

Abstract
Acquired uniparental disomy (aUPD) is a common and recurrent molecular event in human cancers, which leads to homozygosity for tumor suppressor genes as well as oncogenes, while retaining diploid chromosomal complement. Due to the lack of copy number change, aUPD is undetectable by comparative genome hybridization so the magnitude of this genetic change was underappreciated in the past. 9p aUPD was first described in 2002 in polycythemia vera (PV) patients. Since then, systematic application of genome-wide single nucleotide polymorphism arrays has demonstrated that 9p aUPD is the most common chromosomal aberration in myeloproliferative neoplasms (MPN) contributing to discovery of PV-defining mutation JAK2V617F. It was also found in other myeloid and lymphoid malignancies, though at a relatively lower frequency. By leading to JAK2V617F homozygosity, 9p aUPD plays a causal role in the development of PV and is also associated with less favorable clinical outcome. It is also possible that new targets other than JAK2V617F are present within 9p aUPD, which may contribute to diversity of PV outcome and phenotype. This review will summarize recent discoveries on 9p aUPD in hematological malignancies; discuss possible underlying mechanisms and potential roles of 9p aUPD in the PV pathogenesis, the relationship between 9p aUPD and JAK2V617F, and the possible new cancer-related targets within 9p aUPD region.
Keywords: acquired uniparental disomy (aUPD), myeloproliferative neoplasm (MPN), JAK2V617F, loss of heterozygosity (LOH), polycythemia vera (PV)
More than decade ago in a search for the molecular basis of somatic mutation leading to polycythemia vera, genome-wide screening found a high frequency of loss of heterozygosity (LOH) spanning about 40 megabases of chromosome 9p in the PV clone (ref. 6). However, the lesion was found to be genetically diploid, and by analogy with inherited uniparental disomy, it was called acquired uniparenteral disomy (aUPD). This discovery, as a cause of LOH, eventually facilitated the detection of the JAK2V617F mutation (ref. 20), a necessary lesion in the development of PV. However there is more to the story as shown by Wang et al. (ref. 10): the conversion of genetic variants from heterozygosity to homozygosity will likely have functional significance in malignant clone. The precise cellular effects of the duplication of rare genetic variants by aUPD remains to be elucidated.
Introduction
Uniparental disomy (UPD) refers to the situation in which both copies of a chromosome, or of part of a chromosome, is inherited from one chromosome of a single parent, instead of one copy each coming from the mother and father. Thus UPD causes no net change in the copy number in the affected genomic region(s) and in the context of somatic cell mutation is also referred to as copy-neutral loss of heterozygosity (cnLOH). Due to the lack of copy number change, UPD is not detectable by traditional cytogenetic methods, such as conventional cytogenetics analysis, array comparative genomic hybridization and fluorescence in situ hybridization. The concept of UPD as a mechanism for human genetic disease was first described in 1980 by Eric Engel1, on the basis of observations made in mice. However, the first case of UPD proven by molecular methods did not come until 1987 when the development of DNA-based polymorphic probes allowed the parental origin of chromosome homologs to be determined2. At that time, UPD was thought to be “an interesting rarity”3. However, today, there are more than 2,800 reports on UPD in inherited developmental disorders4 and UPD has been considered as an important diagnostic or prognostic factor for these syndromes5.
UPD not only can occur as a germ line lesion (constitutional UPD) but also can be acquired in somatic cells, referred to as acquired UPD (aUPD). The first observation of aUPD was made by Kralovics and Prchal in 2002 in a hematological malignancy, polycythemia vera (PV)6, in which 9p aUPD was observed in (33%) of PV subjects. During the past decade, significant advances in the genome-wide high-density single nucleotide polymorphism (SNP) arrays provided finer resolution and greatly facilitated assay of UPD. Numerous studies have demonstrated that aUPD is a common feature in a variety of human cancers7,8. Since the first description of 9p aUPD in PV6, a number of studies have indicated that the 9p aUPD appears to be the most common chromosomal aberration in PV patients9–11. It is also found in other MPN disorders12–15 as well in various myeloid and lymphoid malignancies7,8. In this review, we will summarize recent discoveries on 9p aUPD in hematological malignancies; discuss the possible underlying mechanisms and potential roles of 9p aUPD in the PV and other hematological malignancies pathogenesis, the relationship of 9p aUPD and other key cancer genes and driver mutations, and the molecular bases of the genetic effects of 9p aUPD on the clinical outcomes.
