

Abstract
The INPPL1 (inositol polyphosphate phosphatase-like 1) gene encodes the inositol phosphatase, SHIP2 (for src homology 2 domain-containing inositol phosphatase 2). SHIP2 functions to dephosphorylate, and negatively regulate, the lipid second messenger phosphatidylinositol (3,4,5)P3. SHIP2 has been well studied in the area of insulin resistance and obesity but has roles in cancer and other disorders. Recently, it was reported that mutations in INPPL1 cause opsismodysplasia, a rare, autosomal recessive severe skeletal dysplasia. This review focuses on the mutations associated with opsismodysplasia and explores the role of INPPL1/ SHIP2 in skeletal development.
INTRODUCTION
Skeletal dysplasias are a large family of disorders characterized by disturbances of cartilage and bone growth and development leading to shape and size abnormalities of the skeleton. The family is markedly heterogeneous with 456 clinically distinct disorders caused by mutations in 226 genes.1 The chondrodysplasias are a subset of skeletal dysplasias and result from the abnormal growth of cartilage of the long bones, which typically manifests as disproportionate short stature. Opsismodysplasia (OPS) (OMIM 258480) is a rare skeletal chondrodysplasia primarily characterized by growth plate defects and delayed bone maturation. Clinical features at birth are variable but include short limbs, small hands and feet, macrocephaly with an anterior fontanel and facial dysmorphism including prominent brow ridge, depressed nasal bridge, anteverted nose and relatively long philtrum (http://omim.org/entry/258480). Typical radiographical features include short long bones with markedly delayed epiphyseal mineralization, metaphyseal cupping, short metacarpals and phalanges, and severe platyspondyly.2–17 Radiographs of an individual with severe OPS are shown in Figure 1. Recurrence in the siblings and consanguinity suggested an autosomal recessive mode of inheritance. In 2013, it was reported that OPS is caused by homozygous or compound heterozygous mutations in the INPPL1 (inositol polyphosphate phosphatase-like 1) gene encoding the SHIP2 protein (for src homology 2 domain-containing inositol phosphatase 2).4,6,7,9,17 However, not all patients have INPPL1 variants suggesting that OPS exhibits genetic heterogeneity.4,7
Figure 1.
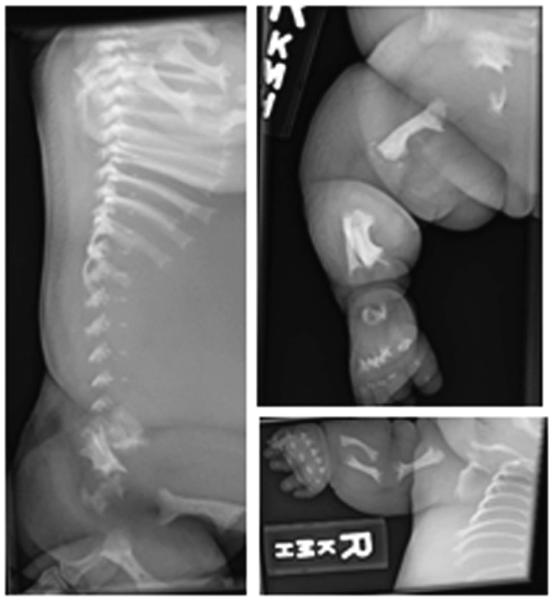
Radiographs of a severely affected opsismodysplasia fetus at 31 weeks gestation.56 Left, lateral view of spine showing severe platyspondyly. Top right, lateral view of right leg showing bowed femur and flared metaphyses. Lower right, anteroposterior view of right arm showing undermineralized bones of the hand with shortened metacarpals and phalanges, and bowing of the humerus, radius and ulna.
INOSITOL POLYPHOSPHATE PHOSPHATASE-LIKE 1/SRC HOMOLOGY 2 DOMAIN-CONTAINING INOSITOL PHOSPHATASE 2
The INPPL1 gene is 14.3 kb in size, composed of 28 exons and located on human chromosome 11q13.4. The INPPL1/SHIP2 open reading frame comprises 1258 amino acids with a predicted molecular weight of 113 kDa. In addition to a catalytic domain, SHIP2 contains several motifs known to be involved in protein–protein interactions and has been shown to function as a docking protein for a variety of intracellular molecules.18,19 These include an N-terminal SRC homology 2 (SH2) domain, potential phosphotyrosine binding sites containing the consensus NPxY, a sterile alpha male domain and C-terminal proline-rich domains with potential for SH3 binding (Figure 2).
