

Abstract
Recent hemophilia B clinical trials using adeno-associated virus (AAV) gene delivery have demonstrated much lower FIX production in patients compared to the high levels observed in animal models and AAV capsid specific CTLs response elicited at high doses of AAV vectors. These results emphasize the necessity to explore effective approaches for enhancement of AAV transduction. Initially, we found that incubation of all AAV vectors with human serum enhanced AAV transduction. Complementary analytical experiments demonstrated that human serum albumin (HSA) directly interacted with the AAV capsid and augmented AAV transduction. The enhanced transduction was observed with clinical grade HSA. Mechanistic studies suggest that HSA increases AAV binding to target cells and that the interaction of HSA with AAV doesn’t interfere with the AAV infection pathway. Importantly, HSA incubation during vector dialysis also increased transduction. Finally, HSA enhancement of AAV transduction in a model of hemophilia B displayed greater than a 5-fold increase in vector derived circulating FIX, which improved the bleeding phenotype correction. In conclusion, incubation of HSA with AAV vectors supports a universal augmentation of AAV transduction and more importantly, this approach can be immediately transitioned to the clinic for the treatment of hemophilia and other diseases.
Keywords: AAV, serum protein, albumin, transduction, gene therapy
Introduction
Adeno-associated virus (AAV) vector has been used in over 100 clinical trials with promising results, in particular, for the treatment of blindness and hemophilia B(1-3). AAV is nonpathogenic, has a broad tissue tropism, and can infect dividing or non-dividing cells. More importantly, AAV vector transduction has induced long-term therapeutic transgene expression in pre-clinical and clinical trials. As of today, there are 12 serotypes of AAV isolated for gene delivery. Among them, AAV8 has been shown to be the best one for mouse liver targeting. Due to extensive studies in pre-clinical animals with FIX deficiency, Phase I/II clinical trials have been carried out using AAV2 and AAV8 in patients with hemophilia B (1-3). The results from these trials are very promising; however, the FIX expression from patients receiving AAV/FIX was not proportional to what has been achieved in animal models even though the same vector dosage/kg was used. When 1×1011 particles of AAV8 encoding FIX were used in FIX knock out mice for systemic administration, 160% of normal level FIX was detected in blood(4). However, when 2×1011 particles of AAV8/FIX were administered, only 40% of FIX was achieved in primates(4) and less than 1% of FIX were found in human(2, 3). The inconsistent FIX expression following AAV vector transduction among these species may be due to altered hepatocyte tropism in different species. Another interesting finding from AAV FIX clinical trials is the capsid specific cytotoxic T lymphocyte (CTL) response that eradicates AAV transduced hepatocytes and, thus, results in therapeutic failure(1, 2). This phenomena has not been demonstrated in animal models following AAV delivery, which points out another variation between preclinical and clinical studies. When a much higher dose of AAV/FIX vector was used, FIX expression was detected in both clinical trials using either AAV2 or AAV8; however the blood FIX level decreased at week 4 or 9 post injection, respectively. Further studies suggested that AAV vector infection elicited a capsid specific CTL response, which appeared to eliminate AAV transduced hepatocytes(1, 2). Therefore, the results from these clinical trials highlight the necessity to explore effective approaches for enhancement of AAV transduction without increasing vector capsid burden. Any vector improvement that reduces AAV capsid antigen will also impact the daunting vector production concerns and be a welcome addition to viable gene therapy drug development.
Many strategies have been explored to increase AAV vector transduction. One strategy is to optimize the AAV vector cassette by utilization of a strong promoter and/or enhancer, codon-optimization of the transgenic cDNA, effective poly-adenylation sequence, and the use of a self-complementary vector genome if possible (5, 6) (7, 8). At the level of the AAV capsid, much attention has been focused on employing natural serotypes that display differential tropisms, rationally designed capsids, or capsids selected or screened from a mutant capsid library(9-11). However, a drawback of this approach is that the relevant experiments cannot be performed in humans and interspecies variation in AAV capsid tropism continues to be observed given the continued collection of human data. A third method to enhance AAV vector transduction relies on altered cellular physiology via pharmacological agents(12-22). Many pharmacological agents have been used to enhance AAV transduction at various levels of infection; however, most of these drugs are used as cancer therapies and have severe side effects. In our previous neutralizing antibody studies, it was found that human serum had an enhanced effect on AAV transduction(23). In this study, we have identified several proteins from human serum which directly interact with AAV virions and have the potential to impact AAV transduction. Among these proteins, the most interesting one is human serum albumin (HSA), a therapeutic agent that is the most abundant protein in the blood and has been widely used in clinical practice. If the interaction of HSA with AAV virions enhances AAV transduction, this approach can be immediately applied in AAV clinical trials. Herein, we demonstrated that the interaction of HSA with AAV vector enhances AAV transduction, and that this enhancement is not restricted to specific cells in vitro or tissues in vivo. Comparable enhancement was achieved regardless of incubation of HSA with AAV vectors before vector freezing or after thawing. Addition of HSA into vector preparations before dialysis did not impact HSA enhancement effect on AAV transduction. Mechanism studies suggest that HSA increased AAV binding to the target cell surface in vitro and resulted in the rapid clearance in blood after systemic administration. Neutralizing antibody (Nab) analysis demonstrated that the interaction of albumin with AAV still enhanced AAV transduction in the presence of Nab and didn’t impact Nab activity. We applied this approach for treating hemophilia in FIX deficient mice. After systemic administration of AAV/FIX incubated with human albumin, increased transgene FIX expression and improved phenotypic correction were achieved.
