

Abstract
Recapitulation of the nuclear auxin response pathway in Saccharomyces cerevisiae (yeast) provides a means to functionally assay the contribution of individual signaling components to response dynamics. Here, we describe a time course assay for characterizing auxin response circuits using flow cytometry. This method allows for quantitative measurements of the dynamic response of up to 12 circuits (strains) at once. We also describe a steady-state assay and how to utilize an R package we developed to facilitate data analysis.
Keywords: Synthetic biology, Budding yeast, Fluorescent reporters, Signaling dynamics
1 Introduction
Auxin influences nearly every aspect of plant growth and development. The core auxin signaling pathway consists of only a handful of components, and yet is capable of eliciting a diverse array of context-specific responses to auxin. Experimental analysis of this small signaling pathway in planta is confounded by the ubiquity of auxin response, integration and feedback from other signaling pathways, and genetic redundancy among the core signaling components. To test the capacity of the auxin signaling pathway to generate diverse responses to auxin, we transplanted each component of the pathway from Arabidopsis to the budding yeast Saccharomyces cerevisiae [1].
A minimal yeast auxin response circuit is composed of a TIR1/AFB auxin receptor, an Aux/IAA (IAA), a TOPLESS (TPL) co-repressor, an ARF transcription factor, and an auxin-responsive promoter driving a fluorescent reporter (Fig. 1). In the absence of auxin, ARF activity is repressed through interaction with an IAA-TPL fusion. Auxin treatment promotes interaction between the IAA and the AFB receptor, an F-box protein, which catalyzes the ubiquitination and subsequent degradation of the IAA. IAA degradation relieves the repression of the ARF transcription factor resulting in the induction of the fluorescent reporter. Flow cytometry is used to monitor levels of the fluorescent reporter over time following auxin treatment.
Fig. 1.
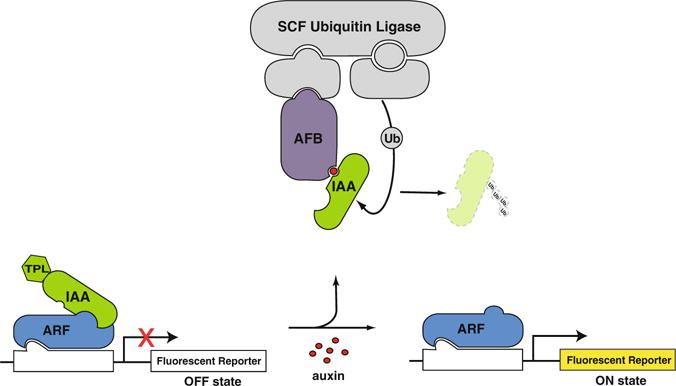
A yeast auxin response circuit. An AFB auxin receptor, an IAA-TPL repressor, an ARF transcription factor, and an auxin -responsive promoter driving a fluorescent reporter are sufficient to recapitulate auxin-induced transcription in yeast
This user-defined auxin response circuit can be used to generate numerous circuit variants by substituting variants of any one of the core components alone or in combination. These component variants can be derived from the naturally evolved gene family members or by altering a functional domain within a component through mutation or truncation [1, 2]. The modularity of the circuit also facilitates decomposition into sub-modules to focus on specific aspects of the pathway, such as auxin -induced degradation of the IAAs. Auxin response circuit composition can therefore be tailored to suit the need of the researcher. As such, we focus here on describing a method for the quantitative characterization of auxin circuits rather than detailing their assembly.
2 Materials
2.1 Yeast Materials
Diploid yeast strains containing circuit variants to be assayed (see Note 1).
YPDA agar plates: Add about 700 mL of water to a glass beaker. Add a magnetic stir bar and set on a stir plate. Add each ingredient while stirring: 10 g yeast extract, 20 g of peptone, 80 mg of adenine. Bring volume up to 900 mL with water. Stir well, and then split into two 450 mL aliquots in 1 L bottles. Add 10 g of agar into each bottle. Autoclave for 25 min at 121 °C. After cooling, bring media up to final volume of 500 mL per bottle containing 2 % glucose by supplementing with 50 mL 20 % sterile filtered glucose. Pour 20 mL molten YPDA into 95 × 15 mm petri dishes as required.
