lower than in mice with wild-type apoB100 (P<0.001, Figure 1E). Fecal cholesterol and triglyceride levels of mice after 2 months on HFW diet were not significantly different (Figure 1F). These data suggest that the apoB100-β mice are also protected from liver steatosis. Under light microscopy, however, liver sections from mice on HFW contained similar degrees of fatty droplets and no remarkable difference was observed between the Apob genotypes (data not shown).
Ldlr−/− Apobec1−/− Mice With apoB100-β Secrete Larger VLDL
A possible source of the disparity in plasma LDL levels is a difference in hepatic VLDL production rates. To estimate the secretion rate of triglyceride-rich lipoprotein (TRL) particles from the liver, we injected Triton WR1339 (Tyloxapol) intravenously into mice to inhibit lipolysis and uptake of TRLs, and measured plasma triglycerides (Figure 2A). Although the basal triglyceride levels differ in the 2 groups of mice, triglyceride secretion rates were nearly identical at 280 to 300 μg/mL/h regardless of whether they have apoB100 or apoB100-β.
Figure 2.

A, Hepatic triglyceride secretion rates. Female mice (5- to 8-month-old fed a HFW, n=11) were injected via tail-vein with Triton WR-1339, and plasma triglyceride levels were measured postinjection. B, 35S Pulse-chase analysis in primary he-patocytes. ApoB100 bands in the Gel were quantified using Image J software. Error represents SEM. *P<0.005, **P<0.001.
We next analyzed VLDL particle size at 2 hours post-Tyloxapol injection. The size of particles in the <1.006 g/mL density fraction was significantly different between the 2 groups of Ldlr−/− Apobec1−/− mice; the mean±SD diameter of apoB100-β VLDL particles were larger (55.0±15 nm) than those with wild-type apoB100 (45.6±14 nm). Based on the difference in the diameter, we estimate that the average apoB100-β VLDL has approximately 46% more surface area and 76% greater volume than that of the normal apoB100 VLDL. Assuming that the triglyceride content of a particle is relative to its volume, this implies that the number of VLDL particles secreted from the Ldlr−/− Apobec1−/− liver with apoB100-β is approximately 60% that from the liver with wild-type apoB100.
To examine the production and degradation of apoB100 proteins, we conducted a pulse-chase experiment with radio-labeled methionine in the primary hepatocytes isolated from the Ldlr−/− Apobec1−/− mice with apoB100 and with apoB100-β (Figure 2B). Immunoprecipitable apoB protein in the apoB100-β cells after the 30-minute pulse was not significantly different from that in apoB100 cells, suggesting that the initial synthesis rates are not different. After 4-hour chase in the medium with excess of cold methionine, however, the immunoprecipitable apoB protein both in the medium and associated with cells was significantly less in the apoB100-β hepatocytes. Thus, the mutated apoB protein is degraded more quickly, leading to the reduced number of VLDL particles secreted from the Ldlr−/− Apobec1−/− liver with apoB100-β.
B100-β VLDL Is Efficiently Cleared From the Circulation
The lack of LDL accumulation in the Ldlr−/− Apobec1−/− mice with apoB100-β is not proportional to the amount of VLDL particles secreted in these mice compared to that in mice with wild-type apoB. To test a hypothesis that apoB100-β VLDLs are cleared more efficiently than apoB100 VLDL, we isolated VLDL from Ldlr−/− Apobec1−/− mice with either apoB100 or apoB100β, labeled them with 125I, and injected them into Ldlr−/− mice via the tail vein. Monitoring plasma clearance of 125I labeled VLDL over a 2-hour period showed that 125I VLDL with apoB100-β are cleared faster than VLDL with wild-type apoB100 (Figure 3A). To determine the specific tissue loci of the cleared VLDL, we repeated the 125I-VLDL turnover, this time measuring radioactivity in various tissues after 20 minutes. Of 125I-VLDL cleared, the majority was found in the liver. The distribution of 125I-VLDL with apoB100β did not differ from that of 125I-VLDL with apoB100 in the five organs measured (Figure 3B).