Mechanism of aUPD
In cancer, aUPD has been detected across entire chromosomes or chromosome segments of varying length. Unlike constitutional UPD, our knowledge about the mechanisms underlying aUPD is far from complete. Several different mechanisms have been suggested for whole-chromosome aUPD and segmental aUPD as previously reviewed7,8. Whole-chromosome aUPD might result from incomplete chromosomal segregation during mitosis, in which one chromosome is lost due to anaphase lag and the remaining chromosome is then duplicated by mitotic non-disjunction7. This represents an attempt to correct for the unbalanced chromosomal loss by using the remaining chromosome as a template. Both anaphase lag and mitotic non-disjunction errors can lead to homozygosity of a chromosome or its part. The mechanism for segmental aUPD, however, is thought to be more complicated and three mechanisms have been suggested. First, mitotic nonallelic homologous recombination between highly homologous, low-copy repeats in the G2 phase of the cell cycle is suggested to be the most likely cause7,8,16,17. Second, it could be a result of double-strand DNA break repair error. Double-strand DNA break is a common event in cancer. In such a context, a compensatory reduplication of the remaining chromosomal fragment takes place as an attempt to repair the break. This mechanism is compatible with the finding that aUPD was detected at an extremely high frequency of the BRCA-associated ovarian carcinomas18 as deficiency in the BRCA1/2 proteins results in the loss if homologous recombination and loss of homologous recombination results in the inappropriate repair of double strand DNA breaks. The third proposed mechanism for aUPD is gene conversion. But since most detectable segmental aUPD events extend beyond 1 kilobase pairs (kb) while gene conversion ordinarily involves only small regions of DNA sequence (300 bp to 1 kb) transforming one allele to the other allele, it seems unlikely that gene conversion to be the cause of segmental aUPD7,19.
Frequency of 9p aUPD in hematological malignancies
The most commonly affected region of aUPD in hematological malignancies is on chromosome 9p. The reported frequencies of 9p aUPD in hematological malignancies vary greatly and largely depend on the disease subtypes and the methodology used for UPD detection (see Figure 1 and Table 1). In 2002, Kralovics and Prchal first reported 9p aUPD in polycythemia vera (PV) in about a third of PV subjects studied6. To identify UPD, the group analyzed 382 autosomal microsatellite markers, which covered the genome at 10 cM density. Using the same technology, Kralovics and others further examined a larger patient cohort in 2003 including not only PV patients but also patients with other MPN diseases including essential thrombocythemia (ET) and primary idiopathic myelofibrosis (PMF)9. They confirmed the high frequency of 9p UPD in PV subjects and further found it in one of fifteen of ET patients9. Subsequently the study was extended to an even larger cohort of MPN patients and found 9p aUPD again in a third of PV patients, 3% of ET patients and 22% of PMF patients20. Besides the low-density polymorphic microsatellite markers6,9,20, researchers also used multiple ligation probe amplification technique for UPD detection but this technique didn’t appear to increase the sensitivity of UPD detection15.
Figure 1. The frequency of 9p aUPD in hematological malignancies.
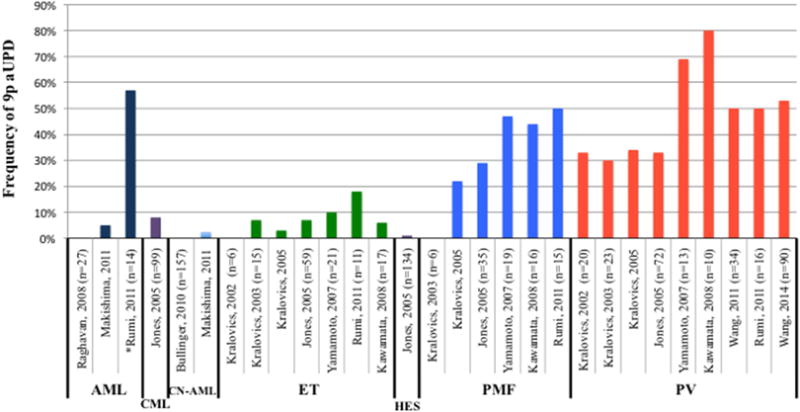
The source data and references of this figure were shown in the Table 1. AML, acute myeloid leukemia; CN-AML, cytogenetically normal AML. CML, chronic myeloid leukemia; ET, essential thrombocythemia; HES, Idiopathic hypereosinophilic syndrome; PMF, primary myelofibrosis; PV, polycythemia vera. *All the AML patients in the study of Rumi et al24. were secondary AML that transformed from PV, ET or PMF.
Table 1.