Synthetic Complete (SC) liquid media: Add about 1 L of water to a glass beaker. Add a magnetic stir bar and set on a stir plate. Add each ingredient while stirring: 2.55 g of yeast nitrogen base without amino acids or ammonium sulfate (Sigma); 2.1 g of yeast synthetic drop-out medium without histidine, leucine, tryptophan, and uracil (Sigma); 7.5 g ammonium sulfate; 150 mg of histidine; 150 mg of leucine; 150 mg of tryptophan; 37.5 mg of uracil; and 120 mg adenine. Bring volume up to 1.35 L with water. Stir well, and then split into two 675 mL aliquots in 1 L bottles. Autoclave for 25 min at 121 °C. After cooling, bring each bottle to a final volume of 750 mL by adding 75 mL 20 % sterile filtered glucose. Store at room temperature and protect liquid media from light.
2.2 Cytometry Assay Materials
17 × 100 mm sterile plastic (translucent polypropylene) culture tubes with snap cap.
50 mL plastic conical centrifuge tubes.
25 mL serological pipettes.
Pipet-Lite™ XLS Adjustable-Spacer Multichannel Manual Pipette, 100–1200 μl (Rainin).
Multiplex culture tube lids (see Note 2).
96-well PCR plates.
Indole-3-acetic acid (auxin).
95 % EtOH.
2000 μl Eppendorf™ Deepwell™ Plates 96.
25 mL reagent troughs.
12-channel pipette with 100 μl capacity.
AeraSeal™ breathable film seals (Excel Scientific).
2.3 Equipment
Flow cytometer with plate capacity configured to detect GFP or YFP. We currently take fluoorescence measurements with a BD Accuri™ flow cytometer with a CSampler 96-well plate adapter using an excitation wavelength of 514 nm and an emission detection filter at 545/35 nm (see Note 3).
Benchtop shaker incubator amenable to rapid and repeat sampling of growing yeast cultures such as a MaxQ™ 4000 Benchtop Oribital Shaker (Thermo Scientific).
2.4 Data Analysis Software
We have developed a package called flowTime for the R programming language to facilitate data analysis for these assays. This package will soon be available via http://bioconductor.org (current and continuing development version available at http://github.com/wrightrc/flowTime). We suggest using RStudio (http://www.rstudio.com) to edit and run the R-code for your data analysis. Numerous resources are available on the internet to gain an introductory knowledge of the R language [3].
3 Methods
3.1 Auxin Time Course Assay
This protocol can be used to assay auxin-induced transcription of up to 12 strains in parallel. This constraint is due to the time required for auto-sampling of multiple wells by the cytometer as well as to minimize the time a yeast culture sits in the sampling plate. We therefore generally limit each read to 12 strains. This assay can also be used for monitoring auxin-induced degradation with only minor adjustments (see Note 4).
Day One
-
1
For each strain to be assayed, inoculate ¼ to ½ of a fresh yeast colony from a YPDA plate into 3 mL of synthetic complete (SC) media. Vortex well to mix. Aliquot 100 μl into a 96-well plate and read on cytometer to estimate cell density (in events per microliter).
-
2
Export data as. FCS files and import into R as a flowSet (see Subheadings 3.3 and 3.4, for example R-code). Create summary statistics for this flowSet using the summary.cyt function with the “only” parameter set to “ yeast” to subset the data for all yeast. The value in the “ conc” column of the resulting data frame is the concentration in yeast events per μL for each sample or flowFrame. Use this value to calculate the volume of the initial 3 mL inoculant required to prepare the requisite dilution.
-
3
Dilute each strain to 0.25 events per μL in 12 mL of SC in a 50 mL conical tube. Mix well by vortexing.
-
4
Split dilutions into duplicate 5 mL aliquots in sterile plastic culture tubes and incubate for 16 h at 30 °C with shaking at 220 rpm.