Figure 2.

Genomic structure of inositol polyphosphate phosphatase-like 1 and location of mutations involved in opsismodysplasia. 1 mark indicates recurrent mutation.
SHIP2 functions to dephosphorylate the lipid second messenger phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3). Phosphatidylinositol (PI) is a membrane phospholipid that can be reversibly phosphorylated at the 3, 4 and 5 positions through the action of PI kinases and PI phosphatases. SHIP2 is one of 10 mammalian inositol polyphosphate 5-phosphatase that dephosphorylates PI(3,4,5)P3 at the 5'-position of the inositol ring to PI(3,4)P2.20,21
SHIP2 is prominent in a range of human diseases with most research focused on diabetes and cancer (reviewed by Suwa et al.22). Single-nucleotide polymorphisms in INPPL1 gene are associated with susceptibility to type 2 diabetes and metabolic syndrome in Japanese, Chinese and European populations.23–27 In vitro studies suggested that several of these variants increase SHIP2 levels24,26 or enhance Akt pathway activation,27 the major downstream target of SHIP2. The role of SHIP2 in diabetes is an active area of research because of the possibility for developing small-molecule inhibitors that modulate SHIP expression or activity for the treatment of diabetes.27–30
For many cancers, increased SHIP2 expression is associated with cancer progression and is being explored as a clinical biomarker for these cancers.31–35 However, SHIP2 may not be a universal marker for cancer as in gastric cancer the reverse occurs where suppression of SHIP2 is associated with tumorigenesis.36 On the basis of these studies, efforts are also underway to develop anticancer drugs with the goal of pharmacological inhibition of SHIP2 activity.37
Clearly, SHIP2 has multiple, and sometimes opposite, roles in the growth and function of cells depending on cell type and pathology. In the majority of these conditions, the pathology is associated with enhanced SHIP2 expression. The remainder of this review will focus on the emerging role of SHIP2 mutations in skeletal development, which appears to be associated with the loss of SHIP2 function.
MUTATIONS IN INPPL1
Overall, 25 mutations spread throughout the molecule have been identified in 20 families; with 3 nonsense mutations, 1 in-frame mutation, 7 missense mutations, 9 frameshift mutations and 5 splice site mutations reported (Table 1).4,6,7,9,16,17 The SHIP2 domain structure and location of mutations are shown in Figure 2.
Table 1.
Inppl1 mutations and outcome in patients with opsismodysplasia
|
Mutation
type |
Status |
Nucleotide
change |
AA change | Location | Domain | Consequence | N | Recurrence | CS | Outcome | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Frameshift | He | c.24-39 del | p.Gly9Trp | Exon 1 | - | Stop codon | 2 | 1 family | No | Alive | 4 |
| Missense | c.753 G>C | p.Gln251His | Exon 6 | - | - | ||||||
| Frameshift | He | c.35dup | p.Ala13Arg | Exon 1 | - | Stop codon | 1 | 1 family | No | Death (newborn) | 4 |
| Frameshift | c.1687-1691 de | l p.Thr563Gly | Exon 14 | Catalytic domain |
Stop codon | ||||||
| Frameshift | Ho | c.94-121 del | p.Glu32Met | Exon 1 | SH2 domain |
Stop codon | 3 | 2 families | Yes | Death (stillborn; newborns) | 6 |
| Splice site | Ho | c.183-8G>A | Intron 1 | New acceptor site, in frame |
1 | 1 family | Yes | Alive | 9 | ||
| Frameshift | He | c.276-280 del | p.Gln93Pro | Exon 3 | SH2 domain |
Stop codon | 1 | 1 family | No | Alive | 6 |
| Missense | c.1975 C>T | p.Pro659Ser | Exon 17 | Catalytic domain |
_ | ||||||
| Splice site | Ho | c.519-3A>G | Intron 4 | New acceptor site, stop codon |
1 | 1 family | Yes | Alive | 6 | ||
| Nonsense | Ho | c.545 C>A | p.Ser182 | Exon 5 | - | Stop codon | 1 | 1 family | Yes | Alive | 4 |
| Frameshift | Ho | c.768-769 del | p.Glu258Ala | Exon 7 | - | Stop codon | 1 | 1 family | No | Alive | 4 |
| Frameshift | He | c.768-769 del | p.Glu258Ala | Exon 7 | - | Stop codon | 1 | 1 family | No | Alive | 4 |
| Splice site | 2415+1G>A | Intron 21 | - | Suppression donor site, stop codon |
|||||||