Results
Human serum enhances AAV transduction
Our previous results demonstrated enhanced AAV transduction solely by the presence of human serum(23). We extended this finding to examine AAV transduction enhancement using 10 human serum samples and found that the interaction of human serum with AAV induced an approximately 4-fold increase in transgene activity in an AAV capsid-independent manner in vitro(Figure 1a). Since AAV8 has been used in several clinical trials in patients with hemophilia, the AAV8 capsid was chosen for following experiments. Although enhanced transgene activity was observed at shorter durations, the greatest effect was achieved following serum incubation with AAV virions for ≥2h (Figure 1b). To determine whether the enhancement effect of human serum on AAV transduction in Huh7 cells held true in vivo, AAV8/luc vectors were incubated with serially diluted serum and then administrated via retro-orbital or muscular injections (contralateral muscle received vector with no serum). As shown in Figure 1c and Figure 1d, even a > 3000-fold serum dilution still enhanced AAV transduction in the liver and muscles by 2-5 fold or 4-16 fold, respectively. The enhancement of AAV transduction was also demonstrated following incubation of AAV vectors with serum from other species including mouse, dog, primate and fetal bovines (Supplementary Figures 1 and 2).
Fig. 1. The enhanced effect of serum on AAV transduction.
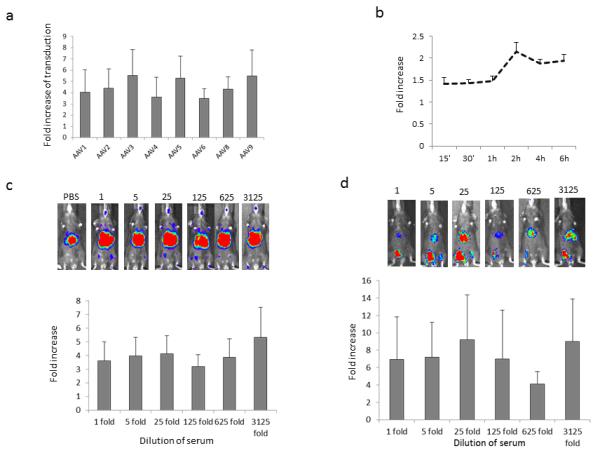
a. Human serum enhances AAV transduction from different serotypes. 1×108 particles of AAV/luc vector were incubated with 1:500 diluted sera or PBS for 2hr at 4°C. The mixture of AAV vector and sera was used to transduce 1×105 Huh7 cells in a 48-well plate in the presence of adenovirus dl309 at MOI of 5. After 24 hr, luciferase activity from the cell lysate was analyzed. The fold increase of transgene expression from sera incubation was calculated by comparison to PBS. b. The effect of incubation time of AAV with human serum on enhanced transduction. 1×108 particles of AAV8/luc were incubated with 1:100 diluted human sera or PBS for different time periods at 4°C in the presence of ad dl309. 24hr later, luciferase expression was measured from the cell lysate. c. Enhanced AAV transduction after systemic administration. 1×1010 particles of AAV8/luc were incubated with human serum at different dilutions for 2hr at 4°C. The mixture was administered into adult female C57BL mice via retro-orbital injection. The imaging was performed for 5min at day 3 after AAV injection. Upper panel: Representative live animal bioluminescent images of luciferase transgene expression profiles. Bottom panel: Quantification of luciferase transgene expression for enhanced AAV transduction from 6 mice after systemic administration. d. Enhanced AAV transduction after muscular injection. The mixture of AAV8/luc with human serum from Figure 1c was diluted to 1×109 particles/200ul in PBS and injected into mouse hind leg muscle. At week 2 post injection, the imaging was taken for 5min. Face up: left leg-AAV8 + human sera, right leg-AAV8 + PBS. Upper panel: Representative imaging. Bottom: Data of enhanced AAV transduction from 6 mice after muscular injection. The fold increase of transduction was calculated by transduction from HSA incubated AAV to that from the PBS treated one.