Day Two
-
5
After 16 h of growth, combine duplicate aliquots to homogenize cultures. Mix gently by pipetting and split back into two 5 mL aliquots.
-
6
Separate duplicate cultures into two groups of tubes while maintaining the strain order between groups. This will be the order samples are read on the cytometer. Return tubes to shaker and replace individual plastic caps with 3D printed multiplex tube lids. Use of these lids facilities rapid sampling of the cultures using the Rainin Adjustable-Spacer Multichannel Pipette.
-
7
Set cytometer settings to measure 10,000 events for each well at a flow rate of 66 μl/min and core size of 22 μm with a time limit of 1 min. Use these settings for the entire experiment.
-
8
Transfer 100 μl of each culture from one replicate group to a 96-well PCR plate by expanding the multichannel pipette to sample from the culture tubes and collapsing to dispense into the PCR plate. Fill an additional well with 200 μl of sheath fluid to serve as a wash between readings (see Note 5).
-
9
Load PCR plate containing samples onto cytometer sampling arm and run the auto-sampling program to flow samples. The density of each culture during this initial read should be around 200 events/μl (see Note 6). Repeat steps 8 and 9 for the second replicate of cultures.
-
10
For each strain, mock treat the first replicate measured with 95 % ethanol. Immediately transfer 100 μl of each mock-treated culture to the next row of the 96-well PCR plate and read samples on the cytometer to record fluorescence. These measurements serve as the 0-min time point.
-
11
Treat each strain of the second replicate of cultures with indole-3-acetic acid (IAA) dissolved in 95 % ethanol to a final concentration of 10 μM. Immediately after auxin addition, transfer 100 μl of each culture to a new 96-well PCR plate. Open a new file with the settings described in step 7 and measure fluorescence for the 0-min time point.
-
12
Use the same plate and file to acquire measurements for auxin-treated samples at 10-min intervals. Prepare new plates and a corresponding cytometer file as needed to acquire 5 h of data.
-
13
Take measurements for mock-treated samples every hour using the same plate and file used in step 9.
-
14
For each plate file, export all wells as a folder of. FCS files. There should be a separate plate file containing the initial and mock-treated reads and several files for the auxin-treated measurements.
3.2 Auxin Steady-State Assay
Day One
-
1
For each strain to be assayed, inoculate ¼ to ½ of a fresh yeast colony from a YPDA plate into a well of a 2000 μl Eppendorf™ Deepwell™ Plate 96 containing 500 μl of SC media.
-
2
Seal plate with a breathable film seal and incubate at 30 °C with shaking at 375 rpm for 16 h.
Day Two
-
3
After 16 h of growth, dilute strains 1:200 into a new well containing 500 μl of fresh SC media in a deep 96-well plate (see Note 7). Prepare two replicate wells for each strain.
-
4
Return plate containing diluted cultures to the shaker for 2 h of additional growth at 30 °C with shaking at 375 rpm (see Note 8).
-
5
After 2 h, mock (95 % EtOH) treat one replicate well and auxin (indole-3-acetic acid) treat the other (see Note 8). To assay more than 12 strains or multiple doses of auxin, treatment additions must be staggered for every 12 wells. This is important to allow time for data acquisition and maintain a consistent treatment time between reads.
-
6
Return plate to shaker for an additional 4 h of growth (see Note 8).
-
7
Set cytometer settings to measure 20,000 events for each well at a flow rate of 66 μl/min and core size of 22 μm with a time limit of 1 min.
-
8
Sample up to 12 strains at once using a multi-channel pipette to transfer 100 μL of culture from the deep-well plate in the shaker to a PCR plate. Fill an additional well in the PCR plate with 200 μl of sheath fluid to serve as a wash between readings.
-
9
Load the PCR plate onto cytometer sampling arm and run the auto-sampling program to measure fluorescence.
-
10
Repeat steps 8 and 9 until all strain/treatment combinations have been measured.
-
11
Typically, data for at least three independent replicates for each strain/treatment is collected.
-
12
Export data as a folder of .FCS files.