Frequency of 9p aUPD in hematological malignancies.
| Study group | Disease | Number of patients | Frequency of 9p aUPD | Method for detecting 9p aUPD | Description of subclonal 9p aUPD | Publication date | Reference |
|---|---|---|---|---|---|---|---|
| Kralovics, et al | PV,ET | 26 | 6/20 (33%) in PV; 0/6(0%) in ET |
382 microsatellite markers | No | 2002 | #6 |
| Kralovics, et al. | PV, ET, PMF | 44 | 7/23 (30%) in PV; 1/15 (7%) in ET; 0/6(0%) in PMF |
3 microsatellite markers | No | 2003 | #9 |
| Kralovics, et al. | PV, ET, PMF, CML | 244 | 34% in PV; 3% in ET; 22% in IMF; |
10 microsatellite markers | No | 2005 | #20 |
| Jones, et al. | PV, ET, PMF, CML, AML, Idiopathic HES, Mastocytosis | 480 | 24/72 (33%) in PV; 4/59 (7%) in ET; 10/35 (29%) in PMF; 2/134(1%) idiopathic HES; 8/99 (8%) in CML |
multiplex ligation probe amplification | No | 2005 | #15 |
| Yamamoto, et al. | PV, ET, PMF, AML, ALL | 138 | 9/13 (69%) in PV; 2/21 (10%) in ET; 9/19 (47%) in PMF; |
Affymetrix 50K SNP arrays | Yes | 2007 | #12 |
| Raghavan, et al. | AML | 27 | 0/27 (0%) | Affymetrix 10K SNP array | No | 2008 | #23 |
| Kawamata, et al. | PV, ET, PMF | 43 | 5/10 (50%, clonal) in PV; 3/10 (30%, subclonal) in PV; l/17 (6%,subclonal)in ET; 7/16 (44%, subclonal) in PMF; |
Affymetrix 50K SNP array | Yes | 2008 | #14 |
| Wang, et al. | PV | 34 | 36% (Complete); 14% (Fractional) |
Illumina Human610 SNP | Yes | 2011 | #11 |
| Rumi, et al. | PV, ET, PMF, AML | 29* | 8/16 (50%) in PV; 2/11 (18%) in ET; 10/15 (50%) in PMF; 8/14 (57%) in AML; |
array Affymetrix SNP 6.0 array Illumina Human610 SNP | No | 2011 | #24 |
| Wang, et al. | PV | 90 | 15/31 (48%, discovery cohort); 34/59 (58%, extended cohort) |
Affymetrix SNP 6.0 array Illumina Human610 SNP | Yes | 2014 | #10 |
| Bullinger, et al. | CN-AML | 157 | 0/157 (0%) | Affymetrix 250K SNP arrays | No | 2010 | #22 |
| Makishima, et al. | AML, ALL | – | 2.6% (CN-AML); 5% (AML); |
– | – | 2011 | #8 |
a total of 69 samples from 29 paients were investigated. Five PV and five PMF were transformed from ET. All 14 AML were transformed from PV, ET or PMF.
Over the past decade, the advent of high-throughput genome-wide single nucleotide polymorphism (SNP) array technology allowed allele-specific measurement of copy-number alterations and provided high resolution maps and with greater sensitivity to assay aUPD, to the extent that even subclonal events10–12 could be detected. In 2007, Yamamoto and others developed a method for a quantitative measure (as opposed to a positive/negative binary measure) as the magnitude of aUPD based on SNP genotyping signal intensities12. This improvement increased the sensitivity of detection of aUPD to as little as 8% of the subclone, according to a recent report from Wang and colleagues10, see Figure 2. With these advances in the SNP-based technology and the improved analytical algorithms, an unexpectedly high frequency of 9p aUPD has been revealed in MPN10–12,14 (Figure 1 and Table 1). In summary, 9p aUPD is found in 48% to 80% of patients in PV; about half the patients with primary myelofibrosis (PMF); 6% to 18% in ET; ~5% in acute myeloid leukemia (AML) that occurs de novo8,22,23, but up to ~57% in AML that had transformed from chronic ET, PV or PMF24.
Figure 2. Quantification of the fraction of cells harboring UPD in PV granulocytes.