| Frameshift | He | c.1150-1151 de | l p.Lys385Gly | Exon 10 | - | Stop codon | 2 | 1 family | No | Termination of pregnancy (17 weeks); death (stillborn) |
17 |
| Splice site | c.2327-1G>C | Intron 20 | Skipping exon 21, stop codon |
||||||||
| Missense | He | c.1201 C>T | p.Arg401Trp | Exon 11 | Catalytic domain |
- | 2 | 1 family | No | Alive; termination of pregnancy (22 weeks) |
6 |
| Missense | c.2164 T>A | p.Phe722Ile | Exon 19 | Catalytic domain |
- | ||||||
| Frameshift | He | c.1328 delins T | A p.Thr443Ile | Exon 12 | Catalytic domain |
Stop codon | 2 | 1 family | No | Termination of pregnancy (15 weeks; 29 weeks) |
6 |
| Missense | c.2064 G>T | p.Trp688Cys | Exon 18 | Catalytic domain |
- | ||||||
| Frameshift | Ho | c.1845 dupT | p.Ile616Tyr | Exon 15 | Catalytic domain |
Stop codon | 3 | 1 family | Yes | Termination of pregnancy (14, 15 and 29 weeks) |
6 |
| Splice site | Ho | c.1951+1G>C | Intron 16 | Suppression donor site, stop codon |
1 | 1 family | Yes | Alive | 6 | ||
| In frame | Ho | c.1960-1962 de | l p.del Glu | Exon 17 | Catalytic domain |
- | 1 | 1 family | No | Alive | 7 |
| Missense | Ho | c.1976 C>T | p.Pro659Leu | Exon 17 | Catalytic domain |
- | 4 | 2 families | Yes/? | Alive | 4,16 |
| Missense | Ho | c.2071 C>T | p.Arg691Trp | Exon 18 | Catalytic domain |
- | 1 | 1 family | Yes | Termination of pregnancy | 4 |
| Nonsense | Ho | c.2719C>T | p.Arg907 | Exon 24 | - | Stop codon | 1 | 1 family | Yes | Alive | 6 |
| Nonsense | Ho | c.2845 C>T | p.Arg949 | Exon 25 | SH3 domain |
Stop codon | 2 | 1 family | Yes | Termination of pregnancy; death (15 months) |
6 |
Abbreviations: AA, amino acid; CS, consanguinity; He, heterozygous; Ho, homozygous; Inppl1, inositol polyphosphate phosphatase-like 1; N, number of patients affected.
The nonsense and frameshift mutations lead to premature stop codons and are expected to result into a truncated protein lacking the proline-rich domain and the sterile alpha male domain and, for most of them, the truncated protein is expected to also lack the catalytic domain. To minimize the potentially deleterious effects of truncated protein exerting a dominant-negative effect on cells and tissues, the vast majority of premature stop codons undergo a process called ‘nonsense-mediated mRNA decay’.38 This is a cell surveillance system that serves to specifically degrade mRNA for the mutant allele containing the premature stop codon leaving the non-mutated allele intact. With the OPS mutations, premature stop codons on both alleles would result in no production of SHIP2 protein. One individual had an in-frame mutation that resulted in the deletion of glutamine at amino acid 654 in the catalytic domain that is predicted to be deleterious for the protein by the PROVEAN algorithm.39 This glutamine residue is conserved between other inositol-5-phosphatase family members. Seven missense mutations have been identified and all are pathogenic according to Sift and PolyPhen pathogenicity algorithms.40,41 Significantly, the proline 659, the tryptophan 688 and the arginine 691 are highly conserved among the inositol-5-phosphatase family members highlighting the notion that their substitution has a deleterious effect on protein function.
All patients reported in the literature have mutations in the homozygous state or are compound heterozygous suggesting that SHIP2 needs to be disabled for complete penetrance of the phenotype to occur. No parents of affected individuals have features of OPS suggesting that 50% levels of wild-type SHIP2 is sufficient to protect from the effects of the mutant allele. The majority of mutations (17/25) either create a stop codon directly or, for the frameshift and splice-site mutations, are predicted to result in a downstream stop codon. These are expected to result in a null for the affected allele by nonsense-mediated mRNA decay. However, even if these mutant transcripts escaped nonsense-mediated mRNA decay the resulting truncated protein would likely not have significant function. Six out of seven missense mutations are located in the catalytic domains and presumably inactivate the phosphatase function of SHIP2.