Figure 3.

A, Clearance of 125I labeled apoB100 or B100-β VLDL in Ldlr−/− Apobec1−/− mice. n>3 for each group. B, Organ uptake expressed as the percentage of total 125I-VLDL in all organs measured. n=3 for each group. C, Conversion of injected 125I-VLDL after 2 hours, expressed as a percentage of injected VLDL. D, FFA release in heparin treated plasma from Ldlr−/− Apobec1−/− mice with apoB100 or apoB100-β measured over 1 hour.
To assess the conversion of VLDL to smaller particles in vivo, plasma samples were isolated from mice 2 hours after 125I-VLDL injection, pooled, and separated into VLDL, IDL, and LDL fractions using ultracentrifugation. Proportions of counts in the density fractions containing each class of lipoproteins were similar between mice received 125I-VLDL with apoB100 and those with apoB100-β (Figure 3C). Furthermore, the apoB100-β VLDL particles incubated with postheparin plasma released FFA at rates of 12±1 nmol FFA/min compared to apoB100 VLDL at 11±1 nmol FFA/ min (Figure 3D), suggesting that the lack of putative LDLR binding domain sequences of apoB does not affect the lipolysis of VLDL in Ldlr−/− Apobec1−/− apoB100-β mice.
Taken together, these experiments suggest that the absence of hyperlipidemia in LDLR-deficient mice having apoB100 without the putative receptor-binding sequences is likely because their VLDL particles are quickly cleared from the circulation before they become small, cholesterol-enriched LDL particles.
Role of ApoE-LRP–Mediated Clearance
To examine the specific roles of the LRP on apoB100-β particle uptake, we measured uptake of DiI labeled VLDL and LDL in mouse fibroblasts deficient in either LDLR (LDLR−/− LRP+/+), in LRP (LDLR+/+LRP−/−), or in both LDLR and LRP (LDLR−/− LRP−/−, negative control cells). Two hours after DiI-labeled apoB100-LDL was added to the medium, the uptake into LDLR+/+LRP−/− cells was significantly higher than in LDLR−/− LRP−/− negative control cells (Figure 4A). The uptake of apoB100-LDL by the LDLR−/− LRP+/+ cells was also higher than in negative control cells. Uptake of apoB100-β LDL was not increased by the expression of either LDLR or LRP. The opposite pattern of uptake was observed in studies with VLDL. In 30 minutes, DiI-labeled apoB100-β-VLDL was efficiently taken up by cells expressing LDLR, and particularly LRP, whereas no such increase over the uptake by negative control cells was found in cells given DiI-labeled-apoB100 VLDL (Figure 4B).
Figure 4.
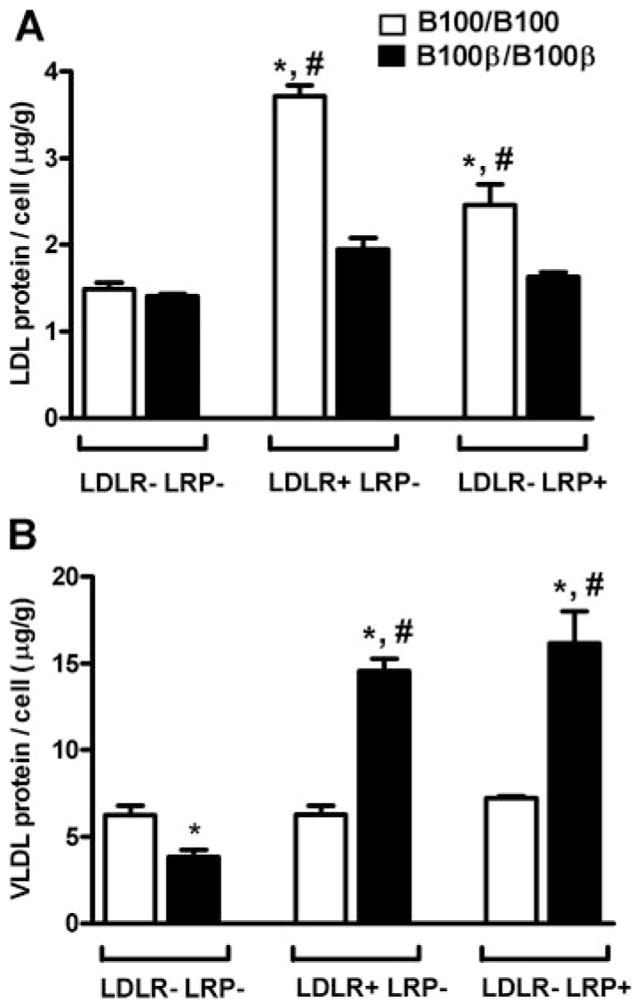
Uptake by fibroblasts of DiI-labeled lipoproteins with apoB100 or apoB100-β. A, Cells were incubated with DiI-labeled LDL 37°C for 2 hours or (B) VLDL for 30 minutes. Error represents SEM. *P<0.05, **P<0.005 against uptake in LDLR-LRP cells. #, P<0.05 between genotypes within the same cell system.