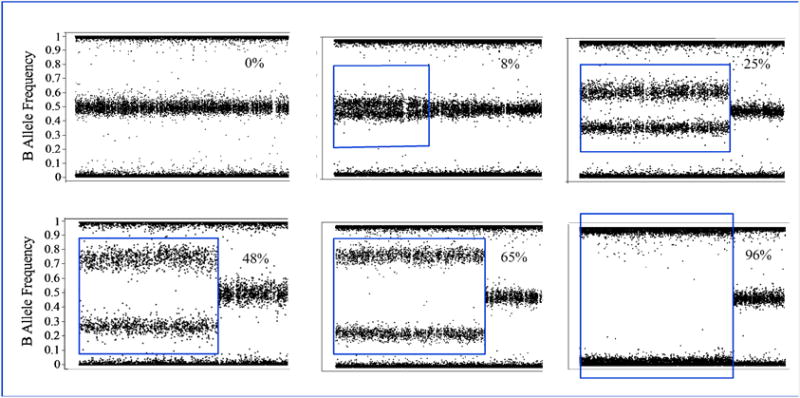
Each panel shows a different patient with increasing level of UPD, from 0 to 96%, from left to right and top to bottom. Y-axis are B-allele frequencies taken from Illumina SNP Array data. Each dot represents a single SNP marker. Markers are arranged in ordinal number order across the p-arm of chromosome 9. The middle band represents heterozygous markers. Homozygous markers are a 0 or 1.0 on the Y axis. The calculated percent of the population harboring LOH is given in the upper right corner of each panel. Red box demarks the region of LOH. Other data must be used to establish the copy number of this LOH to distinguish diploid from hemizygous state. In PV, the LOH is nearly always diploid or, occasionally, triploid. Data taken from Wang et al.10
In 2006, Scott and colleagues analyzed over one thousand hematopoietic colonies from 17 patients with PV who had a heterozygous sequence pattern. Surprisingly, they were able to detect homozygous mutant erythroid burst-forming units (BFU-Es) in all the patients13 suggesting small homozygous mutant subclones were present in all patients. Hence, the reported frequencies on 9p aUPD could be underestimated since the minimum detection level of fractional aUPD by SNP array was 8% based on a most recent report10. These findings should be followed up with evaluation of chromosome copy number in single cells by next generation sequencing25.
The pathogenic consequences of 9p aUPD in hematological malignancies
Recurrent aUPD regions and chromosomes have been identified in a variety of human cancers and many render homozygous mutations in key genes that function in the initiation and/or progression of cancers7,8,26. The most common reported are JAK2 and CDKN2A on 9p, FLT3 on 13q, TP53 in 17p, in addition to other less common instances including WT1, CBL, RUNX1, TET. aUPD can lead to regions that are homozygous for somatically mutated, deleted, methylated, or imprinted genes. The pathogenic consequences of aUPD depend on the functions of these key genes residing within the aUPD region, and whether or not the pathogenic alleles are duplicated. If the affected region includes a tumor suppressor gene, such as TP53, which is frequently seen in lymphomas and chronic lymphocytic leukemia (CLL), aUPD can lead to homozygosity of the somatic mutations that inactivate TP53. The biological impact is considered to be equivalent to cases with TP53 mutation plus 17p deletion27. Thus, aUPD may convey a growth advantage to the cell8. If the affected region includes an oncogene, such as JAK2 in 9p aUPD and FLT3 in 13q aUPD, the aUPD duplicates the activating oncogenic mutations and increases the oncogenic activity through a double dose of the mutated gene product8. In addition to homozygosity of a mutation, aUPD can also lead to homozygous deletion. For example, chromosome 13q aUPD is a common event found in early-stage CLL resulting in homozygous deletion of the micro-RNA-15a/16-1 microRNA cluster, which function as a tumor suppressor and controls B cell proliferation28. Numerous studies have suggested aUPD is a frequent genetic mechanism of enhancing tumor suppressor inactivation or oncogene activation7,8,26.
aUPD was suggested to be associated with tumor types and/or subtypes7,8,26. Studies of aUPD in MPN showed that it is almost exclusively present on chromosome 9p and is often associated with a gain-of-function mutation in JAK2, the JAK2V617F mutation20. JAK2V617F mutation is the most common somatic event reported in MPN patients. It is found in over 95% of PV patients, 55% of ET patients and 65% of PMF patients20,21,29,30. The most intriguing question that arose after the discovery of JAK2V617F mutation is how a single mutation gives rise to three clinically distinct diseases. The underlying mechanisms are still unclear but three hypotheses have been suggested based on the clinical biological and pathological data31. One hypothesis was called “gene-dosage hypothesis”, which suggests a correlation between disease phenotype and the JAK2V617F allele burden31. Interestingly, JAK2V617F mutational burden varies greatly from zero to 100% (homozygosity) in MPN patients. Homozygous JAK2V617F, resulted from 9p aUPD is present in 30–50% of PV patients whereas it is rarely seen in ET10,11,15,20,21,29,32,33, leading to the thoughts that homozygous JAK2V617F may play an essential role in determining PV phenotype.