There does not appear to be a clear genotype–phenotype correlation at the present time and additional mutations need to be described to identify a correlation, if one exists.
Of all the surviving subjects described in the literature, the oldest is now 24 years old.42 This individual is compound heterozygous for a frameshift and the single-missense mutation (p.Gln251His) located outside the catalytic domain. It would be interesting to establish the level of SHIP2 catalytic activity in this patient. Indeed, even a small amount of enzyme function may be enough to ameliorate the severity of the phenotype. It is also possible that other genetic factors modify severity of the phenotype in this individual.
Due to the extreme demineralization and renal phosphate wasting seen in one family with two severely affected OPS children carrying the same homozygous missense mutation in exon 17 of INPPL1 (p. Pro659Leu), bisphosphonate therapy was used to improve bone mineral density.16 Both children responded well to treatment with the more severely affected child markedly improving lumbar and hip Z-score. After 7 years of pamidronate treatment this child is now 10 years of age and is no longer wheelchair-bound suggesting that bisphosphonates may be useful for OPS patients with severe undermineralization.
It is unknown why some OPS patients have phosphate wasting but it is worth noting that mutations in another member of the inositol polyphosphate 5-phosphatase family, INPP5F, have been involved in the oculocerebrorenal syndrome of Lowe that is characterized by phosphate wasting.43
Recently, a homozygous INPPL1 variant was described in Schneckenbecken dysplasia (SD), a severe, autosomal recessive, perinatal lethal dysplasia.44 SD presents some features of OPS notably very-short long bones and platyspondyly, but is distinguished from other severe skeletal dysplasias by the presence on radiographs of a medial projection from the ilia that resembles the shape of a snail. SD demonstrates locus heterogeneity and had previously been shown to be caused by mutations in SLC35D1, encoding an ER-resident sugar transporter essential for glycosaminoglycans synthesis.45,46 However no evidence suggests that SHIP2 could be involved in this process and the identification of INPPL1 in additional SD families is needed to confirm a role for SHIP2 in SD.
MECHANISM OF ACTION IN SKELETAL DEVELOPMENT
Single-nucleotide polymorphisms in SHIP2 are associated with insulin resistance and the development of type 2 diabetes, and there is an emerging role for SHIP2 in cancer where elevated SHIP2 levels are associated with poor prognosis in certain cancers (for review see Suwa et al.22). The majority of these single-nucleotide polymorphisms when tested functionally appear to increase SHIP2 mRNA or protein expression, or activation of Akt, a critical downstream effector. On the other hand, skeletal disease seems to result from the loss of SHIP2 activity due to catalytic domain mutations or no SHIP2 protein due to premature stop codons. Indeed, several INPPL1 mutations are located within the catalytic domain suggesting that the phosphatase activity of SHIP2 is critical for normal chondrogenesis further suggesting that knocking out the catalytic function is sufficient to cause OPS. This is consistent with what is known about PI signaling pathways. SHIP2 dephosphorylates lipid second messenger PI (3,4,5), also known as PIP3, to PIP2. PIP3 is known to participate in bone formation via its role in Akt signaling where it is able to recruit PDK1, which in turn activates Akt.47 Akt and its downstream effectors have critical roles in endochondral ossification; Akt1 knockout mice are smaller and show delayed secondary ossification48 and double Akt1/Akt2 knockout mice display severe dwarfism and delayed bone ossification among other defects.49 Inactive or absent SHIP2 would serve to increase PIP3 levels and promote signaling through Akt and other downstream effectors suggesting the downregulation of the PIP3-Akt1 pathway is not the cause of the skeletal pathology characteristic of OPS.