We next blocked hepatic LRP function using Ad-RAP to determine the role of the LRP in the clearance of apoB100-β VLDL in vivo. While basal cholesterol and triglyceride levels in plasma are significantly lower in Ldlr−/− Apobec1−/− mice with apoB100-β than those with normal apoB100, the levels 5 days after Ad-RAP injection were not different between the 2 groups of mice. FPLC analyses showed a similar accumulation of cholesterol in the VLDL fraction after 5 days in both groups (supplemental Figure I). These data imply that apoB100-β VLDL is removed by receptors inhibited by RAP, such as LRP. We also examined the contribution of HSPG binding by incubating LDLR−/− LRP−/− cells at 4°C with DiI-VLDL. The amount of VLDL released from the surface by heparinase after 2 hours was not significantly different (supplemental Figure II), suggesting that apoB100-β does not affect the binding ability of VLDL to proteoglycans.
To gain further insight into the apparent enhancement of lipoprotein clearance in Ldlr−/− Apobec−/− mice with apoB100-β, we analyzed the expression of the Apob, Apoe, and Lrp1 genes in the liver by real-time PCR. Although there was no Apob genotype effect on the mRNA levels for Apob and Lrp1, liver expression of Apoe was approximately twice as high in mice with apoB100-β as in mice with wild-type apoB100 (supplemental Figure III). These data, combined with the higher apoE:apoB protein ratio, suggest that the increased production of apoE protein may be contributing to the accelerated clearance of apoB100-β containing VLDL-remnants and the resistance to hyperlipidemia in the apoB100-β mice even in the absence of LDLR.
Ldlr−/− Apobec1−/− Mice With ApoB100-β Are Protected From Development of Atherosclerosis
High levels of LDL are a well-documented risk factor for atherosclerosis. Ldlr−/− Apobec−/− female mice with apoB100 had a significant size of lesions (34±6×103μm2), even when they were on NC diet and were as young as 4 to 5 months old (Figure 5A). In marked contrast, the apoB100-β mutation demonstrated a significant atheroprotective effect. Although 3 of 5 Ldlr−/− Apobec−/− mice heterozygous for apoB100-β had visible plaques (16±7×103μm2), there were absolutely no plaques seen in mice homozygous for the apoB100-β mutation. The overall effect of the Apob genotype on plaque development was P<0.002 by ANOVA. Feeding a HFW diet for 2 months accelerated the plaque development in the Ldlr−/− Apobec−/− mice with apoB100 (mean lesions size 54±8 x103μm2; n=5). In contrast, there were virtually no lesions present in apoB100-β mice but only very small foam cell aggregations (1.7±0.8×103μm2, P<0.002).
Figure 5.

A, Atherosclerotic plaque sizes at the aortic roots of 4-month-old female Ldlr−/− Apobec1−/− mice. Number of animals is in parentheses. B, Macrophage VLDL uptake. Ldlr−/− Apobec1−/− macrophages were incubated with DiI-labeled VLDL with apoB100 or with apoB100-β. Cellular florescence is expressed as Arbitrary Units (AU) per cell gram of cell protein.
