

Abstract
A variety of mutations in lentiviral vector expression systems have been shown to generate a non-integrating phenotype. We studied a novel 12 base-pair U3-LTR integrase attachment site deletion (U3-LTR att site) mutant and found similar physical titers to the previously reported integrase catalytic core mutant IN/D116N. Both mutations led to a greater than two log reduction in vector integration; with IN/D116N providing lower illegitimate integration frequency, while the U3-LTR att site mutant provided a higher level of transgene expression. The improved expression of the U3-LTR att site mutant could not be explained solely based on an observed modest increase in integration frequency. In evaluating processing, we noted significant differences in unintegrated vector forms, with the U3-LTR att site mutant leading to a predominance of 1-LTR circles. The mutations also differed in the manner of illegitimate integration. The U3-LTR att site mutant vector demonstrated integrase-mediated integration at the intact U5-LTR att site and non-integrase mediated integration at the mutated U3-LTR att site. Finally, we combined a variety of mutations and modifications and assessed transgene expression and integration frequency to show that combining modifications can improve the potential clinical utility of non-integrating lentiviral vectors.
Keywords: non-integrating lentiviral vectors, HIV-1 vectors, integrase mutants, LTR attachment
INTRODUCTION
The ability to integrate stably within target cells has been a distinct advantage of retroviral and lentiviral vectors. However, these vectors have been associated with a risk of insertional mutagenesis (IM)1–3. To address this concern, non-integrating lentiviral vectors (NILV) have been developed for applications where integration is not required4–7. Such vectors would be most suitable for applications when transient expression of the transgene is desired8,9, transducing non-proliferating cells5,10–12, or as donor templates for use in zinc finger nuclease and transcription activator-like effector nuclease systems13–16.
HIV-1 primarily relies upon integrase (IN) to catalyze the integration of viral DNA into the target cell DNA. The reaction involves an episomal template along with three well-defined catalytic steps: 3’-end processing, DNA strand transfer, and a disintegration reaction. This process requires a complete reverse transcription reaction, an intact IN molecule and available U3 and U5 attachment sites for IN within the viral long terminal repeats (LTRs)17–19. HIV-1 infected cells are known to contain integrated provirus, unintegrated linear virus DNA as well as 1-LTR or 2-LTR circular forms. The 1- and 2-LTR circles arise through homologous recombination (HR) or non-homologous end-joining (NHEJ), respectively20–22. In this paper, insertions of vector DNA into a cell genome by non-integrase mediated methods are referred to as illegitimate integrations.
IN mutations at the highly conserved amino acid positions D64, D116 and E152 disable the catalytic activity of IN by interfering with 3’-end processing, DNA strand transfer activities, and disintegration23. NILV generated using these mutant IN proteins are both infectious, express the vector transcript, and are associated with a marked reduction in vector integration4–6,10,24–27. A second approach is to induce aberrant reverse transcription by deletion of the 3’ polypurine tract (PPT) within the vector backbone11. A third, less well studied approach, is altering the LTR attachment (att) sites by mutagenesis of the CA dinucleotide recognition sequence within the U3 and U5 att sites; these mutations have been shown to inhibit integration while retaining infectivity at approximately 40% and 10% of wild-type virus, respectively18.
In the current study, a novel non-integrating vector with an ablation of the LTRs entire 12 base-pair U3 att site was evaluated, as compared with IN catalytic core mutants, in order to inhibit integration while reducing the risk of reversion mutations. How different mutations alter the processing of vector transcripts, as well as their effect on NHEJ and HR, was also evaluated. Finally, the rate of illegitimate integration and vector expression was assessed for IN, LTR att, and PPT mutations alone or in combination.
RESULTS
Evaluation of integrase and LTR attachment site mutant non-integrating lentiviral vectors
Three novel non-integrating lentiviral vectors were generated: one with an 11 base-pair deletion within the U5 region of the 5’ LTR att site, a second with a 12 base-pair deletion within the U3 region of the 3’ LTR att site, and a third containing an extended mutation in the C-terminus of IN (Figs. 1A, B). The novel vectors were compared to previously reported point mutations in IN23,24,28,29. While the mutations created were not predicted to significantly alter virion assembly24, this was tested experimentally by determining the physical titer (p24) of the vector. As shown in Figs. 1C and 1D, the level of p24 in vector supernatants from mutant vectors was similar to that measured from integration-competent vectors.
Figure 1.
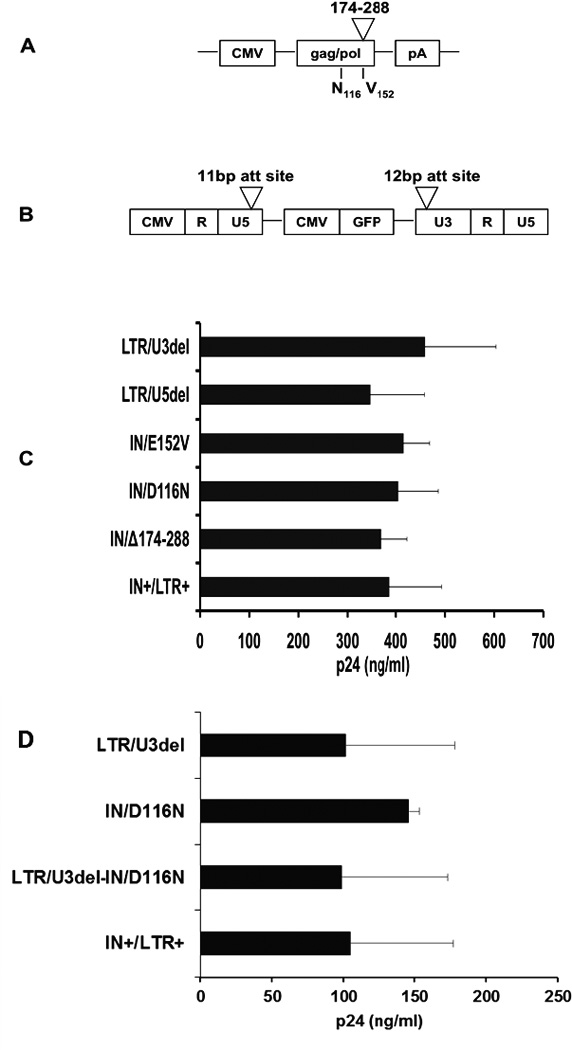
Integrase and LTR attachment site mutant vectors.(A) Integrase mutants derived upon mutation of the HIV-1 pol gene within the packaging plasmid, pMDL. Depicted are the locations of the c-terminal deletion and core domain point mutations. (B) LTR attachment site mutants derived upon deletion of the LTR attachment sites within the transfer vector, pcDNA-CS-CGW. Depicted are the locations of the 12 bp U3 (5’-ACTGGAAGGGCT-3’) and 11 bp U5 (5’-TCTCTAGCAGT-3’) attachment (att) site deletions. (C, D) Gag capsid protein (p24) was measured by ELISA in supernatants from 293T cells transfected with a GFP (C) or bleomycin resistance transgene containing transfer plasmid (D). Data is represented as mean ± SD.
To determine if the mutants were infectious, lentiviral vectors containing eGFP were generated and used to transduce HEK293 cells. As shown in Figs. 2A and 2B, deletions within the C-terminus of IN and at the U5-LTR att site did not provide significant gene transfer while the IN mutants IN/D116N and IN/E152V and the novel U3-LTR att site deletion mutant demonstrated similar vector expression in HEK293 cells. Based upon this data, subsequent studies utilized these latter three constructs.
Figure 2.