3.3 Time Course Data Analysis in R
Here, we demonstrate how to import the .FCS files into R, gate and annotate this data with experimental metadata (e.g., the strain and treatment for each sample), generate summary statistics for each sample and time point, and finally plot the time course data (see Note 9).
3.3.1 Importing and Annotating Data
-
1Import your flow cytometry data using read.flowSet. Here, we will import an example flowSet:
plate1 <- read.flowSet (path = system.file ("extdata", "tc_example/", package = "flowTime"), alter.names = T) # Next, add plate numbers to the sampleNames sampleNames (plate1) <- paste (‘1_’, sampleNames (plate1), sep = “”) -
2If you have several plates, this code can be repeated and each plate can be combined (using rbind2) to assemble the full dataset:
plate2 <- read.flowSet (path = paste (experiment, "_2/", sep = ""), alter.names = T); sampleNames (plate2) <- paste ("2_", sampleNames (plate2), sep = ""); dat <- rbind2 (plate1, plate2) -
3Import the table of metadata for each well. The “ sample-Names” field of the assembled flowSet (“ dat” in this example) must match that of a unique identifier column in the table of metadata. In this example and as a default name is the unique identifier column. One can also create this column from a flowSet and attach the annotation columns, as demonstrated in the code below. The order of the unique identifier column does not matter, as annotateFlowSet will join annotation to “dat” by matching identifiers, as specified by the “mergeBy” parameter:
annotation <- read.csv (system.file ("extdata", "tc_example.csv", package = "flowTime")); sampleNames (dat) #view the sample names sampleNames (dat) == annotation$id #replace 'id' with the unique identifier column to test if this column is identical to the sample names of your flowset annotation <- cbind (annotation, names = sampleNames (dat)) #If the sampleNames and unique identifiers are in the correct order this command will add the sampleNames as the identifier -
4Attach this metadata to the flowSet using the annotateFlowSet function:
adat <- annotateFlowSet (dat, annotation); head (rownames (pData (adat))) #> [1] "0_A08.FCS" "0_B08.FCS" "0_C08.FCS" "0_D08.FCS" "0_E08.FCS" "0_F08.FCS" head (pData (adat)) #>name X strain RD ARF AFB treatment #> 0_A08.FCS 0_A08.FCS 0_A08 3 TPLRD1 19 AFB2 0 #> 0_B08.FCS 0_B08.FCS 0_B08 3 TPLRD1 19 AFB2 0 #> 0_C08.FCS 0_C08.FCS 0_C08 3 TPLRD1 19 AFB2 0 #> 0_D08.FCS 0_D08.FCS 0_D08 3 TPLRD1 19 AFB2 0 #> 0_E08.FCS 0_E08.FCS 0_E08 3 TPLRD1 19 AFB2 0 #> 0_F08.FCS 0_F08.FCS 0_F08 3 TPLRD1 19 AFB2 0
3.3.2 Compiling and Plotting Time Course Data
-
11. Use the summary.cyt function to compile the summary statistics from the raw data in this flowSet. This function will gate each flowFrame in the flowSet and compile and return a dataframe of summary statistics in the specified channel for each flowFrame:
# load the gate set for BD Accuri C6 cytometer loadGates (gatesFile = "C6Gates.RData"); dat_sum <- summary.cyt (adat, ploidy = "diploid", only = "singlets", channel = "FL1.A") #> [1] "Gating with diploid gates…" #> [1] "Summarizing singlets events…"
-
2Use the resulting dataframeto plot mean fluorescence for each mock- and auxin-treated sample over time to generate induction curves (Fig. 2):
qplot (x = time, y = FL1.Amean, data = dat_sum, linetype = factor (treatment)) + geom_line () + xlab ("Time post Auxin addition (min)") + ylab ("Reporter Fluorescence (AU)") + scale_color_discrete (name = expression (paste ("Auxin (", mu, "M)", sep = ""))) + theme_classic (base_size = 14, base_family = "Arial")
Fig. 2.