Histological studies on bone from affected OPS individuals illustrate the negative effects of SHIP2 mutations on developing cartilage revealing a disorganized growth plate with a lack of columnar organization in the proliferative zone and a reduced hypertrophic zone with fewer hypertrophic chondrocytes.5,6,12 However, knockout mice studies provide little in the way of new insights. Inppl1 is expressed at embryonic day E14.5 in the developing limb. Mice deficient for Inppl1 have multiple developmental defects including a resistance to dietary obesity and subtle skeletal phenotype. Inppl1−/− mice have diminished growth and craniofacial abnormalities, however, these are mild in severity compared with humans with null SHIP2 mutations. Both sexes showed diminished longitudinal growth compared with wild-type littermates although this was more pronounced in males compared with females. The only skeletal abnormality detected by imaging was a shortened facial profile. No mineralization defects were apparent and the mice developed to adulthood.50 It is not clear why the lack of Inppl1/Ship2 does not reflect the dramatic mineralization defect seen in humans and result in a milder phenotype overall. However, several mouse models show a disorganized growth plate including the conditional Piga knockout mouse. Piga encodes the catalytic subunit of an essential enzyme in the glycosylphosphatidylinositol anchor pathway. Mice lacking Piga expression in hindlimbs present with a disorganization of the growth plate and a delay in ossification due to the defect of cell polarity at embryonic day E14.5.51 Interestingly, several studies suggest that SHIP2 is involved in cell polarity.52,53 Production of PI (3,4)P2 at cell-extracellular matrix contacts of epithelial cells by SHIP2 is necessary for the recruitment of Dlg1, master regulator of basolateral polarity.52 Also SHIP2 interacts directly with RhoA, one of the major regulators of front-rear polarity in glioma cells.53 One hypothesis is that loss of SHIP2 dysregulates cell position and organization of growth plate chondrocytes leading to undermineralization.
Several studies suggest a role for SHIP2 in fibroblast growth factor (FGF) signaling pathways. Morpholino knockdown of maternal Ship2a in zebrafish leads to dorso-ventral patterning defects in the early embryo due to expanded expression of FGF-mediated outputs including FGF-dependent gene expression and MAPK signaling.54 The authors conclude that modulation of FGF signaling may be a principal function of SHIP2 in mammals. In humans, elevated FGF signaling via gain-of-function mutations in FGFR3 leads to achondrodysplasia (most common form of dwarfism), hypochondroplasia and thanatophoric dysplasia. Disturbances in FGFR3 signaling disrupt the MAPK ERK and p38 pathways, which are important for regulating post-embryonic skeletal development (for review see Narayana and Horton55). These data suggest a role for SHIP2 in FGF signaling, which opens up the exciting possibility of utilizing therapies used in the treatment of FGFR3-related disorders for OPS. For example, C-type natriuretic peptide, which antagonizes the MAP kinase pathway downstream of FGFR3 analogues, has been used to treat achondroplasia.
FUTURE RESEARCH DIRECTIONS
The finding that SHIP2, an inositol phosphatase previously known to be involved in metabolic processes, is a major regulator of extracellular matrix mineralization was an unexpected observation and one that is poorly understood. Future efforts should be directed toward investigating an early growth plate phenotype in SHIP2 knockout mice with attention paid to early growth plate organization and cell polarity, and subsequent mineralization. In cell culture systems using chondrocyte- and osteoblast-related cells lines, it should be possible to define the mechanism of SHIP2 action in skeletally relevant setting. For example, specific SHIP2 inhibitors currently in clinical trials for the metabolic effects of SHIP2 perturbations could be used to define the downstream consequences of SHIP2 deficiency. This could be complemented using CRISPR-Cas9 gene editing technologies to specifically delete or modify the INPPL1 gene. The CRISPR-Cas9 method is well suited for investigating single-gene defects because of the relative ease and speed in generating gene variants.