To examine whether direct VLDL scavenging by macrophages rather than LDL accumulation is responsible for the dramatic differences in atherosclerosis, we isolated peritoneal macrophages from Ldlr−/− mice and incubated with equal amounts of DiI-labeled VLDL in the medium. Uptake of the DiI-VLDL with apoB100 by the macrophages was a little more enhanced compared to that with apoB100-β, but the differences were not statistically significant (Figure 5B). A similar result was obtained in the macrophages isolated from wild-type mice (data not shown), indicating that the scavenging by macrophages of the VLDL is not affected by the apoB100-β mutation.
Taken together our data demonstrate that, even in the absence of LDLR, a mutation in the putative receptor binding domains of apoB prevents LDL accumulation, and dramatically reduces atherosclerosis.
Discussion
LDL is generated in the circulation from VLDL produced by the liver after lipolysis and exchange of surface apolipoproteins. During this conversion, conformational changes occur in its structural protein, apoB100, allowing for the exposure of domain(s) that interact with LDLR.15–17 Exposure of the receptor-binding domain and subsequent binding of apoB100 to the LDLR is the major pathway for the clearance of LDL cholesterol by the liver, as illustrated by the marked accumulation of LDL in plasma of patients and in animals lacking LDLR.18 –21 Particles that lack full-length apoB, such as apoB48-containing chylomicron remnants, can acquire apoE which mediates efficient clearance of these particles by the LDLR, LRP, and other receptors which may act in concert with proteoglycans.22
To investigate the mechanisms for the uptake of apoB48-and apoB100-containing lipoproteins by the LDLR and by the LRP, Veniant et al previously characterized plasma lipoproteins in the Ldlr−/− mice homozygous for an “apoB48-only” allele or homozygous for an “apoB100-only” allele.24 The authors concluded that the LDLR plays a significant role in the clearance of both apoB100- and apoB48-containing lipoproteins, and that the LRP is important for apoB48-containing lipoproteins but has little if any capacity to remove apoB100-containing lipoproteins from the plasma. The “apoB100-only” Ldlr−/− mice are phenotypically identical to the Ldlr−/− Apobec1−/− mice with wild-type apoB100 we used in the current study. Interestingly, the plasma lipids and lipoprotein distribution in Ldlr−/− Apobec1−/− mice with mutant apoB100-β are very similar to “apoB48-only” Ldlr−/− mice, despite that apoB100-β retains the full length of the apoB protein. Both strains of mice have no substantial accumulation of LDL particles, suggesting that apoB100-β remnants, like apoB48-only remnants, are cleared via LRP in the absence of LDLR and that the length of apoB protein does not influence this process. However, whereas Veniant et al showed that LRP inhibition with RAP of “apoB48-only” Ldlr−/− mice leads to higher VLDL levels than in “apo-B100-only” Ldlr−/− mice,23 VLDL accumulation in the Ldlr−/− Apobec1−/− mice with apoB100 and those with apoB100-β were similar after LRP inhibition, suggesting that additional mechanisms may present in the protective effects noted in the apoB100-β–producing mice.
The Ldlr−/− Apobec1−/− mice with apoB100-β do not accumulate substantial LDL particles in plasma even when fed a HFW diet. This lack of LDL accumulation occurs despite the inability of apoB100-β-containing LDL to be cleared in vitro. Importantly, the livers of Ldlr−/− Apobec1−/− mice with apoB100-β appear to produce a smaller number of larger VLDL particles than the livers of the Ldlr−/− Apobec1−/− mice with apoB100, despite equal expression of the Apob gene and protein synthesis. However, the reduction of LDL cholesterol in the Ldlr−/− Apobec1−/− mice with apoB100-β is more than the reduction of apoB secreted. This is in contrast to the report by Crooke et al that Ldlr−/− mice treated with an apoB antisense oligonucleotides had a reduction of apoB mRNA by 74% but still had 48% levels of LDL-cholesterol compared to the pretreatment levels.24 It has long been recognized that the larger VLDL particles are removed faster and less likely converted to LDL than smaller VLDL, and a larger surface area of apoB100-β VLDL may allow more apoE to associate with the particle and facilitate LRP mediated uptake.25,26 The Ldlr−/− Apobec1−/− mice with apoB100-β have plasma lipoproteins containing 4-fold higher apoE protein per particle, and 2-fold higher Apoe gene expression in the liver than mice with apoB100. All together, these changes favor the enhanced clearance of apoB100-β containing particles via the LRP.