Human serum albumin exerts enhancement effect on AAV transduction
The data from the above experiments strongly suggest that some component(s) of serum enhances AAV transduction in vitro and in vivo. To examine whether the enhancement on transduction requires direct interaction of the AAV virion with a serum protein(s), we designed 5 cohorts: 1. Huh7 cells in complete medium and AAV incubated with PBS, 2. Huh7 cells in complete medium and AAV incubated with human serum, 3. Huh7 cells in serum free medium and AAV incubated with PBS, 4. Huh7 cells in serum free medium and AAV incubated with PBS, and then the same amount of serum was added to culture medium just before application of virus on cells, 5. Huh7 cells in serum free medium and AAV incubated with human serum. Similar to the methodology described above, enhanced transduction was achieved in cohort 2 when compared to cohort 1. Interestingly, no increase of AAV transduction was observed in cohort 4 with a high dilution of serum when compared to group 3, while increased transduction was obtained in cohort 5 (Figure 2a). These results suggest that the human serum mediated enhancement of AAV transduction requires the direct interaction of the human serum protein(s) with AAV virions. It is interesting to note that the fold increase was apparently much larger in the "serum free" group than in the "complete medium" group at 4 to 16 fold dilutions of human serum. This is because complete medium contains fetal bovine serum (FBS) which enhances AAV transduction. When AAV vectors incubated with PBS are added to cells maintained in complete medium, AAV vectors will interact with FBS proteins which induce higher transduction than that AAV vectors are applied to cells in the serum free medium (data not shown). To identify which serum proteins augment AAV transduction, human serum was incubated with AAV8 vectors and then an antibody that recognizes intact AAV8 virions was used to pull down AAV8 binding proteins for mass spectrometry analysis(24). Among the proteins identified, the most interesting one is human serum albumin (Supplementary Table 1). Serum albumin is the most abundant protein in the circulation and has been widely used in many clinical settings, and therefore, the primary objective of this study is to investigate the effect of HSA on AAV transduction. To further confirm the mass spectrometry data of HSA binding to AAV8, we incubated AAV8 particles with HSA and then used the human albumin antibody to pull down albumin bound AAV particles. The AAV genome copy number was then quantitated by Q-PCR. As shown in Figure 2b, the immunoprecipitation with the albumin specific antibody (A80-129A, Bethyl Lab, INC) resulted in twice as many genomes pulled down compared to the controls (isotype IgG or PBS). To examine whether the interaction of human albumin with AAV virion impacted AAV transduction, we incubated AAV8 particles with HSA depleted serum (> 99% depletion, Supplementary Figure 3) or recombinant HSA. It was demonstrated that the transduction from human albumin depleted serum was lower than the complete serum treated vector (Figure 2c). AAV vectors incubated with recombinant HSA (rHSA) also resulted in higher transduction, but to a lower extent compared to human whole serum (Figure 2d). To explore whether HSA enhances AAV transduction in vivo, AAV8/luc vectors were incubated with different concentrations of rHSA and then administered into mice via retro-orbital or muscular injection. Following systemic administration, vectors pre-treated with HSA demonstrated increased liver transduction (1.5- to 8-fold (Supplementary Figure 4a)). Consistent with the stimulation of AAV transduction by human serum in muscle, higher transduction in muscle was observed (2.1- to 11.5-fold) following incubation of AAV8 vectors with rHSA (Supplementary Figure 4b). These results implicate that human serum albumin increases AAV transduction in vitro and in vivo.
Fig. 2. The effect of human albumin on AAV8 transduction.
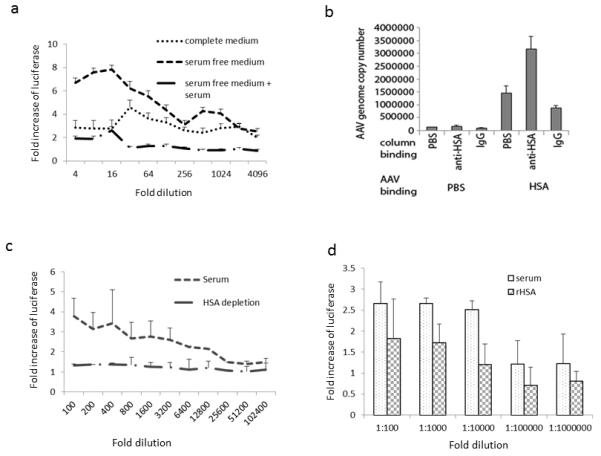
a. Transduction enhancement is related to direct interaction of AAV with serum. AAV8/luc viruses were incubated with human serum or PBS at 1:100 dilution for 2 hr at 4°C, then the mixture was used to transduce Huh7 cells either in medium with FBS, serum free medium, or serum free medium plus human serum just before addition of AAV8 pre-incubated with PBS. 24 hr later, fold increase of transgene expression was calculated. b. AAV8 interaction with human albumin. 1×1010 particles of AAV8/luc were incubated with human sera or PBS for 2hr at 4 °C, then the mixture of virus and human serum or PBS was applied to pre-Ig bound column. After washing, the column binding proteins were eluted for AAV8 genome copy number analysis. c. AAV8 transduction with albumin depleted serum. 1×108 particles of AAV8/luc were incubated with human serum or albumin depleted serum at different dilutions or PBS for 2hr at 4°C. Then the mixture was used to infect Huh7 cells in serum free medium. Two days later, luciferase was detected from the cell lysate, and the fold increase of transgene expression was calculated while compared to PBS. d. Recombinant human albumin enhances AAV8 transduction. AAV8/luc was incubated with recombinant human albumin (50mg/ml) or human serum at different dilutions or PBS. Transgene expression was detected 48hr later, and the fold increase of transgene expression was calculated when compared to PBS.
Enhancement effect of clinical grade HSA on AAV transduction
Since HSA has been widely applied in clinic, we then tested whether clinical grade HSA also has the ability to enhance AAV transduction. When 5% clinical grade HSA, which is identical to the serum albumin concentration in the blood of normal subjects, was incubated with AAV vectors at different dilutions, increased AAV transduction was observed in vitro even at a dilution of 20,000-fold (Figure 3a). Next, we incubated AAV8/luc with 25% HSA at different fold dilutions prior to retro-orbital or muscular injection. A one-fold dilution is defined as 1×1012 AAV particles incubated with 10 ul of 25% HSA in 1ml solution. As shown in Figure 3b and 3c, clinical grade HSA significantly increased AAV8 transduction by about 3- or 5-fold in the liver and muscle, respectively. Next, the long-term effect of HSA on AAV transduction was documented at weeks 1, 2, 4, and 7 after muscular injection (Supplementary Figure 5). These results indicate that clinical grade HSA enhances AAV transduction in the muscle for sustained transgene expression. Also, we observed that incubation of HSA with AAV2 or AAV9 induced much higher transduction in vitro and in vivo (Supplementary Figure 6).
Fig. 3. The effect of clinical grade human albumin on AAV8 transduction.