The ultimate goal is to understand mechanisms that are responsible for the mineralization defect and to translate these insights into new treatments for OPS and potentially other conditions of undermineralization.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. A. 2015;167A:2869–2892. doi: 10.1002/ajmg.a.37365. [DOI] [PubMed] [Google Scholar]
- 2.Al Kaissi A, Chehida FB, Ghachem MB, Grill F, Klaushofer K. Atlanto-axial segmentation defects and os odontoideum in two male siblings with opsismodysplasia. Skeletal Radiol. 2009;38:293–296. doi: 10.1007/s00256-008-0623-4. [DOI] [PubMed] [Google Scholar]
- 3.Beemer FA, Kozlowski KS. Additional case of opsismodysplasia supporting autosomal recessive inheritance. Am. J. Med. Genet. 1994;49:344–347. doi: 10.1002/ajmg.1320490321. [DOI] [PubMed] [Google Scholar]
- 4.Below JE, Earl DL, Shively KM, McMillin MJ, Smith JD, Turner EH, et al. Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 2013;92:137–143. doi: 10.1016/j.ajhg.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cormier-Daire V, Delezoide AL, Philip N, Marcorelles P, Casas K, Hillion Y, et al. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J. Med. Genet. 2003;40:195–200. doi: 10.1136/jmg.40.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber C, Faqeih EA, Bartholdi D, Bole-Feysot C, Borochowitz Z, Cavalcanti DP, et al. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am. J. Hum. Genet. 2013;92:144–149. doi: 10.1016/j.ajhg.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iida A, Okamoto N, Miyake N, Nishimura G, Minami S, Sugimoto T, et al. Exome sequencing identifies a novel INPPL1 mutation in opsismodysplasia. J. Hum. Genet. 2013;58:391–394. doi: 10.1038/jhg.2013.25. [DOI] [PubMed] [Google Scholar]
- 8.Lewis LE, Ramesh Bhat Y, Naik P, Sethi K, Girisha KM. Opsismodysplasia. Indian J. Pediatr. 2010;77:567–568. doi: 10.1007/s12098-010-0043-z. [DOI] [PubMed] [Google Scholar]
- 9.Li B, Krakow D, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian Genomics. Chang Y, et al. Opsismodysplasia resulting from an insertion mutation in the SH2 domain, which destabilizes INPPL1. Am. J. Med. Genet. A. 2014;164A:2407–2411. doi: 10.1002/ajmg.a.36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maroteaux P, Stanescu V, Stanescu R, Le Marec B, Moraine C, Lejarraga H. Opsismodysplasia: a new type of chondrodysplasia with predominant involvement of the bones of the hand and the vertebrae. Am. J. Med. Genet. 1984;19:171–182. doi: 10.1002/ajmg.1320190117. [DOI] [PubMed] [Google Scholar]
- 11.Santos HG, Saraiva JM. Opsismodysplasia: another case and literature review. Clin. Dysmorphol. 1995;4:222–226. [PubMed] [Google Scholar]
- 12.Tyler K, Sarioglu N, Kunze J. Five familial cases of opsismodysplasia substantiate the hypothesis of autosomal recessive inheritance. Am. J. Med. Genet. 1999;83:47–52. doi: 10.1002/(sici)1096-8628(19990305)83:1<47::aid-ajmg9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Zeger MD, Adkins D, Fordham LA, White KE, Schoenau E, Rauch F, et al. Hypophosphatemic rickets in opsismodysplasia. J. Pediatr. Endocrinol. Metab. 2007;20:79–86. doi: 10.1515/jpem.2007.20.1.79. [DOI] [PubMed] [Google Scholar]
- 14.Zeman J, Baxova A, Houstkova H, Kozlowski K. Opsismodysplasia: a case report. Australas. Radiol. 1997;41:35–37. doi: 10.1111/j.1440-1673.1997.tb00465.x. [DOI] [PubMed] [Google Scholar]
- 15.Zonana J, Rimoin DL, Lachman RS, Cohen AH. A unique chondrodysplasia secondary to a defect in chondroosseous transformation. Birth Defects Orig. Artic. Ser. 1977;13:155–163. [PubMed] [Google Scholar]
- 16.Khwaja A, Parnell SE, Ness K, Bompadre V, White KK. Opsismodysplasia: phosphate wasting osteodystrophy responds to bisphosphonate therapy. Front. Pediatr. 2015;3:48. doi: 10.3389/fped.2015.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feist C, Holden P, Fitzgerald J. Novel compound heterozygous mutations in inositol polyphosphate phosphatase-like 1 in a family with severe opsismodysplasia. Clin. Dysmorphol. 2016;25:152–155. doi: 10.1097/MCD.0000000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erneux C, Edimo WE, Deneubourg L, Pirson I. SHIP2 multiple functions: a balance between a negative control of PtdIns(3,4,5)P(3) level, a positive control of PtdIns(3,4)P(2) production, and intrinsic docking properties. J. Cell Biochem. 2011;112:2203–2209. doi: 10.1002/jcb.23146. [DOI] [PubMed] [Google Scholar]