We also observed an enhanced degradation of apoB100-β in primary hepatocytes from the Ldlr−/− Apobec1−/− mice with apoB-100β in culture. Whether the accelerated degradation of apoB100-β results from its abnormal protein folding or is the consequence of enhanced turnover has yet to be determined. Although a limited apoB protein available for lipoprotein assembly could account for the larger size of VLDL, a question remains as to whether apoB100-β fails to form subsets of VLDL particles that are predestined to form LDL particles. Studies have demonstrated that a substantial amount of newly synthesized apoB protein is degraded rather than secreted, and that its interaction with LDLR channels apoB toward presecretory degradation.27–30 Reuptake of newly synthesized lipoproteins by LDLR can also attenuate VLDL secretion, and both apoE and apoB are important for this process.31,32 Loss of these regulations results in an increased secretion of apoB proteins and smaller, underlipidated VLDL particles in humans and mice that lack functional LDLR.33
The metabolism of lipoproteins with apoB100-β mutation is consistent with other observations. For example, truncations of apoB on the C-terminal side of amino acid 3500 result in more efficient clearance of VLDL.34 Individuals heterozygous for a R3480P mutation in apoB exhibit hypo-betalipoproteinemia because of a reduced conversion of VLDL to LDL, despite that this mutation caused reduced binding of LDL to the LDLR.35 Similarly, milder than expected hyperlipidemia in individuals with familial defective apolipoprotein B-100 attributable to mutations at R3500 has been attributed to an enhanced removal of apoE-containing VLDL and decreased production of LDL.36,37 The apoB100-β mutation may also affect a process of structural/ conformational change of apoB100 that is important for the in vivo generation of LDL particles as well as for LDLR binding, although interpretation is complex because amino acid changes disrupting amphipathic helices represented by sites A and B likely cause additional conformational changes of apoB on LDL and VLDL. Systematic replacements of the basic LDLR binding sequences with acidic or neutral residues would provide a potentially less disruptive and comprehensive approach. Chatterton et al hypothesized that the apoB100 “bow”, where a segment of apoB100 crosses over itself between amino acid residues 3000 and 3500, inhibits interaction of apoB100 protein with LDLR, hence inhibiting clearance.38 Because the apoB100-β mutation at amino acids 3147 to 3157 and 3359 to 3367 are within the proposed bow crossing structure, a mutation in these sequences may physically block or otherwise disrupt “bow” structure formation.
There is little doubt that the exposure of the positively charged domains of apoB100 to the lipoprotein surface after conformational changes is required for the effective clearance of LDL through LDLR. Considering the overall consequences of the mutations in the second half of the apoB100, however, it is tempting to speculate that the exposure of the positively charged domains of apoB100 may also inhibit the accumulation of apoE on the particles required for their apoE-mediated uptake via LDLR or LRP. This is consistent with the hypothesis raised by Veniant et al that the presence of the carboxyl half of apoB100 (amino acids 2153 to 4536) on the surface of the lipoprotein prevents the lipoprotein particle from binding a “sufficient dose of supplemental apoE” that is necessary for the lipoprotein particle to escape circulation via uptake by the LRP.23 Lack of the putative LDLR binding domains in apoB100-β may also prevent the secretion of newly packaged but underlipidated particles by enhancing the degradation of apoB through enhanced interactions between apoE and LDLR/LRP.