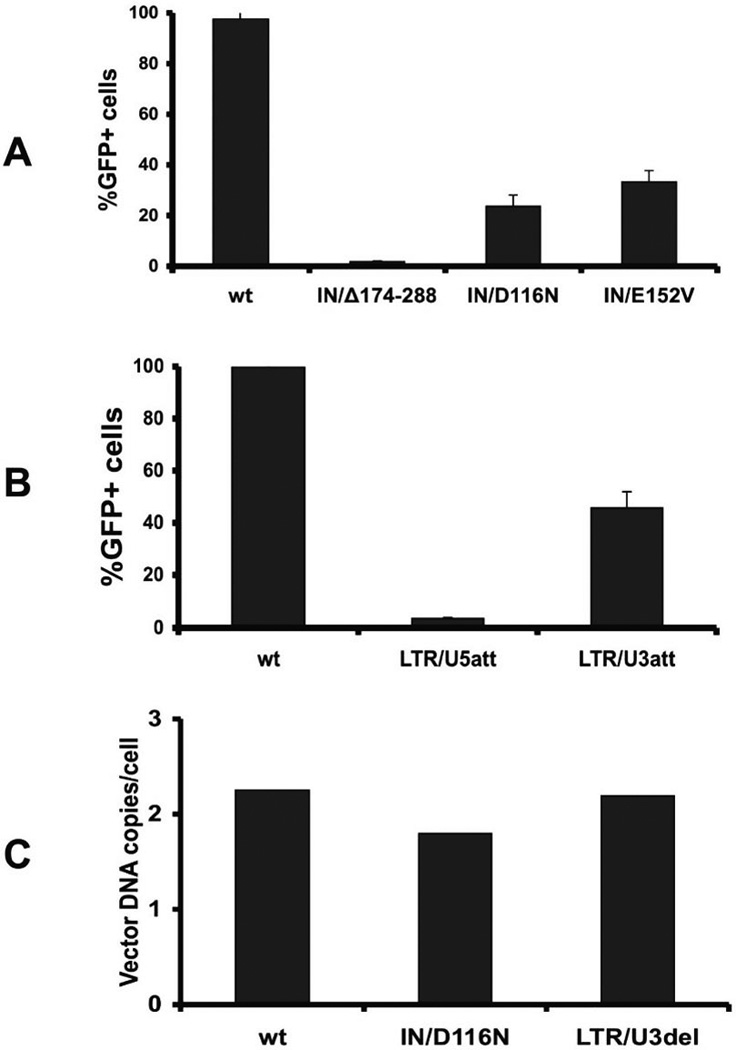
Transduction of HEK293 cells.The percentage of GFP expressing cells (%GFP+ cells) determined by flow cytometry 3 days after transduction with (A) three integrase mutant vectors, IN/Δ174–288, IN/D116N, and IN/E152V, and (B) two LTR attachment site mutant vectors, LTR/U3del and LTR/U5del. For all transductions, mock transduced cells served as a negative control and integration-competent vector (IN+/LTR+) transduced cells served as a positive control. (C) Vector DNA copy number 4 hours after transduction, as determined by quantitative PCR. Data is represented as mean ± SD.
In proliferating cell cultures, the average vector copy per cell from non-integrating vectors decreases with time. Therefore, determining infectious titer by traditional titer methods is problematic. To assess whether physical titer (as measured by p24 ELISA) reflects infectious titer, the amount of vector was normalized using p24, then vector DNA was measured four hours after transduction. As shown in Fig. 2C, physical titer correlated with vector copy number. Based on this finding, p24 levels were used to normalize the amount of vector product in subsequent experiments.
Gene expression with non-integrating vectors
To evaluate transgene expression with non-integrating vectors, firefly luciferase expression was assessed in HEK293 cells. Interestingly, after 3 days the LTR/U3 vector demonstrated higher transgene expression than the IN mutants relative to integrase competent vector (Fig. 3). Additionally, GFP expression was assessed in transduced HEK293 cells over a twenty-one day period. As would be predicted for NILV in dividing cells, the percentage of GFP expressing cells and the vector DNA content declined overtime (Figs. 4A, B). LTR/U3del transduced cells maintained a level of gene transfer that was approximately 20% of integration-competent vector at 21 days, while expression from IN mutants was approximately 5% of integration-competent vector. Fluorescence at day 21 was not due to residual GFP protein alone; GFP mRNA was still detectable twenty-one days after transduction (Fig. 4C). Similar to the observations with luciferase, the LTR/U3del vector provided higher and more persistent GFP expression than the IN mutants (Fig. 4A) despite similar levels of vector DNA (Fig 4B).
Figure 3.
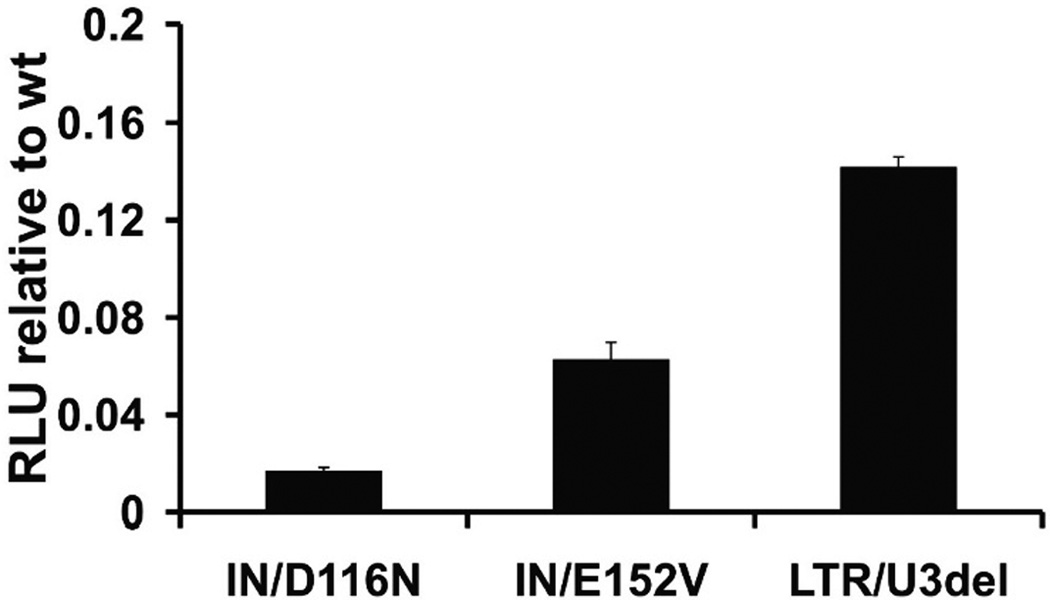
Luciferase expression from transduced HEK293 cells.Firefly luciferase activity (relative light units, RLU) was measured 3 days after transduction. Luciferase activity from cells transduced with IN and LTR att site mutant vectors is shown relative to that achieved from integration-competent vector (IN+/LTR+). Data is represented as mean ± SD.
Figure 4.
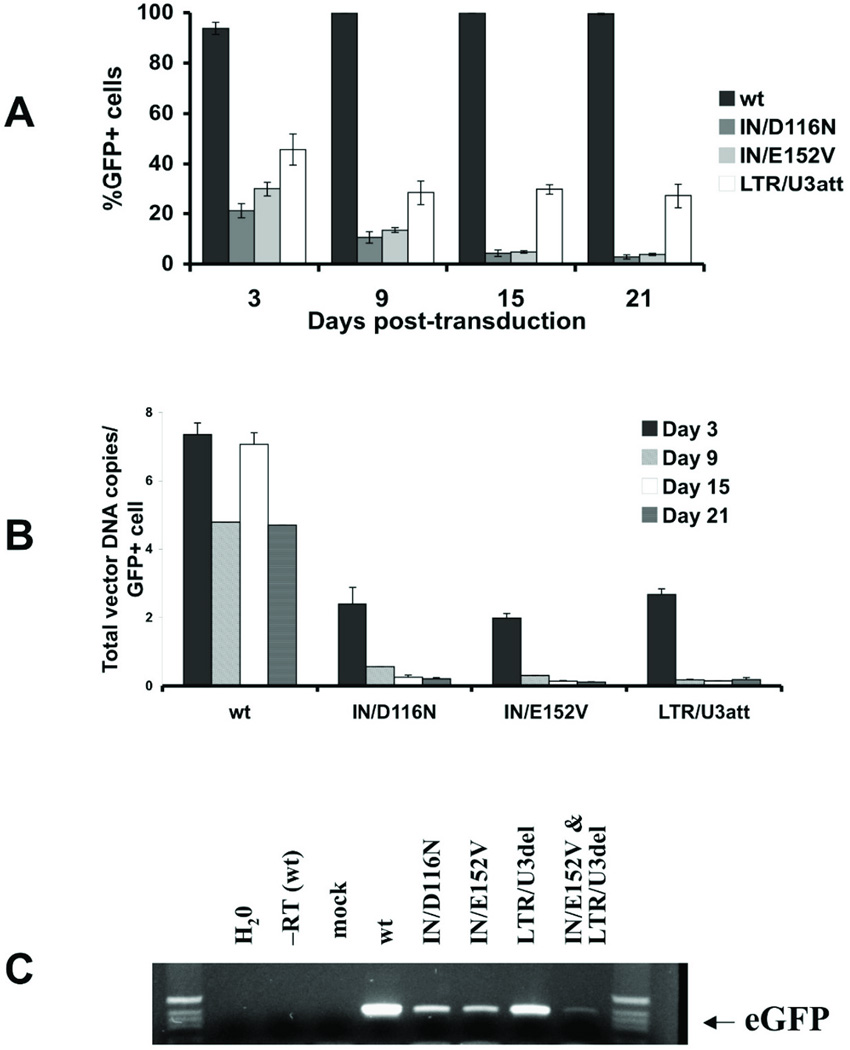
GFP expression from transduced HEK293 cells.(A) The percentage of GFP expressing (% GFP+) cells 3–21 days after transduction. GFP expression was determined at 3, 9, 15, and 21 days by flow cytometry. Data is represented as mean ± SD. (B) Total vector DNA copies estimated per GFP-expressing (GFP+) cell. Total vector DNA copy number was determined by quantitative PCR 3, 9, 15, and 21 days after transduction. Data is represented as mean ± SD. (C) GFP mRNA expression from vector DNA 21 days after transduction, as determined by RT-PCR.