Indeed, a number of studies on JAK2V617F transgenic mice models have suggested that the ratios of mutant to wild-type JAK2 and the levels of JAK2V617F expression are critical factors for the phenotypic manifestation in mice34–41. Low ratios of JAK2V617F to wild-type JAK2, i.e., low levels of JAK2V617F load (heterozygous JAK2V617F) are able to induce ET-, PMF- and PV-like disease38, but high levels of JAK2V617F load (nearly homozygous JAK2V617F) developed only PV-like disease38. A critical role of gene dosage effect is also indicated by a recent study on erythroid colonies13. Scott and others demonstrated that homozygous JAK2V617F mutant erythroid colonies are present in almost all patients with PV but are very rare in patients with ET. As it is well known that PV, ET, and PMF are closely related but clinically distinct disorders, each defined by different cell lineage increase; i.e. erythrocytes in PV, platelets in ET and splenomegaly and marrow fibrosis in PMF. MPN patients are at variable risk of disease progression, including evolution from ET to PV, from both ET and PV to PMF, and ET, PV and PMF to acute myeloid leukemia (AML). Li and colleagues recently demonstrated in mice that JAK2V617F homozygosity drives a switch from ET-like to PV-like phenotype42, direct genetic evidence that homozygosity of JAK2V617F modulates tumor phenotype.
In addition to the critical role in modulating PV phenotype, homozygous JAK2V617F has also been suggested to be associated with prognostically less favorable clinical profiles. Compared with the heterozygous JAK2V617F, homozygous expression of JAK2V617F in hematopoietic cells in mice resulted in a PV-like disease exhibited significantly greater reticulocytosis, leukocytosis, neutrophilia and thrombocytosis, marked expansion of erythroid progenitors and erythropoietin-independent erythroid colonies, larger spleen size, and accelerated bone marrow fibrosis41, reduced numbers of platelets and reduced platelet survival42. This is consistent with the clinical observation that patients bearing homozygous JAK2V617F tend to have a higher hemoglobin levels, increased incidence of pruritus and are more likely to progress to post-PV myelofibrosis24,43–45.
The relationship of 9p aUPD and JAK2V617F
9p aUPD is thought to be a later genetic event that occurs after the acquisition of JAK2V617F to provide a growth advantage to cells by rendering homozygous JAK2V617F. However, it has been recently recognized that 9p aUPD can occur before JAK2V617F and at an early step of MPN development10,46. In 2011, Vilaine and colleagues reported two PV cases where the fractions of cells with 9p aUPD in their granulocytes were much higher than that of the JAK2V617F mutant46 indicating that the acquisition of aUPD preceded JAK2V617F in both patients. The authors proposed a model of MPN certain haplotypes, specifically the “46/1 haplotype” or other nonidentified genetic events, may predispose carriers to early homozygous recombination of wild-type JAK2 and JAK2V617F facilitates additional homozygous recombination of the JAK2 mutant allele and eventually leads to PV phenotype46. This was the first suggestion that that 9p aUPD could have preceded JAK2 mutation.
More recently, Wang and her colleagues systematically investigated the relationship between JAK2V617F and 9p aUPD in a large PV cohort by whole-exome sequencing (WES), high-resolution SNP array and deep targeted sequencing10. Four PV subgroups were defined based on the quantitative relationship between JAK2V617F allelic fraction and the fraction of genomes harboring 9p aUPD. They found that the granulocytes of at least 10% PV patients harbored 9p aUPD at twice the levels of the JAK2V617F allelic burden (designated group III, see Figure 3), which they interpreted as aUPD with heterozygous JAK2V617F (ref. 10). This observation was validated in two additional independent cohorts of PV patients (Figure 3).
Figure 3. Relationship between JAK2V617F and 9p aUPD.
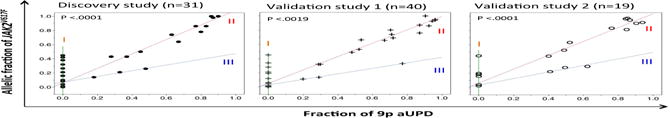
Groups I, II and III are defined by the ratio of JAK2V617F to aUPD. From Wang, et al. ref. 10, Figure 1.
Although any single patient in subgroup-III taken in isolation could be explained as a mixture of granulocytes from Subgroups I (JAK2V617F without detectable 9p aUPD) and II (homozygous JAK2V617F), the best explanation for the consistent pattern of Subgroup III observed in all 3 independent cohorts was 9p aUPD and JAK2V617F reside in the same cell with heterozygous JAK2V617F in all such patients10. This represented a novel genotype, stable over time (see below) not previously described. The forth subgroup consisted of rarer patients with trisomy 9p in which two copies of JAK2V617F were present. At a frequency of less than 3%, this configuration is the most enigmatic. Further study in large cohorts will be needed to elucidate the relevance of these genetic patterns to the clinical profiles.