In conclusion, we have demonstrated that the mutation in the LDLR binding domains of apoB100 dramatically protects mice from both hypercholesterolemia and atherosclerosis that develop in the absence of LDLR. Our observations raise an intriguing possibility that an interference of the exposure of the putative LDLR-binding domains to the lipoprotein surface may indeed enhance remnant clearance through apoE-mediated mechanisms. This may be applicable as a potential therapeutic approach for preventing LDL accumulation in patients with familial hypercholesterolemia.
Supplementary Material
Acknowledgments
The authors thank Shinja Kim, Jennifer Wilder, and Svetlana Zhilcheva for technical help and José Arbonés-Mainar and Avani Pendse for critically reading the manuscript.
Sources of Funding
This work was supported by a grant HL42630 (to N.M.) and Texas AgriLife Research Project #8738 (to R.L.W.).
Footnotes
Disclosures
None.
References
- 1.Davidson NO, Anant S, MacGinnitie AJ. Apolipoprotein B messenger RNA editing: insights into the molecular regulation of post-transcriptional cytidine deamination. Curr Opin Lipidol. 1995;6:70–74. doi: 10.1097/00041433-199504000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Greeve J, Altkemper I, Dieterich JH, Greten H, Windler E. Apolipoprotein B mRNA editing in 12 different mammalian species: hepatic expression is reflected in low concentrations of apoB-containing plasma lipoproteins. J Lipid Res. 1993;34:1367–1383. [PubMed] [Google Scholar]
- 3.Boren J, Lee I, Zhu W, Arnold K, Taylor S, Innerarity TL. Identification of the low density lipoprotein receptor-binding site in apolipoprotein B100 and the modulation of its binding activity by the carboxyl terminus in familial defective apo-B100. J Clin Invest. 1998;101:1084–1093. doi: 10.1172/JCI1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toth LR, Smith TJ, Jones C, de Silva HV, Smithies O, Maeda N. Two distinct apolipoprotein B alleles in mice generated by a single ‘in-out’ targeting. Gene. 1996;178:161–168. doi: 10.1016/0378-1119(96)00360-5. [DOI] [PubMed] [Google Scholar]
- 5.Rauh G, Keller C, Schuster H, Wolfram G, Zollner N. Familial defective apolipoprotein B-100: a common cause of primary hypercholesterolemia. Clin Investig. 1992;70:77– 84. doi: 10.1007/BF00422946. [DOI] [PubMed] [Google Scholar]
- 6.Hirano K, Young SG, Farese RV, Ng J, Sande E, Warburton C, Powell-Braxton LM, Davidson NO. Targeted disruption of the mouse apobec-1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J Biol Chem. 1996;271:9887–9890. doi: 10.1074/jbc.271.17.9887. [DOI] [PubMed] [Google Scholar]
- 7.Morrison JR, Pászty C, Stevens ME, Hughes SD, Forte T, Scott J, Rubin EM. Apolipoprotein B RNA editing enzyme-deficient mice are viable despite alterations in lipoprotein metabolism. Proc Natl Acad Sci U S A. 1996;93:7154–7159. doi: 10.1073/pnas.93.14.7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folch J, Lees M, Slaone Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 9.Li X, Catalina F, Grundy SM, Patel S. Method to measure apolipoprotein B-48 and B-100 secretion rates in an individual mouse: evidence for a very rapid turnover of VLDL and preferential removal of B-48- relative to B-100-containing lipoproteins. J Lipid Res. 1996;37:210–220. [PubMed] [Google Scholar]