Frequency of illegitimate integration with non-integrating vectors
The disparities observed with the varied levels of transgene expression suggested there may also be significant variation in the frequency of illegitimate integration by NILV. To determine the frequency of integration, a colony formation assay was performed using vectors containing an antibiotic resistance transgene and drug selection of transduced HEK 293 cells. As shown in Fig. 5, the non-integrating vectors significantly reduced the frequency of illegitimate integration at least 2 logs below that of an integration-competent vector. LTR/U3del integrated at a significantly higher frequency than the integrase mutant IN/D116N, but the difference is smaller than predicted when considering the differences in transgene expression (Figs. 3, 4A). Combining the IN and U3-LTR att site deletion mutations resulted in a significantly reduced frequency of integration as compared to either mutation independently.
Figure 5.
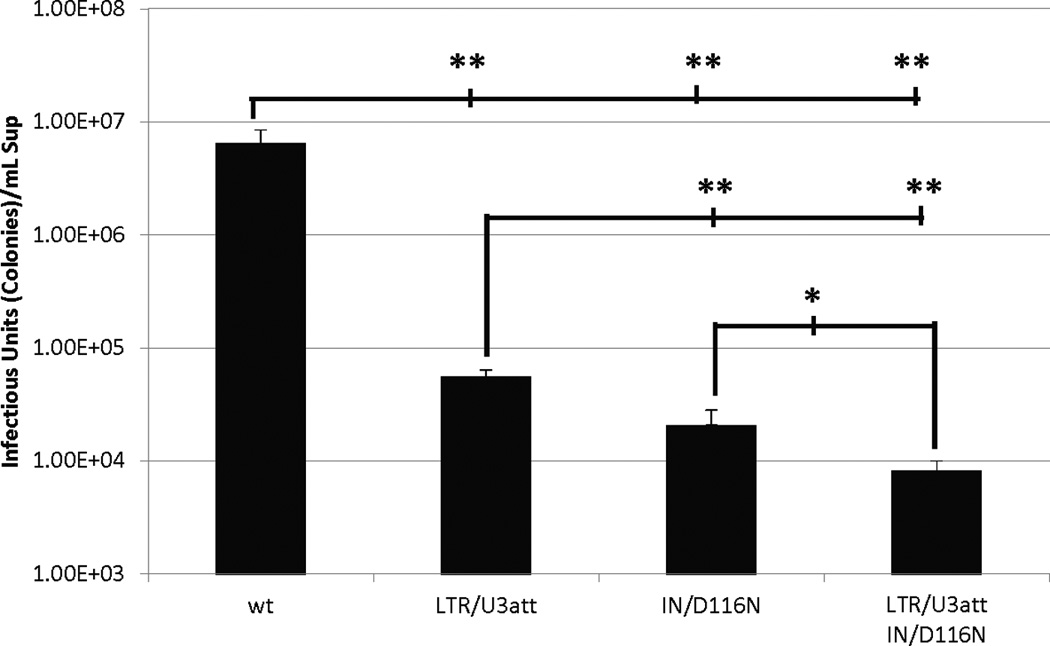
Frequency of integration in transduced HEK293 cells. Average antibiotic resistant colony formation titer assay results from two independent experiments comparing the frequency of integration among an integration competent vector (wt), a U3 LTR integrase attachment site mutant NILV (LTR/U3att), an integrase deficient NILV (IN/D116N), and a double mutant NILV (LTR/U3att-IN/D116N). Y-axis represents infectious units/ml of vector supernatant. Error bars indicate standard deviation of the mean. * - p < 0.05; ** - p < 0.005
Insertion-site analysis of non-integrating vectors
To further evaluate vector integration and expression, vector insertion sites were evaluated. The vector-genome junctions were amplified, sequenced and analyzed for the canonical features associated with integrase-mediated insertion as well as other means of integration, including NHEJ and/or HR. The results of this analysis are depicted in Fig. 6. As expected, the integration competent control vectors insertions demonstrated a 5 bp repeat of genomic DNA flanking the vector and end-processing of the integrase attachment sites as indicated by the presence of terminal CA dinucleotides. Integrase mutant vectors (IN/D116N) lack these findings and demonstrate insertions and/or deletions at the vector-genome junctions. Interestingly, LTR/U3del vectors demonstrate insertions and deletions at the 5’ LTR vector-genome junction but display the canonical features associated with integrase-mediated insertion at the 3’ LTR. Combining the LTR deletion with the IN mutation was able to recapitulate the insertions or deletions observed with IN/D116N at both LTR junctions, indicative of non-integrase mediated insertions.
Figure 6.
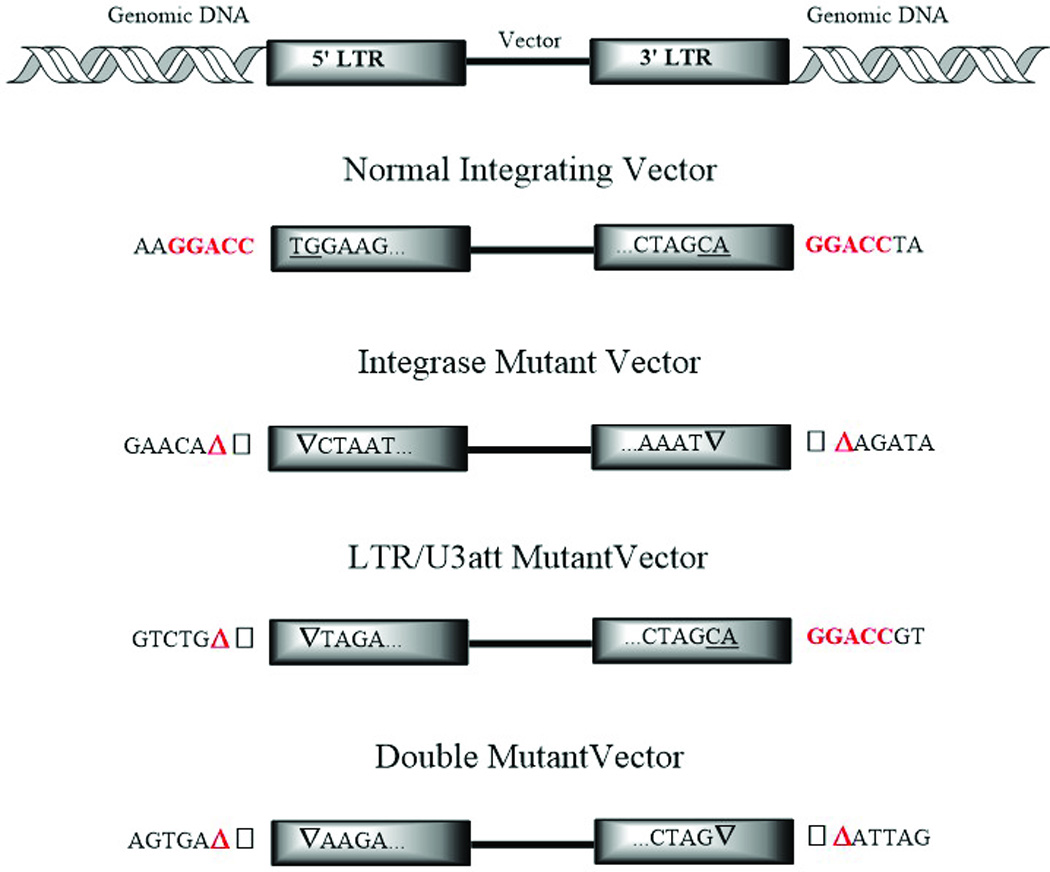
Vector-genome junctions of transduced HEK293 clones. Summary of results obtained from sequencing of Zeocin selected clones transduced with a normal integrating vector (CS-CZW), an integrase catalytic core mutant NILV (IN/D116N), a U3 LTR integrase attachment sited deleted NILV (LTR/U3att) and a double mutant (LTR/U3att-IN/D116N).Red highlighted base-pairs indicate flanking 5-bp repeat of genomic DNA by integrase-mediated strand-transfer; underlined TG/CA dinucleotides represent end-processing by a functional integrase protein; inverted black triangles represent LTR truncations; red highlighted triangle represents deletions of genomic DNA; black squares indicate insertions at the vector-genome junction.