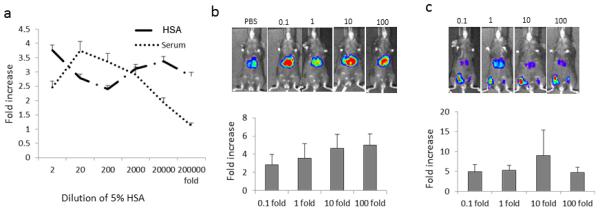
a. Enhanced AAV8 transduction in Huh7 cells from clinical grade HSA. 1×108 particles of AAV8/luc were incubated with 5% HSA or human serum at different dilutions or PBS for 2hr at 4°C. Then the mixture was used to transduce Huh7 cells; 48hr later, luciferase expression was assayed. b. Enhanced AAV8 transduction from clinical grade HSA after systemic administration. 1×1010 particles of AAV8/luc were incubated with 25% HSA at different dilutions and then injected into adult female C57BL mice via retro-orbital. Imaging was taken at day 7. Upper panel: Representative animal image. Bottom panel: Data of enhanced AAV transduction from 6 mice after systemic administration. c. Enhanced AAV transduction from clinical grade HSA after muscular injection. 1×109 particles of AAV8/luc were incubated with 25% HSA at different dilutions and then injected into muscles in C57BL mice. One week later, the imaging was performed. Upper panel: Representative animal image. Bottom: Data of enhanced AAV transduction from 6 or 7 mice after muscular injection.
The enhancement of AAV transduction by HSA is not altered by freeze/thaw
In the above experiments, AAV preparations were thawed and incubated with HSA before being administered to cells or mice. In the clinical setting, it may not be practical for the medical staff to perform this incubation immediately prior to injection. Therefore, incubation of HSA with AAV vectors prior to storage at −80 °C would simplify the translation of HSA-enhanced AAV vector transduction. To investigate this, we first incubated AAV vectors with clinical grade HSA for 2 hours at 4 °C. Half of the solution was stored in −80 °C for three days, while the other aliquot of AAV virus was immediately used to infect Huh7 cells at a dose of 1×103 particles/cell. After thawing the frozen HSA-AAV preparation, vector transduction was analyzed in Huh7 in the same manner. As shown in Figure 4a, a similar increase in luciferase activity was observed regardless of HSA-vector cryopreservation. HSA’s enhancement of AAV transduction following incubation and cryopreservation was also observed after muscular injection (Figure 4b and 4c).
Fig. 4. Incubation of AAV vector with HSA pre-freezing or post-thawing of viruses has the similar enhanced effect.
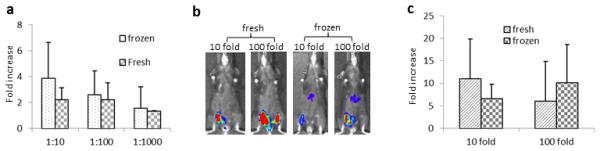
a. Enhanced transduction in Huh7 cells. 1×108 particles of AAV/luc were incubated with clinical grade HSA at different dilutions for 2hr at 4°C before virus freezing or after virus thawing, and then added to Huh7 cells. 48 hr later, luciferase activity in the cell lysate was measured. b and c. Enhanced muscle transduction. 1×109 particles of AAV8/luc were directly injected into muscles of mice. At day 7 post injection, the mouse imaging (b) was carried out (left panel) and the fold increase (c) of transgene expression was calculated (right panel, n=6). Face up: left leg-HSA, right leg-PBS.
AAV transduction following HSA addition to AAV preparations before dialysis
During vector production, it is necessary to perform vector dialysis to remove high concentrations of salt regardless of methods used for purification (CsCl or column chromatography). To determine if the incubation of AAV vectors in HSA during dialysis impacts AAV transduction, AAV8/luc vectors purified by CsCl gradient ultra-centrifugation or by anion exchange column were mixed with 10ul of 25% HSA or PBS in 1 ml of 1012 particles just before dialysis. Then, these formulations were dialyzed against PBS and AAV transduction was analyzed in mice via retro-orbital or direct muscular injection. As shown in Figure 5, HSA incubation during dialysis still increased vector transduction in the liver and muscle by greater than 2-fold or 4-fold, respectively, as compared to the PBS incubation control. The enhancement effect was similar for different approaches of purification. This observation implicates that human albumin could be added to AAV preparations before dialysis of vectors purified in different manners in order to enhance gene delivery.
Fig. 5. Addition of HSA to virus preparation before dialysis does not compromise transduction enhancement.
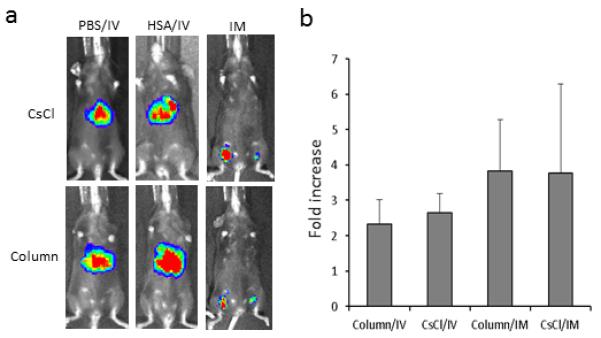
AAV8/luc viruses purified either from CsCl or column were mixed with 1% of 25% HSA, and then applied for dialysis against PBS. After dialysis, AAV viruses were frozen; two days later, the in vivo transduction assay was performed. For liver transduction, 1×1010 particles of AAV/luc were administered via retro-orbital injection; the imaging was taken at day 3 after AAV injection for 5 mice (a). For muscle transduction, 1×109 particles of AAV/luc were used; imaging was performed at day 7 post injection for 4 mice (a). Face up: left leg-HSA, right leg-PBS. Quantitation of imaging (b) was also performed.