- 10.Véniant MM, Sullivan MA, Kim SK, Ambroziak P, Chu A, Wilson MD, Hellerstein MK, Rudel LL, Walzem RL, Young SG. Defining the atherogenicity of large and small lipoproteins containing apolipoprotein B100. J Clin Invest. 2000;106:1501–1510. doi: 10.1172/JCI10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altenburg M, Arbones-Mainar J, Johnson L, Wilder J, Maeda N. Human LDL receptor enhances sequestration of apoE4 and VLDL remnants on the surface of hepatocytes but not their internalization in mice. Arterioscler Thromb Vasc Biol. 2008;28:1104–1110. doi: 10.1161/ATVBAHA.108.164863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farkas MH, Swift LL, Hasty AH, Linton MF, Fazio S. The recycling of apolipoprotein E in primary cultures of mouse hepatocytes. Evidence for a physiologic connection to high density lipoprotein metabolism. J Biol Chem. 2003;278:9412–9417. doi: 10.1074/jbc.M208026200. [DOI] [PubMed] [Google Scholar]
- 13.Stephan ZF, Yurachek EC. Rapid fluorometric assay of LDL receptor activity by DiI-labeled LDL. J Lipid Res. 1993;34:325–330. [PubMed] [Google Scholar]
- 14.Atsma DE, Kempen HJ, Nieuwenhuizen W, van ’t Hooft FM, Pauwels EK. Partial characterization of low density lipoprotein preparations isolated from fresh and frozen plasma after radiolabeling by seven different methods. J Lipid Res. 1991;32:173–181. [PubMed] [Google Scholar]
- 15.Chen GC, Lau K, Hamilton RL, Kane JP. Differences in local conformation in human apolipoprotein B-100 of plasma low density and very low density lipoproteins as identified by cathepsin D. J Biol Chem. 1991;266:12581–12587. [PubMed] [Google Scholar]
- 16.Chen GC, Zhu S, Hardman DA, Schilling JW, Lau K, Kane JP. Structural domains of human apolipoprotein B-100. Differential accessibility to limited proteolysis of B-100 in low density and very low density lipoproteins. J Biol Chem. 1989;264:14369–14375. [PubMed] [Google Scholar]
- 17.Bradley WA, Hwang SL, Karlin JB, Lin AH, Prasad SC, Gotto AM, Jr, Gianturco SH. Low-density lipoprotein receptor binding determinants switch from apolipoprotein E to apolipoprotein B during conversion of hypertriglyceridemic very-low-density lipoprotein to low-density lipoproteins. J Biol Chem. 1984;259:14728–14735. [PubMed] [Google Scholar]
- 18.Brown MS, Goldstein JL. Familial hypercholesterolemia: genetic, biochemical and pathophysiologic considerations. Adv Intern Med. 1975;20:273–296. [PubMed] [Google Scholar]
- 19.Powell-Braxton L, Veniant M, Latval RD, Hiran KL, Won WB, Ross I, Dybdal N, Zlot CH, Young SG, Davidson NO. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998;4:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- 20.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883– 893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Havel RJ, Kita T, Kotite L, Kane JP, Hamilton RL, Goldstein JL, Brown MS. Concentration and composition of lipoproteins in blood plasma of the WHHL rabbit. An animal model of human familial hypercholesterolemia. Arteriosclerosis. 1982;2:467– 474. doi: 10.1161/01.atv.2.6.467. [DOI] [PubMed] [Google Scholar]
- 22.Mahley RW, Huang Y. Atherogenic remnant lipoproteins: role for proteoglycans in trapping, transferring, and internalizing. J Clin Invest. 2007;117:94–98. doi: 10.1172/JCI30889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veniant MM, Zlot CH, Walzem RL, Pierotti V, Driscoll R, Dichek D, Herz J, Young SG. Lipoprotein clearance mechanisms in LDL receptor-deficient “Apo-B48-only” and “Apo-B100-only” mice. J Clin Invest. 1998;102:1559–1568. doi: 10.1172/JCI4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crooke RM, Graham MJ, Lemonidis KM, Whipple CP, Koo S, Perera RJ. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. 2005;46:872– 884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Abrams JJ, Grundy SM, Ginsberg H. Metabolism of plasma triglycerides in hypothyroidism and hyperthyroidism in man. J Lipid Res. 1981;22:307–322. [PubMed] [Google Scholar]