The distribution of circular vector DNA transcripts amongst cells transduced with integrase and LTR attachment site mutant vectors
To further evaluate NILV processing, the relative distribution of unintegrated vector was determined by Southern blot analysis of low molecular weight DNA (Fig. 7A). Three days after transduction, circular forms were the predominant episomal form for wild-type, IN mutant, and LTR att site mutant vectors (Fig. 7A). While 1-LTR circles were detected amongst all three populations of transduced cells, integrase mutants have equal or greater number of 2-LTR circles in comparison with 1-LTR circles. In support of this finding, quantitative PCR analysis found that IN mutant vectors have a four-fold increase in 2-LTR circles compared to the LTR/U3att vector (Fig. 7B).
Figure 7.
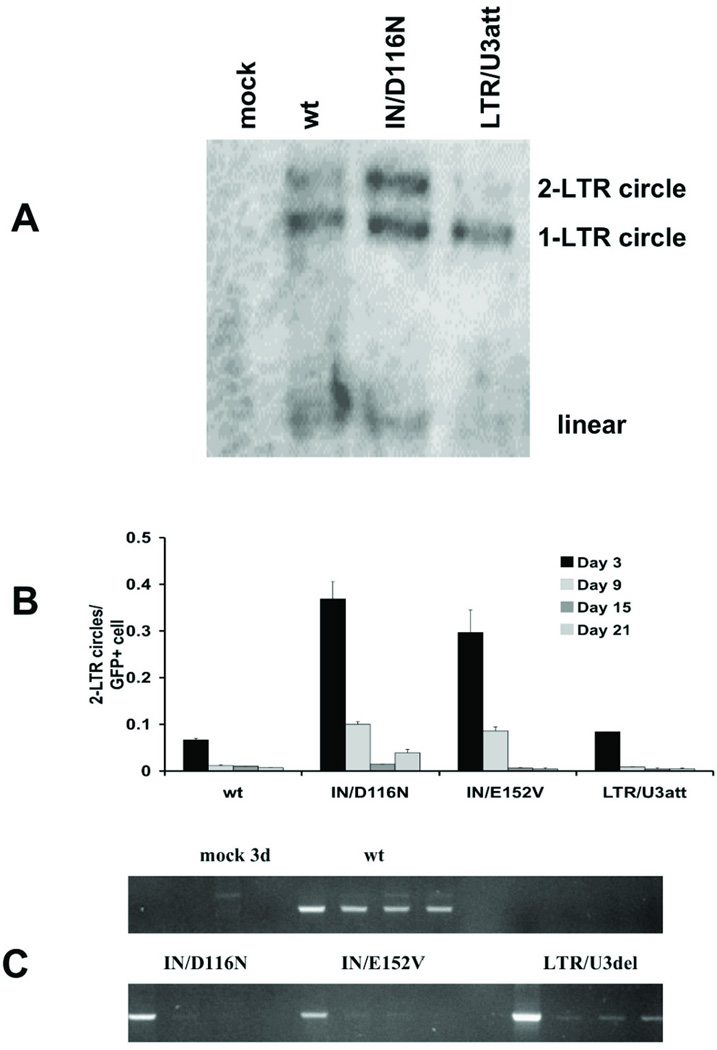
Unintegrated linear and circular vector DNA in transduced HEK293 cells.(A) Southern blot analysis of unintegrated vector DNA from low molecular weight (Hirt) DNA prepared from transduced cells 3 days after transduction. Linear (2667 bp), 1-LTR circular (c1-LTR, 4185 bp), and 2-LTR circular (c2-LTR, 4688 bp) vector DNA forms are detected upon hybridization of a radiolabeled eGFP probe. (B) 2-LTR circles estimated per GFP-expressing (GFP+) cell. 2-LTR circular vector DNA copy number was determined by quantitative PCR 3, 9, 15, and 21 days after transduction. Data is represented as mean ± SD. (C) Amplification of 1-LTR circles 3, 9, 15, and 21 days after transduction by standard PCR.
To assess the relative amount of 1-LTR circles, standard PCR was utilized as quantitative PCR methods cannot distinguish 1-LTR circles and linear transcripts. Three days after transduction, 1-LTR circles could be detected in cells transduced with wild-type, IN mutant, and LTR att site mutant vectors (Fig. 7C). However, 1-LTR circles persisted during the twenty-one day observation period in cells transduced with wild-type and LTR/U3del vectors but not in the IN mutants.
The fate of episomal vector DNA in cells deficient for non-homologous end-joining process
As 2-LTR circle formation is reported to involve NHEJ, wild-type and DNA-PKcs-deficient MO59J human glioma cell lines were used to evaluate the distribution of unintegrated vector DNA. DNA-PKcs is a necessary component of several DNA damage repair pathways, including NHEJ30. As determined by Southern blot analysis and quantitative PCR, wild-type MO59K cells transduced with the LTR/U3del vector had less 2-LTR circles than wild-type and IN/D116N vectors (Figs. 8 A, B), with a relative distribution similar to that observed in HEK293 cells (Figs. 7 A, B). In contrast, 2-LTR circles did not predominate in DNA-PKcs-deficient cells regardless of vector type (Figs. 8 A, B). This suggested that 2-LTR circles in cells transduced with IN mutant vectors are formed, at least in part, by DNA-PKcs-dependent NHEJ activities.
Figure 8.
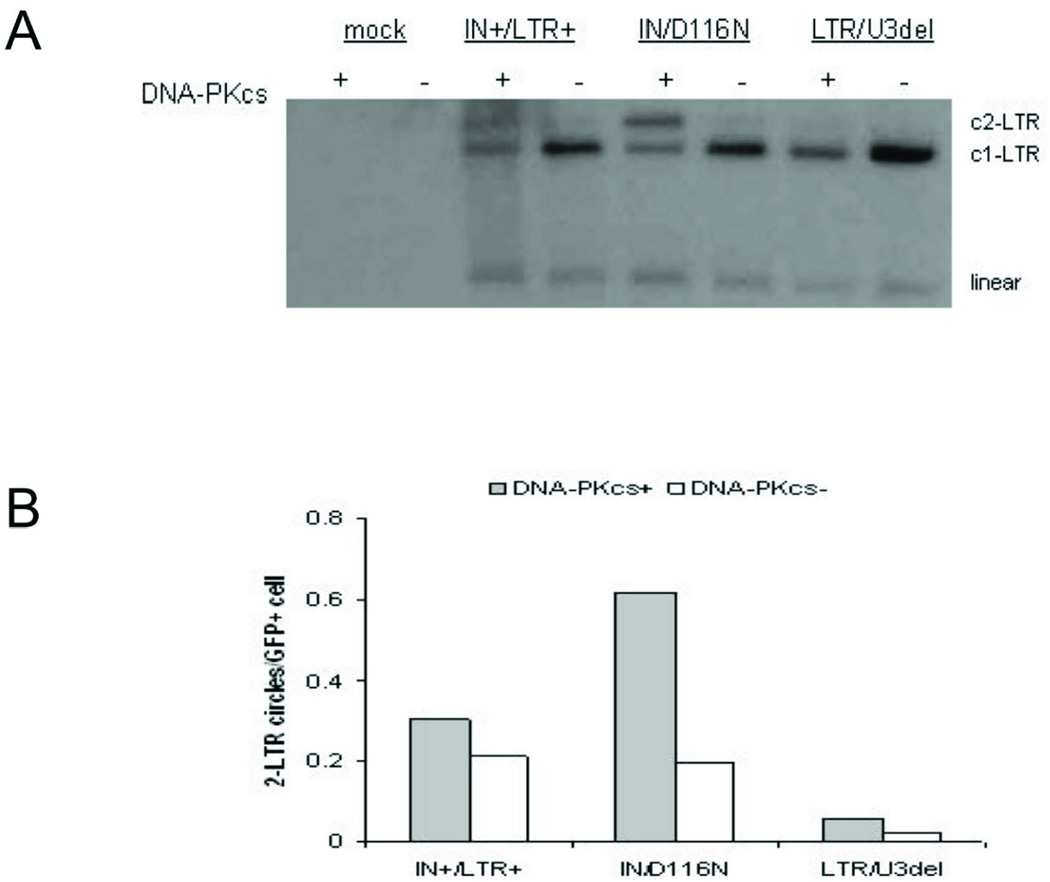
Vector DNA in transduced MO59K (DNA-PKcs+) and MO59J (DNA-PKcs−) cells. (A) Southern blot analysis of unintegrated vector DNA from low molecular weight DNA prepared from transduced cells 3 days after transduction. Vector DNA forms are detected upon hybridization of a radiolabeled eGFP probe. (B) 2-LTR circles estimated per GFP-expressing (GFP+) cell by quantitative PCR 3 days after transduction.