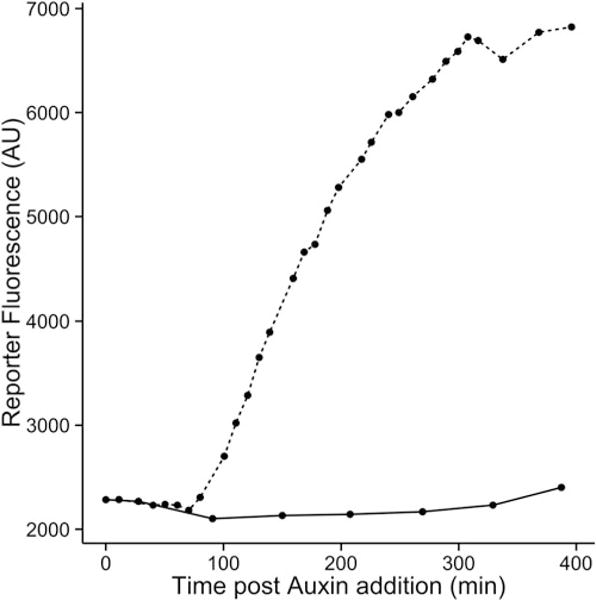
Example data from a time course assay. Induction of transcription in response to auxin is shown for a mock- (solid line) and auxin-treated (dashed line) circuit
3.4 Steady-State Assay Data Analysis
For steady-state assays, the entire dataset (all measured events/sample) can be readily plotted. Data import and annotation remains the same.
3.4.1 Compiling and Plotting Steady-State Data
-
1Use the steadyState function to gate each flowFrame in the flowSet and compile and return a dataframe of the relevant data and metadata for each event:
loadGates (gatesFile = "SORPGates.RData") dat.SS <- steadyState (flowset = adat, ploidy = "diploid", only = "singlets") #> [1] "Gating with diploid gates…" #> [1] "Converting singlets events…"
-
2Use the dataframe to plot the fluorescence for all events captured for each sample (Fig. 3):
p <- ggplot (dat.SS, aes (as.factor (treatment), FL2.A, fill = AFB)) + geom_boxplot (outlier.size = 0) + facet_grid (IAA ~ AFB) + theme_classic (base_family = "Arial", base_size = 16) + ylim (c (-1000, 10000)) + xlab (expression (paste (" Auxin (", mu, "M)", sep = ""))) + ylab ("Fluorescence (AU)") + theme (legend.position = "none"); p
Fig. 3.
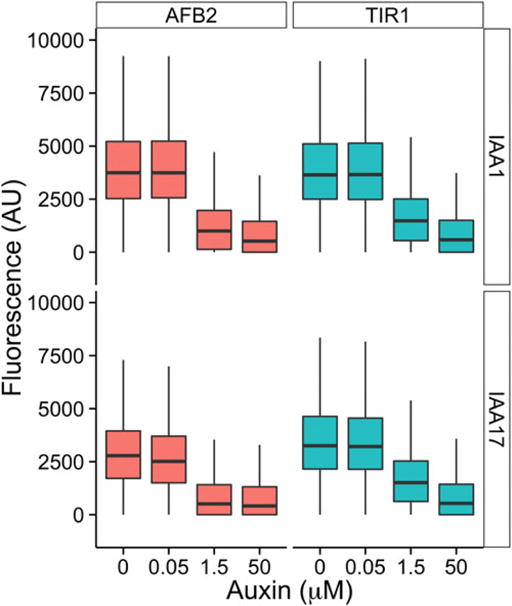
Example data from a steady-state assay. Each sub-panel represents a different circuit built to assay auxin-induced degradation. Box plots display the fluorescence distribution of IAA protein levels following a dose response assay
Footnotes
Strain construction for auxin response circuits has been previously described [1]. Briefly, we use laboratory strains W814-29B MATα and W303-1A MATa which are both auxotrophic for uracil, histidine, tryptophan, and leucine. Each component of the pathway is singly integrated into the yeast genome at a different auxotrophic locus. Typically, a TIR1/AFB receptor is integrated at LEU2 and an IAA-TPL fusion is integrated at TRP1 in W814-29B MATα. An ARF is integrated at HIS3 and the auxin-responsive fluorescent reporter (i.e., pIAA19∷GFP) is integrated at URA3 in W303-1A MATa. The two strains can then be mated to generate a diploid strain containing the entire auxin circuit. For auxin-induced degradation assays, only introduction of a TIR1/AFB receptor and a fluorescently tagged IAA is required [2].