Albumin increases AAV binding capacity to target cells
The first step for effective AAV transduction is AAV virion binding on the target cells via primary and secondary receptors. To examine whether incubation of albumin with AAV vectors increases cellular binding, AAV8/luc vectors were incubated with HSA or PBS. Then, Huh7 cells were added at 4 °C to prevent vector internalization, as shown in our previous study(25). After extensive washes, total DNA was recovered and AAV genome copy number was determined by Q-PCR. As shown in Figure 6a, incubation with HSA significantly increased AAV vector binding to Huh7 cells by 3-fold. To determine if HSA increases vector binding and uptake by the liver, 1×1011 particles of AAV8/luc, pre-incubated in HSA or PBS, was administered via retro-orbital injection and luciferase activity was determined 24 hours later. Consistent to the result observed in Huh7 cells, higher transduction was achieved in the liver (Figure 6b and 6c). Forty eight hours post-injection, mice were sacrificed and the liver was harvested for quantitation of luciferase activity and AAV genome copy number. Similar to live imaging analysis, higher luciferase activity and AAV genome copy number were found in the livers of mice administered with AAV vectors pre-treated with HSA compared to those given AAV vectors incubated in PBS alone (Figure 6d and 6e). The result of more AAV vector uptake by the liver with HSA pre-incubation was correlated to vector clearance from the blood. After administration of AAV vector, there was a marginal decrease in AAV genome copy number per microliter of plasma in mice receiving HSA treated AAV vector, as compared to control mice at 15min and 24hr post injection (p>0.05). However, a significant reduction of AAV genome copy number was observed in the HSA cohort at 2hr after AAV administration (p<0.05). These results suggest that enhanced AAV vector transduction by HSA results from increased particle binding to target cells.
Fig. 6. Human albumin increases AAV binding ability.
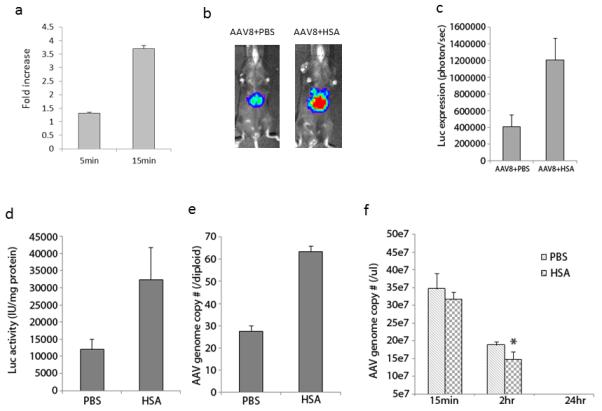
a. HSA increases AAV virus binding to Huh7 cells. AAV viruses were incubated with HSA for 2hr at 4°C, and then added to 1×106 Huh7 cells for 5 or 15 min at 4°C. After washing 5 times, total DNA was extracted for AAV genome copy number analysis by q-PCR. b. Imaging of liver transduction. 1×1011 particles of AAV8/luc were administered into mice via retro-orbital vein. Twenty four hr later, the imaging was carried out and the quantitation of imaging was calculated (c). Forty-eight hr later, mice were euthanized and liver tissue was harvested; the luciferase activity in liver tissue lysate was measured (d) and the AAV genome copy number was analyzed (e). During the first 24hr, plasma from blood was collected at 15min, 2hr, and 24hr post AAV injection, and AAV genome copy number was analyzed (f). The data represented the average of 4 mice and standard deviations. (*) indicates statistically significant difference with p<0.05 when the AAV genome copy number in the liver with HSA treatment was compared to that with PBS.
Interaction of albumin with AAV does not interfere with neutralizing antibody activity
To investigate whether the interaction of human albumin with AAV virions blocks AAV neutralizing antibody (Nab) activity, we performed the Nab assay in vitro. IVIG is the pooled sera from over 1000 subjects and contains AAV Nab against different serotypes(23). We first incubated AAV8/Luc virions with 100-fold dilution of HSA or PBS, and then added IVIG at different concentrations. After transduction in Huh7 cells, the Nab titer was calculated. As shown in Figure 7a, the same Nab titer (1:200 of IVIG) was obtained regardless of AAV vector pre-incubated with HSA. We also studied whether HSA is still able to enhance AAV transduction in the presence of AAV Nab, and found that the incubation of HSA with AAV increased AAV transduction with similar efficiency in the presence of different amount of IVIG (Figure 7b). These results suggest that interaction of HSA with AAV does not impact AAV virus infection mechanism.
Fig. 7. Interaction of human albumin with AAV doesn’t block Nab activity.
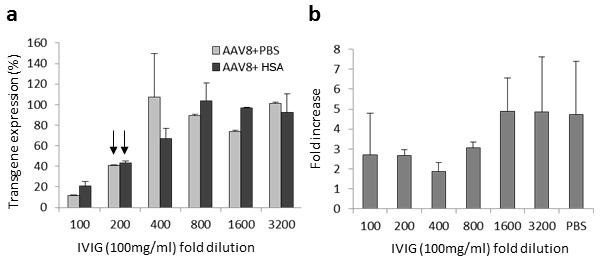
AAV8/luc vector was first incubated with human albumin for 2hr at 4°C, then human IVIG at different dilution was added for another 2hr at 4°C. The mixture was added to Huh7 cells. At 48hr, the transgene expression from cell lysate was measured and Nab titer was calculated. a. The effect of interaction of human albumin with AAV virions on Nab activity. b. The effect of IVIG on human albumin enhancement of AAV transduction.