Optimizing NILV vectors for expression while minimizing illegitimate integration
In order to identify clinically useful NILV designs, the integration frequency of various combinations of NILV mutations were compared. As Kantor and colleagues have shown, there is decreased integration with a deletion in the 3’ PPT11. We evaluated pairing LTR/U3att with the 3’ PPT deletion with and without IN/D116N. Similar to results reported by Kantor et al. and Tareen et al.7,11, pairing ΔPPT with IN/D116N provided a modest reduction in integration as compared to either mutation individually (Fig. 9), indicating that each modification works independently to inhibit integration. Combining the ΔPPT modification with LTR/U3att did not alter the frequency of integration compared to either modification alone. In contrast, combining the IN/D116N mutation with LTR/U3att did decrease integration suggesting the mutations work independently. Combining all three mutations (ΔPPT, IN/D116N and LTR/U3att) did not offer any additional improvement to integration frequency reduction.
Figure 9.
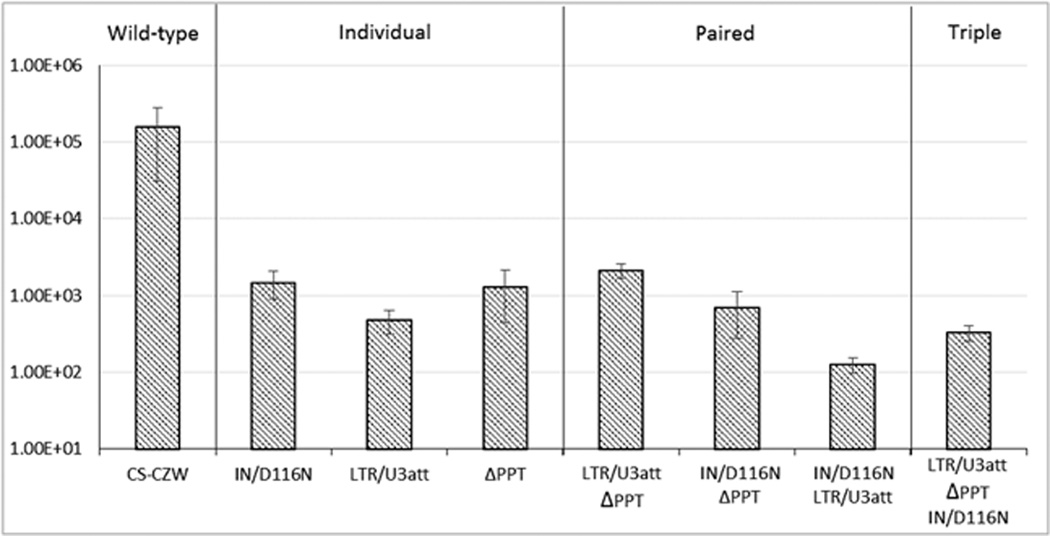
The frequency of integration in NILV with combined modifications for inhibiting integration.Average bleomycin resistant colony formation assay results from two experiments comparing the frequency of integration among combined mutations to LV design. The horizontal axis designates the independent modification or combining it with the indicated mutation(s). CS-CZW - an integration competent vector; ΔPPT - an NILV with a deletion of the 3’ polypurine tract; IN/D116N - an integrase deficient NILV; LTR/U3att - a U3 LTR integrase attachment site mutant NILV. Data is represented as mean ± SD.
NILV have also been associated with significantly reduced levels of transgene expression relative to integration competent vectors. The vectors studied here (ΔPPT, IN/D116N and combined ΔPPT/IN/D116N) show similar levels of expression (Fig. 10) consistent with previous reports5,11,27,31,32. Pairing the LTR/U3att and ΔPPT mutations did not affect expression. However, pairing the IN/D116N mutation with LTR/U3att modification resulted in a significant decrease in vector expression (Fig. 10).
Figure 10.
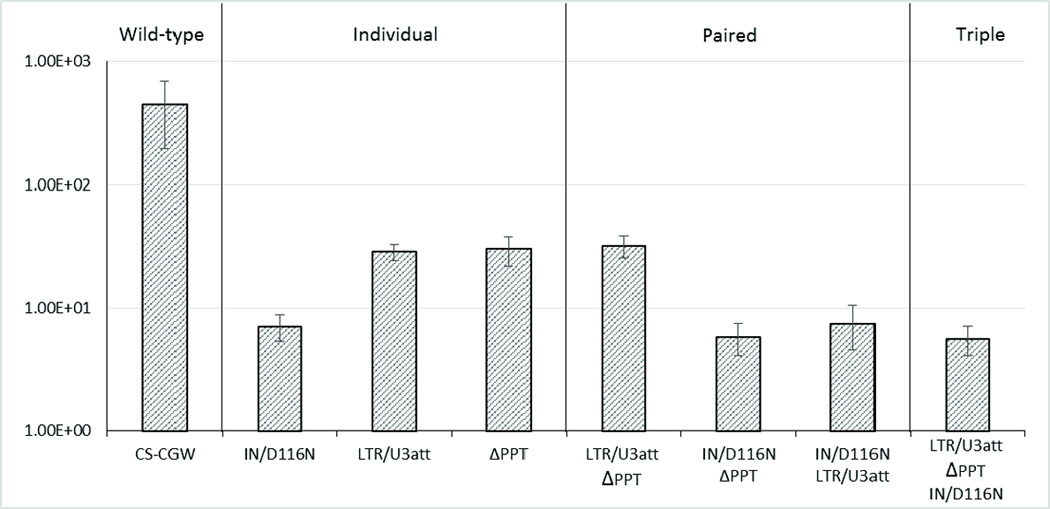
The effect of combining modifications to reduce integration frequency on transgene expression.Average flow cytometry results for GFP expression from two experiments comparing the levels among combined modifications to lentiviral vector design. Bars indicate the geometric mean fluorescence intensity (MFI) for the indicated vector on the x-axis. CS-CGW - normal integrating vector (wild-type); ΔPPT - NILV with a deletion of the 3’ polypurine tract; IN/D116N - an integrase deficient NILV; LTR/U3att - a U3 LTR integrase attachment site mutant NILV. Data is represented as mean ± SD.
We created a vector containing the ΔPPT, LTR/U3att, and IN/D116N mutations to investigate if transgene expression is affected. The triple mutant vector produced levels of transgene expression similar to those observed with all vectors containing the IN/D116N mutation. These findings suggest that the decreased illegitimate integration associated with IN/D116N is at the expense of expression.
DISCUSSION
The goal of this study was to evaluate a novel LTR att site mutation for transgene expression and illegitimate integration. The 12 base-pair deletion in this novel LTR/U3del vector is larger than the point mutants described by Nightingale et al.4 in order to decrease the chances of reversion mutations. Overall the LTR/U3del had higher and more persistent gene expression compared to IN mutations, but also was associated with a higher rate of illegitimate integration. Differences in processing appeared to contribute to the differences observed in vector expression and integration frequency. A novel mutant consisting of an 11 base-pair deletion of the U5-LTR att site was also evaluated that generated vector particles but failed to provide significant expression in transduced cells. This mutation was likely unsuccessful due to defective reverse transcription (RT), attributed to overlap of the U5-LTR att site and the RT primer binding site necessary for first strand synthesis33,34,35.
The novel LTR/U3att vector was compared to the previously published IN mutation IN/D116N5,6,26,27. Vector integration frequency was similar to that reported by Yanez-Munoz et al. using a D64 IN mutant (reversion rate of 1 in 815)6 but there was a modest 2.7 fold increase in integrations with the LTR/U3att vector versus the IN/D116N. Interestingly, combining the LTR and IN mutations significantly reduced the frequency of integration as compared to either mutation independently, in contrast to previous studies combining IN and LTR att site point mutations4,10. The previous LTR/U3 att mutations were point mutation and it is possible the relatively large deletion used in our study provided for this difference by reducing the chance of any latent binding of integrase to the attachment site.
Insertion site analysis revealed non-integrase mediated insertion at the ablated U3 att site of the 3’LTR-genome junction (Fig. 6), similar to what has been observed with IN mutants4,36–38. However the intact U5 att site at the 5’LTR-genome junction displayed features associated with integrase mediated insertion, consistent with the ability of intact integrase to interact with each attachment site independently18. This difference in processing LTR/U3att mutants presumably contributes to the increased illegitimate integration observed.