Multiple levels of aUPD in the Vilaine et al.46 patients, who were both under treatment, suggested multiple PV subclones. Wang et al.10 followed 17 of their patients for 2–6 years and observed clonal changes in 6 of them. Half increased the dosage of JAK2 mutant relative to aUPD and half decreased. Since all patients were under treatment it was unclear whether these changes reflected the natural progression of the disease or not.
These observations, based on a small patient cohort, has not shed yet light on the phenotypic consequences of aUPD alone and thus large-scale studies are needed to provide more insights into this. One hypothesis is that the presence of aUPD before acquisition of JAK2 mutation may confer some selective advantage over the wildtype cells and this selective advantage could determine clonal heterogeneity in a given patient. Nonetheless, these results are consistent with the possibility of a genetically heterogeneous reservoir of mutant cells and hint at complexity in managing these patients. By normalizing the level of JAK2V617F to the level of UPD, it has been possible to genetically dissect PV and more further reveal the subclonal architecture of the disease.
The evolution of 9p aUPD during disease progression
Both the length of aUPD and the fraction of cells carrying aUPD can change during treatment and disease progression10,11,24. Wang and others first described the stepwise extension of 9p aUPD in the granulocytes of a PV patient11 wherein three separate segments were identified on chromosome 9p, each in decreasing cellular fraction, suggesting UPD extended itself in three consecutive salutatory events. Similarly, the stepwise pattern of 9p aUPD was observed in another PV patient as shown in Figure 4 by Wang et al10. In addition, Rumi et al24. describe the evolution of 9p aUPD over the course of disease progression, from PMF to PV and then to AML. In one case, they detected the same stepwise pattern of 9p aUPD as shown in Figure 4, with the telomeric 9p aUPD present in the majority of cells but the centromeric 9p aUPD present only in a subpopulation of the cells24. Upon transformation to PV, the fraction of cells carrying the centromeric 9p aUPD extended to the entire population of cells and the size of 9p aUPD extended from the telomeric segment to the whole 9p arm. Interestingly, it seems like the genomic location of the telomeric breakpoints of the stepwise 9p aUPD reported in all these cases are very close. It is still unclear why 9p breaks more often at that location, why segmental aUPD occurs first and extends to the whole-arm aUPD.
Figure 4. Stepwise expansion aUPD on chromosome 9p.
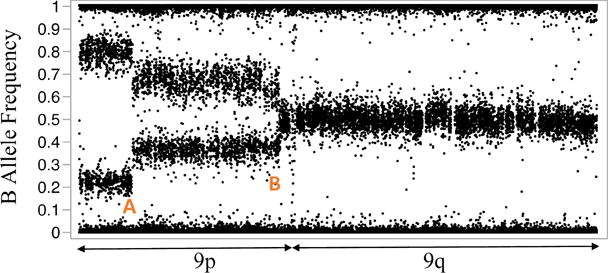
Data from granulocytes from a PV patient with evolving 9p aUPD. Y-axis is the B-allele frequency (see Figure 2.) generated from SNP array data. If a subpopulation of cells harbors LOH, the heterozygous alleles split into two bands separated symmetrically apart from 0.5. Distinct breakpoints observed at positions A and B, indicate the presence of distinct subclonal populations. Data taken from Wang et al.10
Wang and co-workers systematically investigated the evolution of JAK2V617F and 9p aUPD in 36 longitudinal samples from 17 PV patients during a 2–6 year period10. They demonstrated that both the allelic fractions of JAK2V617F and 9p aUPD could change, increasing or decreasing, during the period of observation. But the JAK2V617F and aUPD ratios remained stable in two thirds of the patients. In the remaining one third of patients, the ratios of JAK2V617F and 9p aUPD significantly changed. Three patients progressed from prognostically more favorable heterozygous JAK2V617F to less favorable homozygous JAK2V617F clones. And 3 other patients transformed their JAK2 genotype and gained new clones10. This was the first report of clonal stability in PV patients. The contributing factors are still unclear but the impact of treatment strategies on clonal stability is clearly an important question.