Improved phenotypic correction of hemophilia B using human albumin to enhance AAV vector transduction
To study the phenotypic correction using AAV vectors incubated with HSA, we used hemophilia B mice as a disease model and AAV8/FIX-OPT, which has been used in Phase I clinical trials in patients with hemophilia B(12). After injection, FIX concentration and function were determined at different time points and phenotypic correction was assessed at week 6. As shown in Figure 8a, over 5-fold higher FIX levels were detected in mice receiving has incubated AAV8/FIX-OPT than those with the same vectors treated with PBS. Similarly, plasma FIX activity was much higher in mice receiving vector treated with HSA (Figure 8b). At 6 weeks post AAV injection, all mice underwent a tail vein transection bleeding challenge to assess in vivo function of the vector-expressed human factor IX. Untreated hemophilia B mice had profound bleeding (30mg of blood/g of mice body weight) following the challenge compared to WT controls. Hemophilia B mice receiving AAV vectors incubated in HSA demonstrated a significant decrease in blood loss compared to AAV vectors incubated in PBS (p<0.05, Figure 8c). In fact, hemophilic mice treated with vectors incubated in HSA demonstrated blood loss similar to that of WT controls. These results demonstrate improved correction of hemophilia B using AAV vectors pre-incubated with HSA, and also suggest possible utilization of this formulation to increase efficacy at lower vector doses for the treatment of hemophilia and other diseases.
Fig. 8. Improvement of phenotypic correction of hemophilia B using human albumin incubated AAV vector.
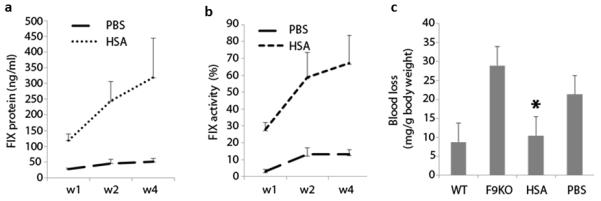
2×109 particles of AAV8/FIX-opt vector were incubated with human HSA or PBS for 2 hr at 4 °C, then AAV vector was administered into adult male FIX deficient mice via tail vein injection. Post AAV injection, blood was collected at indicated time points for FIX expression (a) and function assay (b). At week 6 post AAV injection, mice were applied for in vivo bleeding assay (c). (*) indicates statistically significant difference for blood loss between HSA treated mice and PBS mice with p<0.05. The data are based on the average and standard deviations from 6 to 8 mice.
Discussion
Observations of lower FIX expression and capsid-specific CTL responses to the AAV capsid at high doses in human AAV FIX trials have emphasized the need for more efficient strategies that maintain efficient gene delivery at lower doses. Our earlier report noted that AAV transduction was enhanced by human serum; however, the precise component(s) was not identified. Therefore, for the search of more efficient AAV vectors, the objective of this study was to identify specific protein(s) from human serum that interact with AAV virions to induce higher transduction. Of AAV8 capsid interacting proteins identified from mass spectroscopy analysis (Supplementary Table 1), further experimentation was pursued with HSA. These investigations demonstrated that incubation of AAV8 with recombinant or clinical grade HSA increased AAV transduction while human albumin depleted serum decreased transduction. Clinical grade HSA significantly enhanced AAV transduction in the liver and skeletal muscles of mice. To facilitate the application of HSA in AAV vector production and clinical trials, our studies demonstrated that freezing AAV vectors after incubation with human albumin or addition of HSA into AAV preparations before dialysis still resulted in enhanced transduction. Mechanism studies suggested that human albumin increased AAV vector binding to the target cell surface and resulted in faster blood clearance after systemic administration but did not impact AAV infection pathway. Finally, in a preclinical mouse model of hemophilia B, AAV vectors incubated with albumin increased human FIX expression and improved the bleeding phenotype to WT levels.
Serum proteins are able to interact with viruses and impact virus infection(26-34). For example, adenovirus has been widely studied for its interaction with serum proteins including coagulation factors and complements for liver targeting(32-34). Our previous Nab study demonstrated that serum at the dilution without Nab activity actually enhanced AAV transduction regardless of serotype(23). Other studies have found that several serum proteins have an effect on AAV transduction via interaction with AAV virions(35-37). Denard, et al have identified galectin 3 binding protein (G3BP) and C-reactive protein (CRP) which interact with AAV(35, 36). They showed that the interaction of G3BP with AAV virions led to the formation of AAV aggregates which block AAV transduction, and that interaction of CRP with AAV resulted in higher transduction. The CRP enhancement of AAV transduction is species specific and AAV serotype specific. In another study, Zaiss, et al demonstrated that the AAV2 capsid binds to C3 complement proteins to enhance macrophage uptake of AAV and induce macrophage activation(37). In this study, incubation of AAV and clinical grade HSA enhances transduction in vitro as well as in vivo. Generally, the enhancement of human albumin on AAV transduction is lower than whole serum. This finding implicates that other proteins in the serum may also play a role to enhance AAV transduction. It will be worthwhile to study how the interaction of these proteins impacts AAV transduction.