Interestingly, the modest increase in illegitimate integration rates between the mutations were not consistent with the larger differences in vector expression (Figs. 4, 5). To investigate this further, we evaluated the processing of non-integrated vector DNA. Despite similar levels of vector DNA, more GFP-expressing cells were detected upon transduction with LTR/U3del than IN mutants. This observation was associated with differences in the ratio of 1-LTR and 2-LTR vector forms. At the point of maximal transgene expression, IN/D116N and IN/E152V transduced cells contained nearly four-to-five times as many copies of 2-LTR circular vector DNA relative to LTR/U3del transduced cells (Fig. 7B). In contrast, 1-LTR circles were the predominate form in cells transduced with LTR/U3del vector (Fig. 7A). Given that LTR/U3del maintains higher transgene expression despite similar total vector DNA, further work is warranted to determine if 1-LTR circles facilitate higher and/or more persistent gene expression than 2-LTR circles. Possible mechanisms meriting further investigation include improved trafficking to the nucleus and resistance to degradation. The presence of a functional integrase protein allows for its binding to the wild-type U5-LTR att site for nuclear localization, while providing steric hindrance protecting the viral DNA ends from exonuclease activity39. The predominance of 1-LTR episomes can also improve vector viability. 2-LTR circles are derived from linear episomes as products of NHEJ of the viral DNAs 5’ and 3’ ends. Linear episomes are readily degraded by exonuclease activity which could lend to reduced transgene expression, whereas circular DNA is protected from exonuclease activity40.
Formation of 2-LTR circles is believed to be dependent upon NHEJ-double strand break DNA repair mechanisms, such as Ku-80, XRCC4, DNA ligase 4, and DNA-PKcs41. NILV transduction of DNA-PKcs-deficient cells suggests that a U3-LTR att site deletion may negatively influence NHEJ-mediated vector DNA circularization thereby limiting the number of 2-LTR circles (Fig. 8). Another explanation could be due to the proximity of the U3 LTR att site deletion which is immediately adjacent to the vectors 3’ PPT. The PPT acts as a primer site for second strand synthesis during RT and Kantor et al. have shown that deletion of this element results in aberrant RT and the preferential production of 1-LTR circles11. Deletion of the juxtaposed U3 attachment site may also affect RT in a similar fashion. The results of this study suggest that IN and LTR att site mutations may differentially influence the circularization of unintegrated vector DNA.
It was observed that combining the IN and LTR mutations led to an improved reduction in integration frequency as compared to either mutation independently (Fig. 5). As NILV have been generated by deleting the 3’ PPT, we evaluated this mutation in combination with IN and LTR/U3att mutations. Combining ΔPPT with IN/D116N provided between a 2 and 3 fold reduction in integration (Fig. 9), similar to previous observations when combined with a D64E IN mutation11. Interestingly, combining the ΔPPT and LTR/U3att mutation provided little or no reduction in integration frequency (Fig. 9). This suggests they evoke similar alterations in the processing of episomal DNA.
Combining ΔPPT and LTR/U3att had no effect on transgene expression as compared to either mutation alone, however, combining ΔPPT, LTR/U3att or both together with IN/D116N resulted in a significant reduction in transgene expression (Fig. 9). The results show a trend of reduced transgene expression when the vector contains the IN mutation (Fig. 10). Hence, many of the vector combinations with the lowest frequencies of integration also demonstrated the lowest levels of transgene expression (Figs. 9, 10). Therefore, design of NILV for clinical applications should balance the risk of insertional mutagenesis and the level of vector expression. The risk of insertional mutagenesis with NILV is dependent on both vector integration rates and vector dose; if a vector with low integration rates requires significantly higher doses the safety benefit could favor a vector with higher expression.
This study suggests that the type of mutation used to generate non-integrating lentiviral vectors can influence cellular processing of vector transcripts. Such data will be important when attempting to design non-integrating vectors that maximize vector expression with low frequencies of integration and when considering transduction of target cells that may be defective in DNA repair pathways.
MATERIALS AND METHODS
Cell culture and reagents
HEK293 and HEK293T cells were purchased from American Type Culture Collection (Manassas, VA). MO59J and MO59K cell lines were kindly provided by D. Gilley (Indiana University School of Medicine, Indianapolis, IN). All cell lines were maintained in DMEM (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C with 5% CO2.
Vector plasmids consisted of the eGFP expressing pcDNA-CS-CGW42 (kindly provided by P. Zoltick, Children’s Hospital of Philadelphia, Philadelphia, PA), the gag-pol expressing pMDL, the VSV-G envelope expressing pMDG, and the rev expressing pRSV-rev plasmid (all kindly provided by Cell Genesys, South San Francisco, CA)43.
Construction of integrase and LTR attachment site mutant vector plasmids
For construction of the integrase deletion mutant IN/Δ174–288, digestion of the pMDL plasmid with AflII and subsequent base fill-in created an early stop codon within the integrase coding sequence. Two integrase missense mutants, IN/D116N and IN/E152V, were generated by site-directed mutagenesis of pMDL. Site-directed mutagenesis was performed using a Quik Change II XL Site-Directed Mutagenesis Kit (Stratagene, Cedar Creek, TX) according to the manufacturer’s specifications.
For generation of LTR/U3del, the 3’-LTR from the pcDNA-CS-CGW was subcloned into pZero2 (Invitrogen, Carlsbad, CA) at an upstream Asp718 and a downstream ApaI restriction sites. Site-directed mutagenesis was performed on the resulting plasmid, allowing for the deletion of the twelve base-pair integrase attachment site. For generation of LTR/U5del, the 5’-LTR from pcDNA-CS-CGW was subcloned into pcDNA3 (Invitrogen, Carlsbad, CA) at an upstream SspI restriction site and a downstream NotI restriction site. Site-directed mutagenesis was performed deleting the eleven base-pair integrase attachment site. Incorporation of a bleomycin resistance transgene in lieu of eGFP was accomplished by insertion using unique AgeI and XhoI restriction enzyme sites. A deletion of the transfer plasmids 3’ polypurine tract (ΔPPT) was incorporated by standard restriction site insertion of the modified region synthesized by GeneArt (Life Technologies, Grand Island, NY). The mutations were confirmed by DNA sequencing.
Vector production and measurement of physical and infectious titer
VSV-G pseudotyped vector particles were produced by a four-plasmid calcium phosphate transient transfection method described previously44. All vector supernatants were treated with Benzonase (Novagen, San Diego, CA) to remove residual plasmid DNA, as previously described45. Triplicates of three dilutions (10−3, 10−4, and 10−5) of supernatant were analyzed for p24 gag capsid protein production using a p24 gag ELISA kit (Beckman Coulter, Fullerton, CA). The infectious titer of integration-competent vectors was determined in HEK293 cells by a method described previously44.
Transduction of cell lines and assessment of GFP or LUC expression
HEK293 cells, seeded at 105 cells per well of a six-well plate or 3 × 105 cells per T25 flask 24 hours prior to transduction, were incubated with vector in the presence of 8 µg/ml of polybrene for 4 hours at 37°C. Transductions, performed in duplicate, utilized vector normalized for p24 (325 ng of p24 corresponding to an estimated MOI of five). GFP expression was assessed using a FACScan cytometer and CellQuest analysis software (Becton-Dickinson, San Jose, CA). For measurement of LUC expression, cell lysates were prepared from transduced cells, upon which 10 µg of protein per sample was added per well of a 96-well plate. LUC activity was measured as light intensity upon the addition of the LUC substrate, luciferin (Luciferase Assay System, Promega, Madison, WI).
MO59J and MO59K cells, seeded at 3 × 105 cells per 100 mm dish 24 hours prior to transduction and transduced with vector supernatant containing 2000 ng of p24.
Measurement of integration frequency by antibiotic resistant colony forming assay
Vectors containing a bleomycin resistance transgene were generated for measurement of integration frequency as described above. HEK293 cells seeded at 105 cells per well, were transduced with vector supernatant normalized by p24 content with 10 replicates per vector. The selection reagent Zeocin™ (Life Technologies; Carlsbad, CA), at a concentration of 500 µg/mL, was added to the media 48 hours post-transduction. The cells were incubated in selection media for 21 days post-transduction and the frequencies of integration (demonstrated by infectious titer) were determined by multiplying the number of viable colonies by their respective dilution factor.