- 26.Stalenhoef AF, Malloy MJ, Kane JP, Havel RJ. Metabolism of apolipoproteins B-48 and B-100 of triglyceride-rich lipoproteins in normal and lipoprotein lipase-deficient humans. Proc Natl Acad Sci U S A. 1984;81:1839–1843. doi: 10.1073/pnas.81.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borchardt RA, Davis RA. Intrahepatic assembly of very low density lipoproteins. Rate of transport out of the endoplasmic reticulum determines rate of secretion. J Biol Chem. 1987;262:16394–16402. [PubMed] [Google Scholar]
- 28.Twisk J, Gillian-Daniel DL, Tebon A, Wang L, Barrett PH, Attie AD. The role of the LDL receptor in apolipoprotein B secretion. J Clin Invest. 2000;105:521–532. doi: 10.1172/JCI8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillian-Daniel DL, Bates PW, Tebon A, Attie AD. Endoplasmic reticulum localization of the low density lipoprotein receptor mediates presecretory degradation of apolipoprotein B. Proc Natl Acad Sci U S A. 2002;99:4337– 4342. doi: 10.1073/pnas.072557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsson SL, Skogsberg J, Björkegren J. The low density lipoprotein receptor prevents secretion of dense apoB100-containing lipoproteins from the liver. J Biol Chem. 2004;279:831– 836. doi: 10.1074/jbc.M303057200. [DOI] [PubMed] [Google Scholar]
- 31.Williams KJ, Brocia RW, Fisher EA. The unstirred water layer as a site of control of apolipoprotein B secretion. J Biol Chem. 1990;265:16741–16744. [PubMed] [Google Scholar]
- 32.Blasiole DA, Oler AT, Attie AD. Regulation of ApoB secretion by the LDL receptor requires exit from the endoplasmic reticulum and interaction with ApoE or ApoB. J Biol Chem. doi: 10.1074/jbc.M710457200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nassir F, Xie Y, Patterson BW, Luo J, Davidson NO. Hepatic secretion of small lipoprotein particles in apobec-1−/− mice is regulated by the LDL receptor. J Lipid Res. 2004;45:1649–1659. doi: 10.1194/jlr.M300505-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Borén J, Ekström U, Agren B, Nilsson-Ehle P, Innerarity TL. The molecular mechanism for the genetic disorder familial defective apolipoprotein B100. J Biol Chem. 2001;276:9214–9218. doi: 10.1074/jbc.M008890200. [DOI] [PubMed] [Google Scholar]
- 35.Benn M, Nordestgaard BG, Jensen JS, Nilausen K, Meinertz H, Tybjaerg-Hansen A. Mutation in apolipoprotein B associated with hypobetalipoproteinemia despite decreased binding to the low density lipoprotein receptor. J Biol Chem. 2005;280:21052–21060. doi: 10.1074/jbc.M413877200. [DOI] [PubMed] [Google Scholar]
- 36.Schaefer JR, Scharnagl H, Baumstark MW, Schweer H, Zech LA, Seyberth H, Winkler K, Steinmetz A, März W. Homozygous familial defective apolipoprotein B-100. Enhanced removal of apolipoprotein E-containing VLDLs and decreased production of LDLs. Arterioscler Thromb Vasc Biol. 1997;17:348–353. doi: 10.1161/01.atv.17.2.348. [DOI] [PubMed] [Google Scholar]
- 37.Gaffney D, Forster L, Caslake MJ, Bedford D, Stewart JP, Stewart G, Wieringa G, Dominiczak M, Miller JP, Packard CJ. Comparison of apolipoprotein B metabolism in familial defective apolipoprotein B and heterogeneous familial hypercholesterolemia. Atherosclerosis. 2002;162:33– 43. doi: 10.1016/s0021-9150(01)00679-7. [DOI] [PubMed] [Google Scholar]
- 38.Chatterton JE, Phillips ML, Curtiss LK, Milne R, Fruchart JC, Schumaker VN. Immunoelectron microscopy of low density lipoproteins yields a ribbon and bow model for the conformation of apolipoprotein B on the lipoprotein surface. J Lipid Res. 1995;36:2027–2037. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.