- 19.Backers K, Blero D, Paternotte N, Zhang J, Erneux C. The termination of PI3K signalling by SHIP1 and SHIP2 inositol 5-phosphatases. Adv. Enzyme Regul. 2003;43:15–28. doi: 10.1016/s0065-2571(02)00043-2. [DOI] [PubMed] [Google Scholar]
- 20.Habib T, Hejna JA, Moses RE, Decker SJ. Growth factors and insulin stimulate tyrosine phosphorylation of the 51C/SHIP2 protein. J. Biol. Chem. 1998;273:18605–18609. doi: 10.1074/jbc.273.29.18605. [DOI] [PubMed] [Google Scholar]
- 21.Pesesse X, Deleu S, De Smedt F, Drayer L, Erneux C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem. Biophys. Res. Commun. 1997;239:697–700. doi: 10.1006/bbrc.1997.7538. [DOI] [PubMed] [Google Scholar]
- 22.Suwa A, Kurama T, Shimokawa T. SHIP2 and its involvement in various diseases. Expert Opin. Ther. Targets. 2010;14:727–737. doi: 10.1517/14728222.2010.492780. [DOI] [PubMed] [Google Scholar]
- 23.Kaisaki PJ, Delépine M, Woon PY, Sebag-Monteflore L, Wilder SP, Menzel S, et al. Polymorphisms in type II SH2 domain-containing inositol 5-phosphatase (INPPL1, SHIP2) are associated with physiological abnormalities of the metabolic syndrome. Diabetes. 2004;53:1900–1904. doi: 10.2337/diabetes.53.7.1900. [DOI] [PubMed] [Google Scholar]
- 24.Ishida S, Funakoshi A, Miyasaka K, Shimokata H, Ando F, Takiguchi S, et al. Association of SH-2 containing inositol 5'-phosphatase 2 gene polymorphisms and hyperglycemia. Pancreas. 2006;33:63–67. doi: 10.1097/01.mpa.0000222317.82231.16. [DOI] [PubMed] [Google Scholar]
- 25.Hao YM, Liu QJ, Wang RY, Cao YP, Zhang Y, Zuo LF. Single nucleotide polymorphisms on SHIP2 is associated with type 2 diabetes mellitus in Chinese Han population. Eur. Rev. Med. Pharmacol. Sci. 2015;19:129–137. [PubMed] [Google Scholar]
- 26.Marion E, Kaisaki PJ, Pouillon V, Gueydan C, Levy JC, Bodson A, et al. The gene INPPL1, encoding the lipid phosphatase SHIP2, is a candidate for type 2 diabetes in rat and man. Diabetes. 2002;51:2012–2017. doi: 10.2337/diabetes.51.7.2012. [DOI] [PubMed] [Google Scholar]
- 27.Kagawa S, Sasaoka T, Yaguchi S, Ishihara H, Tsuneki H, Murakami S, et al. Impact of SRC homology 2-containing inositol 5'-phosphatase 2 gene polymorphisms detected in a Japanese population on insulin signaling. J. Clin. Endocrinol. Metab. 2005;90:2911–2919. doi: 10.1210/jc.2004-1724. [DOI] [PubMed] [Google Scholar]
- 28.Suwa A, Yamamoto T, Sawada A, Minoura K, Hosogai N, Tahara A, et al. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. Br. J. Pharmacol. 2009;158:879–887. doi: 10.1111/j.1476-5381.2009.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishihara H, Sasaoka T, Ishiki M, Wada T, Hori H, Kagawa S, et al. Membrane localization of Src homology 2-containing inositol 5'-phosphatase 2 via Shc association is required for the negative regulation of insulin signaling in Rat1 fibroblasts overexpressing insulin receptors. Mol. Endocrinol. 2002;16:2371–2381. doi: 10.1210/me.2002-0083. [DOI] [PubMed] [Google Scholar]
- 30.Annis DA, Cheng CC, Chuang CC, McCarter JD, Nash HM, Nazef N, et al. Inhibitors of the lipid phosphatase SHIP2 discovered by high-throughput affinity selection-mass spectrometry screening of combinatorial libraries. Comb. Chem. High Throughput Screen. 2009;12:760–771. doi: 10.2174/138620709789104870. [DOI] [PubMed] [Google Scholar]
- 31.Prasad NK, Tandon M, Badve S, Snyder PW, Nakshatri H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis. 2008;29:25–34. doi: 10.1093/carcin/bgm213. [DOI] [PubMed] [Google Scholar]
- 32.Prasad NK. SHIP2 phosphoinositol phosphatase positively regulates EGFR-Akt pathway, CXCR4 expression, and cell migration in MDA-MB-231 breast cancer cells. Int. J. Oncol. 2009;34:97–105. [PubMed] [Google Scholar]
- 33.Prasad NK, Tandon M, Handa A, Moore GE, Babbs CF, Snyder PW, et al. High expression of obesity-linked phosphatase SHIP2 in invasive breast cancer correlates with reduced disease-free survival. Tumour Biol. 2008;29:330–341. doi: 10.1159/000172970. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Fu M, Ding Y, Weng Y, Fan W, Pu X, et al. High SHIP2 expression indicates poor survival in colorectal cancer. Dis. Markers. 2014;2014:218968. doi: 10.1155/2014/218968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou X, Liu Y, Tan G. Prognostic value of elevated SHIP2 expression in laryngeal squamous cell carcinoma. Arch. Med. Res. 2011;42:589–595. doi: 10.1016/j.arcmed.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 36.Ye Y, Ge YM, Xiao MM, Guo LM, Li Q, Hao JQ, et al. Suppression of SHIP2 contributes to tumorigenesis and proliferation of gastric cancer cells via activation of Akt. J. Gastroenterol. 2015;51:230–240. doi: 10.1007/s00535-015-1101-0. [DOI] [PubMed] [Google Scholar]