Amplification of vector-genome junctions
Illegitimate integration was quantified by isolating drug-resistant clones after transducing HEK 293 cells with a Bleomycin resistance transgene. Transduced cells were selected for three weeks prior to isolation, after which genomic DNA was extracted using the Puregene DNA Purification Kit (Gentra Systems, Inc., Minneapolis, MN). The vector-genome junctions were then amplified by linear amplification mediated PCR (LAM-PCR) off of the 3’ LTR as previously described46 in two separate experiments (CS-CZW n =12; IN/D116N n=7, 12; LTR/U3att n=8, 11; LTR/U3att-IN/D116N n=4, 9) and/or by a modified technique adapted from Ravin, et al47 and Zhou, et al48 (IN/D116N n=12; LTR/U3att n=18; LTR/U3att-IN/D116N n=22). Briefly, to perform the modified technique for surveying insertion sites, the genomic DNA was first sheared to an approximate size of 1,000 bp using Covaris sonication (Covaris, Wohurn, MA). Starting with 1000 ng of sheared fragments, the ends were repaired using the NEBNext End Repair Module (NEB, Ipswich, MA) and 3’ dA-tailed with the NEBNext dA-Tailing Module (NEB, Ipswich, MA). Products greater than ~300 bp were selected and purified using a 0.8:1 ratio of AMPure XP beads (Beckman Coulter, Brea, CA) to dA-tailed product. A partially double stranded linker cassette with a 5’ T-overhang (LamTlinkerL: 5’ GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTTAGGT 3’; LamTlinkerS: 5' CCTAACTGCTGTGCCACT 3’) was then ligated to the ends of the purified products using the Fast-Link DNA Ligation kit (Epicentre, Madison, WI). Exponential PCR was performed using the ligation product as a template with a biotinylated vector-specific primer for the 5’ or 3’ end of the vector and a linker-specific primer (LentiLAM-LC1: 5’ GACCCGGGAGATCTGAATTC 3’). Vector-specific PCR products were then enriched using Streptavidin-coated M-280 Dynabeads (Invitrogen, Carlsbad, CA). Next, nested PCR using a second indexed LTR-specific primer for the 5’ or 3’ end and another linker-specific primer (LentiLAM-LC2: 5’ AGTGGCACAGCAGTTAGG 3’) was performed using the enriched product as a template. The final PCR products were then purified using a 1.8:1 ratio of AMPure XP beads and quantified using a Qubit Fluorometer (Life Technologies, Grand Island, NY).
In order to confirm the insertion sites when LAM-PCR was performed solely off the 3’ LTR, conventional PCR was performed in order to amplify the 5’ vector-genome junction using the insertion site predicted by the 3’ LAM-PCR results.
Vector insertion site analysis
Samples from the first LAM-PCR experimental group were analyzed by pyrosequencing (Roche 454 FLX Titanium) at the Indiana University Center for Genomics and Bioinformatics, Bloomington IN and analyzed using the SeqMap 2.0 web server platform49. Subsequent samples were analyzed using the Illumina MiSeq platform at the Genomics Core Facility of the University of Notre Dame. Paired-end reads of 250 base-pair were generated and in-house scripts were used to de-multiplex the reads and to retain paired-end reads that have both perfectly matched viral vector-specific primer and linker primer used in the last round of PCR. MiSeq adapter and linker sequences were trimmed off with Cutadapt version 1.550. Paired-end reads were merged with PEAR version 0.9.5–6451. For the non-overlapping paired-end reads, the reads containing the viral vector sequence were kept. The viral vector sequences in the merged reads were located with cross-match version .9909329 (http://www.phrap.org). Reads with less than 30 base-pair viral vector sequence and less than 10 base-pair remaining genomic sequence were discarded. The remaining reads were further clustered with usearch7.0.1090_i86linux3252. Representative reads of each cluster were mapped to human reference genome hg19 using bwa version 0.7.5a53. Alignments demonstrating 100% sequence identity were designated as integration sites. The cross-match output, cluster and mapping information were compiled together in R54 and further analyzed.
Detection of vector DNA by PCR
Vector and 2-LTR circular DNA was detected using quantitative PCR assay based on a method previously described by Butler et al55. Total cellular DNA was prepared using a Puregene DNA Purification Kit (Gentra Systems, Inc., Minneapolis, MN), from which a 250 ng or 500 ng sample was used for each reaction. To determine total, integrated, and 2-LTR circular vector DNA copy number, standard curves were generated by preparing serial dilutions of pcDNA-CS-CGW, genomic DNA from wild-type vector transduced HEK293 cells, or a 2-LTR circle reference plasmid that was generated upon the introduction of a 2-LTR fragment into a cloning vector, pCR4-TOPO (Invitrogen, Carlsbad, CA), respectively. Each sample was run in duplicate and data analysis was performed using Sequence Detection Systems 1.9.1 software (Applied Biosystems, Foster City, CA).
A 250 ng sample of total cellular DNA from transduced HEK293 cells at 3 days post-transduction was used for detection of 1-LTR circular vector DNA by standard PCR. 1-LTR circles were amplified using the following primers: 5’-GACCAATGACTTACAAGGCAGC-3’ (forward) and 5’-GCAAGCCGAGTCCTGCGTCG-3’ (reverse). The cycling conditions were 95°C for 10 min, thirty cycles of 92°C for 1 min, 56°C for 2 min, 72°C for 1.5 min, and 72°C for 7 min.
Detection of unintegrated vector DNA Southern blot analysis
Low molecular weight (Hirt) DNA (5–10 µg) purified from wild-type and mutant vector transduced HEK293, MO59J, and MO59K cells 3 days post-transduction, was digested overnight with BamHI and separated on an agarose gel. Briefly, the Hirt DNA extraction protocol was as follows. Transduced cells were resuspended in extraction buffer (0.6% SDS, 0.01 M EDTA, and 0.01 M Tris) and 5M NaCl overnight at 4°C. The supernatant was collected and the DNA was isolated by phenol-chloroform extraction. Upon transfer to a nylon membrane, the DNA was probed for GFP using a 32P-labeled eGFP probe prepared with a NEB Blot Kit (New England Biolabs, Ipswich, MA).
Detection of GFP mRNA by RT-PCR
Total cellular RNA was prepared from transduced HEK293 cells using a Qiagen RNeasy Micro Kit (Valencia, CA). For first strand cDNA synthesis, 200 ng of RNA was used in a reverse transcription reaction which included 0.5 µg of oligo(dT)12–18 primer and 2.5 units of HIV-1 reverse transcriptase (Ambion, Austin, TX). The reaction was incubated at 48°C for 30 minutes. About 5 µl of cDNA was used in a subsequent PCR reaction for amplification of GFP using the following primers: 5’-TGACCCTGAAGTTCATCTGCACCA-3’ (forward) and 5’-TGTGGCGGATCTTGAAGTTCACCT-3’ (reverse). The cycling conditions were 94°C for 5 min, thirty cycles of 94°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec, and 72°C for 7 min.
Statistical analysis
Quantitative data are represented as mean ± standard deviation of the mean. Data were analyzed using a two-tailed student’s t-test or analysis of variance (ANOVA). Differences were considered statistically significant for P-values less than 0.05.
Acknowledgments
FUNDING
The study was funded in part by a grant from the NHLBI (P40HL11621). GJ was supported by Edward T. Harper Scholar – funded by NIH NIGMS on 1R25 GM079657-01, Indiana University Initiative for Maximizing Graduate Student Diversity. AS was supported by the Joe and Shirley Christian Graduate Student Education Fund.
Dr. Cornetta is a consultant for Cook Regentec but there are no collaborative or financial relationships with the work contained in this manuscript.
The authors thank Teresa Johnson for her assistance with vector production, David Gilley who provided the MO59K and MO59J cell lines and Troy Hawkins and Hongyu Gao for assistance with bioinformatics analysis.
Footnotes
CONFLICT OF INTEREST STATEMENT
Dr. Shaw, Dr. Joseph, Mrs. Jasti, Dr. Sastry and Dr. Witting declare no potential conflict of interest.