The.stl design file for the 3D printed multiplex lid is publically available on Thingiverse (http://www.thingiverse.com/thing:893490). Each lid can accommodate six culture tubes in a 6 × 6 or 6 × 12 tube rack.
Our published work utilized a standard BD Accuri™ C6 flow cytometer with a CSampler 96-well plate adapter configured with a 488 nm laser and an emission detection filter at 533 nm to measure GFP and YFP fluorescence levels. Our current 514 nm laser configuration optimizes YFP excitation but no longer excites GFP. We tested several bench-top cytometers for YFP and GFP detection in our strains and found the Accuri to be the most sensitive.
For auxin-induced degradation assays, follow time course protocol with the following modifications. The first day, dilute strains to 0.5 events/μl in 10 mL of SC and split into two 4 mL aliquots. Culture density during the initial reads the next day should be ~500 events/μl. After treatments, samples are measured at 10-min intervals for 3 h.
A wash well should be prepared for every read containing up to 12 strains. We typically prepare the full PCR plates containing a wash well for each row so that only cultures need to be transferred to the PCR plate at 10-min intervals.
A starting density of ~200 events/μl after 16 h is based on the growth rate of our particular strains. We found this starting density to be optimal to ensure that yeast are in log phase for the duration of the experiment. If starting density is too low, it is best to allow the yeast to grow for an additional 30 min to an hour as fluorescence readings can be more variable.
For haploid strains, dilute cultures 1:100 in order to maintain the time frame outlined.
We typically treat with 10 μM IAA but auxin treatment can range from 0.05 to 50 μM. Dimethyl sulfoxide can be used as a solvent in place of 95 % ethanol. To conduct a dose response assay, replicate wells for each dose must be prepared in step 3. For auxin-induced degradation assays, treatment should be added 5 h post-dilution and fluorescence measured after 1 h.
To minimize noise in yeast cytometry data we typically present fluorescence measurements from only singlet yeast cells (i.e., cells without a significantly sized bud), as budding increases cell size and decreases the effective concentration of the circuit components. Unfortunately, gates isolating populations of singlet-yeast cells for both haploid and diploid strains are unique for each yeast strain, shaker setup, and flow cytometer. Determining the parameters of these gates is currently done by trial and error in R. More visual programs such as flowJo may be helpful. See the flowCoredocumentation [4] for more information about using R to create gates, and refer to [5] for an example of how to identify live and singlet cell populations.
References
- 1.Pierre-Jerome E, Jang SS, Havens KA, Nemhauser JL, Klavins E. Recapitulation of the forward nuclear auxin response pathway in yeast. Proc Natl Acad Sci U S A. 2014;111(26):9407–9412. doi: 10.1073/pnas.1324147111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Havens KA, Guseman JM, Jang SS, et al. A synthetic approach reveals extensive tunability of auxin signaling. Plant Physiol. 2012;160:135–142. doi: 10.1104/pp.112.202184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. http://www.R-project.org/ [Google Scholar]
- 4.Ellis B, Haaland P, Hahne F, Le Meur N, Gopalakrishnan N, Spidlen J, Jiang M. flowCore: basic structures for flow cytometry data. R package version 1. 36:9. https://www.bioconductor.org/packages/release/bioc/html/flowCore.html. [Google Scholar]
- 5.Baxter SK, Lambert AR, Scharenberg AM, Jarjour J. Flow cytometric assays for interrogating LAGLIDADG homing endonuclease DNAbinding and cleavage properties. Methods Mol Biol. 2013;978:45–61. doi: 10.1007/978-1-62703-293-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]