Other cancer-related genes in 9p aUPD
Analyzing aUPD regions may pinpoint novel candidate genes for mutation screening and it may also help to distinguish cancer driver genes from the passengers. An illustrative example is the discovery of JAK2V617F mutation, which was made by analysis of the 9p aUPD region. To identify if there are genes other than JAK2 that could also contribute to PV pathogenesis, Wang and colleagues integrated the WES mutation data with aUPD data10. Since aUPD can lead to homozygosity of either the common or the rare allele, they sought, genes with recurrent loss of common alleles (i.e., the wild-type allele) within the 9p aUPD region. Among the list of 48 genes with recurrent loss of a common allele, 6 of the top 10 are involved in regulation of cell division. Nearly half of the 48 genes play a role in either cell division, transcriptional and epigenetic regulation, or tumor suppression. The latter includes KDM4C and SMARCA210. In the case of 9p aUPD, multiple genes may be working together in a coordinated fashion in order to modulate the tumorigenic effect. This suggests that the aUPD region may contribute to the tumor phenotype, beyond its impact on JAK2. A larger number of patients from the type III subgroup are needed to test this hypothesis. Thus these early results suggested that many more genes than previously thought may be involved in PV pathogenesis and their discovery could provide valuable insights and important clinical applications.
Conclusions and perspectives
Loss of heterozygosity may be arrived at in two ways: either loss of one chromosome, to produce a hemizygous region, or loss of one with a duplication of the other to retain the diploid copy number. In some tumors, regions of the genome consistently exhibit hemizygous loss, e.g., 3p in 90% of clear cell renal cell carcinoma. Even 9p can exhibit large regions of hemizygous loss in other hematologic cancers, such as ALL47. In the myeloid lineage LOH, with many examples now, appears to be always copy number neutral on 9p, suggesting the interesting possibility of lineage-specific haploinsufficiency in the region. Possibly studies of syntonic loci in mouse models could shed light on this.
Apparently there is precise balancing of the copy number in various tumors contributing to tumors’ evolution or definition of its phenotype. The 9p aUPD might only target the mutant JAK2; however this seems unlikely as described above. First, once the distal arm of 9p has been duplicated, the region of duplication may further extend toward the centromere, suggesting further selection occurs as the region of aUPD expands. Second, as discussed above, there are dozens of genes that play a possible role in tumorigenesis that loose the common allele more often than expected by chance in the aUPD event, suggesting rare, possibly deleterious alleles are advantageous to the tumor. Third, aUPD occurs prior to mutation of JAK2 in about 10% of PV patients, demonstrating that mutant JAK2 do not need to be homozygous to define PV phenotype. These observations are consistent with the hypothesis of “cancer gene island” proposed by Elledge and coworkers48, which posits that recurrent regions of LOH consist of many genes contributing collectively to the tumor phenotype. The manner in which the maintenance of rare alleles is balanced by the haploinsufficiency remains to be elucidated. In any case, the forces of selection and the loci upon which they act that lead to 9p aUPD represent an important model for the development and maintenance of cancer, and an important area of research going forward.
Highlights.
Highlight 1: The most commonly affected region of aUPD in hematological malignancies is on 9p
Highlight 2: 9p aUPD can precede JAK2V617F though the pathogenic consequences are still unclear
Highlight 3: Both the size and the fraction of aUPD can change during disease progression
Highlight 4: 9p aUPD may contribute to the tumor phenotype beyond its impact on JAK2
Acknowledgments
DW and LW were supported by U54HG003273 from NHGRI and NIH-P01CA108671) by JP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors indicated no potential conflicts of interest.
References
- 1.Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980;6:137–143. doi: 10.1002/ajmg.1320060207. [DOI] [PubMed] [Google Scholar]
- 2.Creau-Goldberg N, et al. Maternal origin of a de novo balanced t(21q21q) identified by ets-2 polymorphism. Hum Genet. 1987;76:396–398. doi: 10.1007/BF00272452. [DOI] [PubMed] [Google Scholar]
- 3.Warburton D. Uniparental disomy: a rare consequence of the high rate of aneuploidy in human gametes. Am J Hum Genet. 1988;42:215–216. [PMC free article] [PubMed] [Google Scholar]
- 4.Liehr T. Cases with uniparental disomy. 2015 http://upd-tl.com/upd.html.