It is noted that the enhancement of AAV transduction with human albumin treated virus in muscle is generally higher than that in liver. An explanation of this phenomenon could be that blood contains a very high concentration of albumin, which enhances transduction to some extent following systemic injections. In contrast, less albumin resides in the muscle tissue, so after muscular injection, the magnitude of increased transduction by albumin is greater than that observed following IV injections. After systemic administration of AAV vector, the AAV virons will immediately interact with albumin. However, our studies demonstrated that the longer incubation of HSA with AAV virions induced higher enhancement of transduction. This result indicates that further enhancement of transduction should be achieved by pre-incubation of HSA with AAV virions following systemic administration. In this study, the transduction enhancement from AAV virions incubated with human serum albumin was achieved in the liver and muscles after systemic administration and direct injection, respectively. Since the liver takes up more AAV virions from circulation after systemic administration, it is possible that less AAV vector will be escaped from blood to transduce other tissues like heart and skeletal muscle. On another hand, AAV vector treated with albumin increases muscular transduction. It is unknown whether the enhanced transduction in heart or skeletal muscle can be achieved after systemic administration of AAV vectors incubated with albumin. To increase AAV transduction in heart and skeletal muscle after systemic administration, several liver detargeted AAV mutants (AAV2i8 and AAV9.45) have been developed (38, 39), it is under way to test whether transduction enhancement in muscle will be achieved after systemic administration of these liver detargeted AAV vectors incubated with albumin.
Albumin is emerging as a versatile protein carrier for drug targeting and for improvement of the pharmacokinetic profile of peptide- or protein-based drugs(40-45). Several albumin receptors have been described that induce endocytosis (46). The pathway for albumin endocytosis is cell type dependent and includes either clathrin- or caveolin-mediated endocytosis(47-51). Although it is unknown how AAV vector interacts with albumin, this study demonstrated that interaction of AAV with albumin increases AAV binding ability of target cells. This can be explained by the fact that albumin, after interaction with AAV, provides another layer for AAV binding on the cell surface via albumin receptors.
Albumin has a prolonged half-life in the blood. It has now become apparent that homeostatic regulation of albumin is controlled by the neonatal Fc receptor (FcRn) (52, 53). FcRn rescues albumin from degradation in cells by binding albumin within intracellular endosomal compartments, which then results in transport of the ternary complex to the cell membrane for release of ligands back into the circulation(52). Due to these properties of albumin, there are some questions about the effect of interaction of albumin with AAV vector on transduction. Since albumin uptake is via either clathrin- or caveolin-mediated endocytosis, and AAV cellular entry is by clathrin-mediated endocytosis, it is unknown whether albumin and AAV compete in the endocytosis pathway. Another question is whether albumin disassociates from AAV in the endosome or trafficking into the nucleus. The third question is whether albumin exocytosis or transcytosis impacts AAV transduction. The fourth one is the effect of enhanced transduction from interaction with albumin and AAV on AAV capsid specific CTL response. Although the interaction of albumin with AAV virions increases AAV binding to target cell surface, it is possible that the interaction may influence AAV trafficking intracellularly. Further elucidation of these issues will help design more effective approaches to the use of albumin in AAV gene therapy.
Taken together, our study demonstrated that AAV capsids interact with human serum albumin, which increases transduction in vitro and in vivo. The transduction efficiency enhancement from clinical grade human albumin also allowed phenotypic correction in a hemophilia B mouse model after systemic administration of an otherwise suboptimal dose AAV vector. For clinical purposes, addition of human albumin into AAV virus preparations before dialysis or freezing AAV virus after incubation with albumin still results in transduction enhancement. Although the exact mechanism of AAV virion interaction with human albumin is unknown, our findings are important for immediate inclusion of HSA into diverse clinical applications suffering from subpar transduction at tolerated vector doses. Therefore, our results from these studies strongly indicate that incubation of clinical grade human albumin with AAV vectors during dialysis should be performed to enhance AAV transduction efficiency in future clinical trials.
Materials and Methods
Cell lines
HEK293 and Huh7 cells (from ATCC) were maintained at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin-streptomycin.
AAV virus production
AAV vector was produced using a standard approach with three-plasmid transfection in HEK293 cells(54). Briefly, AAV transgene plasmid pTR/CBA-luc or pTTR/FIX-opt was co-transfected with AAV helper plasmid and adenovirus helper plasmid pXX6-80 into HEK293 cells. Sixty hr later, cells were harvested and lysed, and cell lysate was applied for ultra-centrifugation against CsCl gradient or purification with column. AAV virions were collected and tittered by dot-blot.
Individual human serum was purchased from Valley Biomedical (Minchester, VA), and aliquoted and stored at −80 °C for future use.
AAV transduction assay in vitro
1×105 Huh7 cells were seeded on a 48-well plate in 300 uL DMEM containing 10% FBS or serum-free medium. AAV/luc was incubated with serum or rHSA (Sigma-Aldrich, St. Louis, MO) or clinical grade HSA (Albuminar, CSL Behring LLC, Kankakee, IL). The mixture was then added to the indicated cells. Forty-eight hr later, cells were lysed with passive lysis buffer (Promega) and luciferase activity was measured with a Wallac1420 Victor 2 automated plate reader. The fold increase of transgene expression was calculated as the transgene expression from serum or albumin treated groups compared to that from PBS.
Animal experiments
All mice were maintained in specific pathogen-free facilities according guidelines instituted by the animal committees of the University of North Carolina at Chapel Hill. All animal experiments were reviewed and approved by the University of North Carolina Institutional Animal Care and Usage Committee. The animal experiments were performed in hemophilia B (FIX−/−) mice or normal C57BL/6 mice (purchased from Jackson Laboratories, Bar Harbor, ME). For systemic administration, 1×1010 particles of AAV/luc vector were incubated with serum or human albumin for 2 hr at 4 fo owed by retro-orbital administration into adult female C57BL mice. At the indicated time points, imaging was performed using a Xenogen IVIS Lumina imaging system (Caliper Lifesciences, Hopkinton, MA) following intraperitoneal injection of D-luciferin substrate at 120 mg/kg (Nanolight, Pinetop, AZ). Bioluminescent images were analyzed using Living Image software. For muscular injection, 1×109 partic es of AAV uc vector were incubated with serum or human a bumin for 2 hr at 4 . Then the mixture was directly injected into the hind leg muscles of 6-8 week old C57BL mice. At the indicated time points, imaging was performed and bioluminescent images were analyzed. For hemophilia B studies, adult male hemophilia B mice were injected with 2×109 particles of AAV8/FIX vectors via the tail vein. At indicated time points, blood was collected from the retro-orbital venous plexus under anesthesia using isoflurane. At week 6 after AAV8/FIX injection, in vivo bleeding analysis was performed.