REFERENCES
- 1.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack M, Wulffraat N, Leboulch Pl, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 2.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein S, Ott M, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 4.Nightingale S, Hollis R, Pepper K, Petersen D, Yu X, Yang C, et al. Transient gene expression by nonintegrating lentiviral vectors. Mol Ther. 2006;13:1121–1132. doi: 10.1016/j.ymthe.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Philippe S, Sarkis C, Barkats M, Mammeri H, Ladroue C, Petit C, et al. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc Natl Acad Sci U S A. 2006;103:17684–17689. doi: 10.1073/pnas.0606197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yanez-Munoz R, Balaggan K, NacNeil A, Howe S, Schmidt M, Smith A, et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. 2006;12:348–353. doi: 10.1038/nm1365. [DOI] [PubMed] [Google Scholar]
- 7.Tareen S, Kelley-Clarke B, Nicolai C, Cassiano L, Nelson L, Slough M, et al. Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol Ther. 2014;22:575–587. doi: 10.1038/mt.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negri D, Michelini Z, Baroncelli S, Spada M, Vendetti S, Buffa V, et al. Successful immunization with a single injection of non-integrating lentiviral vector. Mol Ther. 2007;15:1716–1723. doi: 10.1038/sj.mt.6300241. [DOI] [PubMed] [Google Scholar]
- 9.Mali P, Ye Z, Hommond H, Yu X, Lin J, Chen G, et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells. 2008;26:1998–2005. doi: 10.1634/stemcells.2008-0346. [DOI] [PubMed] [Google Scholar]
- 10.Apolonia L, Waddington S, Fernandes C, Ward N, Bouma G, Blundell M, et al. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol Ther. 2007;15:1947–1954. doi: 10.1038/sj.mt.6300281. [DOI] [PubMed] [Google Scholar]
- 11.Kantor B, Bayer M, Ma H, Samulski J, Li C, McCown T, et al. Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Mol Ther. 2011;19:547–556. doi: 10.1038/mt.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahim A, Wong A, Howe S, Buckley S, Acosta-Saltos A, Elston K, et al. Efficient gene delivery to the adult and fetal CNS using pseudotyped non-integrating lentiviral vectors. Gene Ther. 2009;16:509–520. doi: 10.1038/gt.2008.186. [DOI] [PubMed] [Google Scholar]
- 13.Lombardo A, Genovese P, Beausejour C, Colleoni S, Lee Y, Kim K, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 14.Lombardo A, Cesana D, Genovese P, Stefano B, Provasi E, Colombo D, et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- 15.Joglekar A, Hollis R, Kuftinec G, Senadheera S, Chan R, Kohn D. Integrase-defective lentiviral vectors as a delivery platform for targeted modification of adenosine deaminase locus. Mol Ther. 2013;21:1705–1717. doi: 10.1038/mt.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osborn M, Starker C, McElroy A, Webber B, Riddle M, Xia L, et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21:1151–1159. doi: 10.1038/mt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown HE, Chen H, Engelman A. Structure-based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site: effects on integration and cDNA synthesis. J Virol. 1999;73:9011–9020. doi: 10.1128/jvi.73.11.9011-9020.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masuda T, Kuroda MJ, Harada S. Specific and independent recognition of U3 and U5 att sites by human immunodeficiency virus type 1 integrase in vivo. J Virol. 1998;72:8396–8402. doi: 10.1128/jvi.72.10.8396-8402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman PA, Dickson ML, Fyfe JA. Human immunodeficiency virus type 1 integration protein: DNA sequence requirements for cleaving and joining reactions. J Virol. 1992;66:3593–3601. doi: 10.1128/jvi.66.6.3593-3601.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farnet CM, Haseltine WA. Circularization of human immunodeficiency virus type 1 DNA in vitro. J Virol. 1991;65:6942–6952. doi: 10.1128/jvi.65.12.6942-6952.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeanson L, Subra F, Vaganay S, Hervy M, Marangoni E, Bourhis J, et al. Effect of Ku80 depletion on the preintegrative steps of HIV-1 replication in human cells. Virology. 2002;300:100–108. doi: 10.1006/viro.2002.1515. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Olvera J, Yoder K, Mitchell R, Butler S, Lieber M, et al. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 2001;20:3272–3281. doi: 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leavitt AD, Shiue L, Varmus HE. Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase functions in vitro. J Biol Chem. 1993;268:2113–2119. [PubMed] [Google Scholar]
- 24.Leavitt AD, Robles G, Alesandro N, Varmus HE. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol. 1996;70:721–728. doi: 10.1128/jvi.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matrai J, Chuah MK, VandenDriessche T. Recent advances in lentiviral vector development and applications. Mol Ther. 2010;18:477–490. doi: 10.1038/mt.2009.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saenz D, Loewen N, Peretz M, Whitman T, Barraza R, Howell K, et al. Unintegrated lentivirus DNA persistence and accessibility to expression in nondividing cells: analysis with class I integrase mutants. J Virol. 2004;78:2906–2920. doi: 10.1128/JVI.78.6.2906-2920.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vargas J, Jr, Gusella GL, Najfeld V, Klotman ME, Cara A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum Gene Ther. 2004;15:361–372. doi: 10.1089/104303404322959515. [DOI] [PubMed] [Google Scholar]
- 28.Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol. 1995;69:2729–2736. doi: 10.1128/jvi.69.5.2729-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heuer TS, Brown PO. Mapping features of HIV-1 integrase near selected sites on viral and target DNA molecules in an active enzyme-DNA complex by photo-cross-linking. Biochemistry. 1997;36:10655–10665. doi: 10.1021/bi970782h. [DOI] [PubMed] [Google Scholar]
- 30.Peddi P, Loftin C, Dickey J, Hair J, Burns K, Aziz K, et al. DNA-PKcs deficiency leads to persistence of oxidatively induced clustered DNA lesions in human tumor cells. Free Radic Biol Med. 2010;48:1435–1443. doi: 10.1016/j.freeradbiomed.2010.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bayer M, Kantor B, Cockrell A, Ma H, Zeithaml B, Li X, et al. A large U3 deletion causes increased in vivo expression from a nonintegrating lentiviral vector. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:1968–1976. doi: 10.1038/mt.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage F, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 33.Das AT, Klaver B, Berkhout B. Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers other than the natural tRNA(3Lys) Journal of virology. 1995;69:3090–3097. doi: 10.1128/jvi.69.5.3090-3097.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aiyar A, Cobrinik D, Ge Z, Kung HJ, Leis J. Interaction between retroviral U5 RNA and the T psi C loop of the tRNA(Trp) primer is required for efficient initiation of reverse transcription. Journal of virology. 1992;66:2464–2472. doi: 10.1128/jvi.66.4.2464-2472.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy JE, Goff SP. Construction and analysis of deletion mutations in the U5 region of Moloney murine leukemia virus: effects on RNA packaging and reverse transcription. Journal of virology. 1989;63:319–327. doi: 10.1128/jvi.63.1.319-327.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaur M, Leavitt AD. Mutations in the human immunodeficiency virus type 1 integrase D,D(35)E motif do not eliminate provirus formation. J Virol. 1998;72:4678–4685. doi: 10.1128/jvi.72.6.4678-4685.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koyama T, Sun B, Tokunaga K, Tatsumi M, Ishizaka Y. DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition. Retrovirology. 2013;10:21. doi: 10.1186/1742-4690-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matrai J, Cantore A, Bartholomae C, Annoni A, Wang W, Acosta-Sanchez A, et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology. 2011;53:1696–1707. doi: 10.1002/hep.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller M, Farnet C, Bushman F. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71:5382–5390. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLenachan S, Sarsero J, Ioannou P. Flow-cytometric analysis of mouse embryonic stem cell lipofection using small and large DNA constructs. Genomics. 2007;89:708–720. doi: 10.1016/j.ygeno.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Kilzer J, Stracker T, Beitzel B, Meek K, Weitzman M, Bushman F. Roles of host cell factors in circularization of retroviral dna. Virology. 2003;314:460–467. doi: 10.1016/s0042-6822(03)00455-0. [DOI] [PubMed] [Google Scholar]
- 42.Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dull T, Zufferey R, Kelly M, Mandel R, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. Journal of virology. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sastry L, Johnson T, Hobson MJ, Smucker B, Cornetta K. Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Ther. 2002;9:1155–1162. doi: 10.1038/sj.gt.3301731. [DOI] [PubMed] [Google Scholar]
- 45.Sastry L, Xu Y, Cooper R, Pollok K, Cornetta K. Evaluation of plasmid DNA removal from lentiviral vectors by benzonase treatment. Hum Gene Ther. 2004;15:221–226. doi: 10.1089/104303404772680029. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I, et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 47.De Ravin S, Su L, Theobald N, Choi U, Macpherson J, Poidinger M, et al. Enhancers are major targets for murine leukemia virus vector integration. J Virol. 2014;88:4504–4513. doi: 10.1128/JVI.00011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou S, Tang C, Rapp S, Bonner M, De Ravin S, Malech H, et al. Molecular Therapy. NEW YORK, NY 10013-1917 USA: NATURE PUBLISHING GROUP 75 VARICK ST, 9TH FLR; 2014. A novel non-restrictive, semi-quantitative method for vector insertion site analysis based on random shearing of genomic DNA; pp. S211–S211. [Google Scholar]
- 49.Hawkins T, Dantzer J, Peters B, Dinauer M, Mockaitis K, Mooney S, et al. Identifying viral integration sites using SeqMap 2.0. Bioinformatics. 2011;27:720–722. doi: 10.1093/bioinformatics/btq722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011;17:10–12. [Google Scholar]
- 51.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 53.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.RCore, T. 2012 (ISBN 3-900051-07-0, URL http://www.R-project.org) [Google Scholar]
- 55.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]