- 5.Liehr T. Cytogenetic contribution to uniparental disomy (UPD) Mol Cytogenet. 2010;3:8. doi: 10.1186/1755-8166-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Experimental hematology. 2002;30:229–236. doi: 10.1016/s0301-472x(01)00789-5. [DOI] [PubMed] [Google Scholar]
- 7.Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med. 2009;15:120–128. doi: 10.1016/j.molmed.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res. 2011;17:3913–3923. doi: 10.1158/1078-0432.CCR-10-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kralovics R, et al. Comparison of molecular markers in a cohort of patients with chronic myeloproliferative disorders. Blood. 2003;102:1869–1871. doi: 10.1182/blood-2003-03-0744. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, et al. The relationship of JAK2(V617F) and acquired UPD at chromosome 9p in polycythemia vera. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2014;28:938–941. doi: 10.1038/leu.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Swierczek S, Hickman K, Hakonarson H, Prchal JT. Convergent mechanisms of somatic mutations in polycythemia vera. Discovery medicine. 2011;12:25–32. [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto G, et al. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007;81:114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108:2435–2437. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- 14.Kawamata N, et al. Genetic profiling of myeloproliferative disorders by single-nucleotide polymorphism oligonucleotide microarray. Experimental hematology. 2008;36:1471–1479. doi: 10.1016/j.exphem.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones AV, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–2168. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 16.van Dartel M, Hulsebos TJ. Amplification and overexpression of genes in 17p11.2 ~ p12 in osteosarcoma. Cancer Genet Cytogenet. 2004;153:77–80. doi: 10.1016/j.cancergencyto.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Stephens K, et al. Interstitial uniparental isodisomy at clustered breakpoint intervals is a frequent mechanism of NF1 inactivation in myeloid malignancies. Blood. 2006;108:1684–1689. doi: 10.1182/blood-2005-11-011486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh CS, et al. Genome-wide loss of heterozygosity and uniparental disomy in BRCA1/2-associated ovarian carcinomas. Clin Cancer Res. 2008;14:7645–7651. doi: 10.1158/1078-0432.CCR-08-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liehr T. Uniparental Disomy (UPD) in Clinical Genetics: A Guide for Clinicians and Patients. Springer Berlin Heidelberg; 2014. [Google Scholar]
- 20.Kralovics R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. The New England journal of medicine. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 21.James C, et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 22.Bullinger L, et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2010;24:438–449. doi: 10.1038/leu.2009.263. [DOI] [PubMed] [Google Scholar]
- 23.Raghavan M, et al. Segmental uniparental disomy is a commonly acquired genetic event in relapsed acute myeloid leukemia. Blood. 2008;112:814–821. doi: 10.1182/blood-2008-01-132431. [DOI] [PubMed] [Google Scholar]
- 24.Rumi E, et al. Identification of genomic aberrations associated with disease transformation by means of high-resolution SNP array analysis in patients with myeloproliferative neoplasm. American journal of hematology. 2011;86:974–979. doi: 10.1002/ajh.22166. [DOI] [PubMed] [Google Scholar]
- 25.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fitzgibbon J, et al. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer research. 2005;65:9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- 27.Zenz T, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114:2589–2597. doi: 10.1182/blood-2009-05-224071. [DOI] [PubMed] [Google Scholar]
- 28.Lehmann S, et al. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia. Cancer. 2008;112:1296–1305. doi: 10.1002/cncr.23270. [DOI] [PubMed] [Google Scholar]
- 29.Levine RL, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 30.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2010;24:1128–1138. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Passamonti F, Rumi E. Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica. 2009;94:7–10. doi: 10.3324/haematol.2008.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baxter EJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(05):1054–1061. 71142–9. doi: 10.1016/S0140-6736. [DOI] [PubMed] [Google Scholar]
- 33.Vannucchi AM, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–846. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 34.Zaleskas VM, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PloS one. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bumm TG, et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer research. 2006;66:11156–11165. doi: 10.1158/0008-5472.CAN-06-2210. [DOI] [PubMed] [Google Scholar]
- 36.Lacout C, et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 37.Wernig G, et al. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shide K, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2008;22:87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 39.Xing S, et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood. 2008;111:5109–5117. doi: 10.1182/blood-2007-05-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiedt R, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–3940. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 41.Akada H, et al. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115:3589–3597. doi: 10.1182/blood-2009-04-215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, et al. JAK2V617F homozygosity drives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustain disease. Blood. 2014;123:3139–3151. doi: 10.1182/blood-2013-06-510222. [DOI] [PubMed] [Google Scholar]
- 43.Passamonti F, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2010;24:1574–1579. doi: 10.1038/leu.2010.148. [DOI] [PubMed] [Google Scholar]
- 44.Tefferi A, et al. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer. 2006;106:631–635. doi: 10.1002/cncr.21645. [DOI] [PubMed] [Google Scholar]
- 45.Barosi G, et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007;110:4030–4036. doi: 10.1182/blood-2007-07-099184. [DOI] [PubMed] [Google Scholar]
- 46.Vilaine M, et al. Homologous recombination of wild-type JAK2, a novel early step in the development of myeloproliferative neoplasm. Blood. 2011;118:6468–6470. doi: 10.1182/blood-2011-08-372813. [DOI] [PubMed] [Google Scholar]
- 47.Sulong S, et al. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood. 2009;113:100–107. doi: 10.1182/blood-2008-07-166801. [DOI] [PubMed] [Google Scholar]
- 48.Solimini NL, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337:104–109. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]