- 37.Fuhler GM, Brooks R, Toms B, Iyer S, Gengo EA, Park MY, et al. Therapeutic potential of SH2 domain-containing inositol-5'-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol. Med. 2012;18:65–75. doi: 10.2119/molmed.2011.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stalder L, Muhlemann O. The meaning of nonsense. Trends Cell Biol. 2008;18:315–321. doi: 10.1016/j.tcb.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 42.Below JE, Earl DL, Shively KM, McMillin MJ, Smith JD, Turner EH, et al. Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 2013;92:137–143. doi: 10.1016/j.ajhg.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV, et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat. 2011;32:379–388. doi: 10.1002/humu.21391. [DOI] [PubMed] [Google Scholar]
- 44.Lee H, Nevarez L, Lachman RS, Wilcox WR, Krakow D, Cohn DH, et al. A second locus for schneckenbecken dysplasia identified by a mutation in the gene encoding inositol polyphosphate phosphatase-like 1 (INPPL1) Am. J. Med. Genet. A. 2015;167A:2470–2473. doi: 10.1002/ajmg.a.37173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furuichi T, Kayserili H, Hiraoka S, Nishimura G, Ohashi H, Alanay Y, et al. Identification of loss-of-function mutations of SLC35D1 in patients with Schneckenbecken dysplasia, but not with other severe spondylodysplastic dysplasias group diseases. J. Med. Genet. 2009;46:562–568. doi: 10.1136/jmg.2008.065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiraoka S, Furuichi T, Nishimura G, Shibata S, Yanagishita M, Rimoin DL, et al. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat. Med. 2007;13:1363–1367. doi: 10.1038/nm1655. [DOI] [PubMed] [Google Scholar]
- 47.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Curr. Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 48.Ulici V, Hoenselaar KD, Agoston H, McErlain DD, Umoh J, Chakrabarti S, et al. The role of Akt1 in terminal stages of endochondral bone formation: angiogenesis and ossification. Bone. 2009;45:1133–1145. doi: 10.1016/j.bone.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 49.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sleeman MW, Wortley KE, Lai KM, Gowen LC, Kintner J, Kline WO, et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 2005;11:199–205. doi: 10.1038/nm1178. [DOI] [PubMed] [Google Scholar]
- 51.Ahrens MJ, Li Y, Jiang H, Dudley AT. Convergent extension movements in growth plate chondrocytes require gpi-anchored cell surface proteins. Development. 2009;136:3463–3474. doi: 10.1242/dev.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Awad A, Sar S, Barré R, Cariven C, Marin M, Salles JP, et al. SHIP2 regulates epithelial cell polarity through its lipid product, which binds to Dlg1, a pathway subverted by hepatitis C virus core protein. Mol. Biol. Cell. 2013;24:2171–2185. doi: 10.1091/mbc.E12-08-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato K, Yazawa T, Taki K, Mori K, Wang S, Nishioka T, et al. The inositol 5-phosphatase SHIP2 is an effector of RhoA and is involved in cell polarity and migration. Mol. Biol. Cell. 2012;23:2593–2604. doi: 10.1091/mbc.E11-11-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jurynec MJ, Grunwald DJ. SHIP2, a factor associated with diet-induced obesity and insulin sensitivity, attenuates FGF signaling in vivo. Dis. Model Mech. 2010;3:733–742. doi: 10.1242/dmm.000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narayana J, Horton WA. FGFR3 biology and skeletal disease. Connect. Tissue Res. 2015;56:427–433. doi: 10.3109/03008207.2015.1051224. [DOI] [PubMed] [Google Scholar]
- 56.Feist C, Holden P, Fitzgerald J. Novel compound heterozygous mutations in inositol polyphosphate phosphatase-like 1 in a family with severe opsismodysplasia. Clin. Dysmorphol. 2016;25:152–155. doi: 10.1097/MCD.0000000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]