Human albumin depletion
A PierceTM albumin depletion kit (Cat # 85160, Pierce Biotechnology, Rockford, IL, USA) was used following the company instruction with slight modification. Briefly, after transferring the resin into the column and centrifugation at 12,000rpm for 1 min, the column was washed and loaded with 50 uL of pre-tested AAV Nab negative serum. After centrifugation, the flow through was applied to new resin treated column and above steps were repeated. To achieve maximum depletion of albumin, the flow through was applied for two more times and a total of 4 columns were used for one sample. 50 uL of binding/washing buffer was added to the column to release unbound proteins and centrifuged. The final flow through was applied for detection of albumin using ELISA kit.
Co-immunoprecipitation
Coimmunoprecipitation of serum proteins was performed with Pierce Co-Immunoprecipitation (Co-IP) Kit (Cat# 26149, Pierce Biotechnology, Rockford, IL, USA). First, antibody immobilization was performed. After addition of the resin slurry into a Spin Column and centrifugation, the column was washed and inserted with the bottom plug. Then, diluted antibodies and the Sodium Cyanoborohydride Solution were directly added to the resin in the spin column sequentially, and incubated for 2hrs at RT. After centrifugation and washing, Quenching buffer was added to the column and centrifuged. Quenching buffer was applied to the resin, followed by addition of Sodium Cyanoborohydride Solution for 15 minutes. After centrifugation and washing, the mixture of AAV virus with human serum or PBS was transferred to the resin and incubated for 2hrs at 4 °C. After centrifugation and washing, elution buffer was added and incubated for 5 minutes and centrifuged; the flow through solution was collected for mass spectrometry analysis or AAV genome number quantitation by Q-PCR.
Mass spectrometry
The proteins were reduced, alkylated, and digested with trypsin using the FASP protocol. The peptides were resuspended in 2% acetonitrile/98% (0.1% formic acid) prior to analysis by LC-MS/MS. Briefly, the peptides were loaded onto a 2 cm long X 360 μm o.d. × 100 μm i.d. microcapillary fused silica pre-column packed with Magic 5 μm C18AQ resin (Michrom Biosciences, Inc.). After sample loading, the pre-column was washed with 95% Solvent A (0.1% formic acid in water)/5% Solvent B (0.1% formic acid in Acetonitrile) for 20 min at a flow rate of 2 uL/min. The pre-column was then connected to a 360 μm o.d. × 75 μm i.d. analytical column packed with 22 cm of 5 μm C18 resin. The peptides were eluted at a flow rate of 250 nL/min by increasing the percentage of solvent B to 40% with a Nano-Acquity HPLC solvent delivery system (Waters Corp.). The LC system was directly connected through an electrospray ionization source interfaced to an LTQ Orbitrap Velos ion trap mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was controlled by Xcalibur software and operated in the data-dependent mode, in which the initial MS scan recorded the mass to charge (m/z) ratios of ions over the range 400–2000. The 10 most abundant ions were automatically selected for subsequent collision-activated dissociation. All files were searched using MASCOT (Matrix Science, Ver. 2.3.02) via Proteome Discoverer (Thermo., Ver. 1.3.0.339) against the database containing human proteins downloaded from Uniprot. The search parameters included peptide mass tolerance of 10 ppm and a fragment ion tolerance of 0.6 mass units. The search allowed for variable modifications of oxidation of Met and carbamidomethyl of Cys. Each sample was run 2 times (R1 and R2). Two fold difference between AAV sample and PBS was considered positive.
Quantitation of luciferase expression in the tissues
Animals utilized for imaging studies were sacrificed two weeks after AAV injection and the following organs were collected: liver, spleen, kidney, heart, lung, skeletal muscle (gastrocnemius), and brain. Tissue was minced and homogenized in passive lysis buffer (Promega, Madison, WI). Tissue lysates were centrifuged at 10,000 rpm for 5 minutes to remove cellular debris. Supernatant was transferred to 96-well plates for luciferase activity analysis as described above. Total protein concentration in tissue lysates were measured using the Bradford assay (BioRad, Hercules, CA).
AAV genome copy number analysis
For determining blood clearance rates of various rAAV vectors, plasma was obtained from mice at 2, 6, 24, and 48 hour after intravenous administration of rAAV vectors. Viral DNA isolation from plasma was performed using the DNeasy Blood & Tissue kit (QIAGEN, CA) fo owing the manufacture’s instruction. Vira genomes were quantified by real-time PCR with forward primer: 5′-AAAAGCACTCTGATTGACAAATAC-3′and reverse primer: 5′-CCTTCGCTTCAAAAAATGGAAC-3′. Real-time PCR was performed on a LightCycler 480 (Roche Diagnostics Cooperation, Indianapolis, IN) instrument. A 10 uL final volume of absolute quantitation reaction was performed using SYBR green (Roche Diagnostics Cooperation, Indianapolis, IN) mix supplemented with 0.2uM primers. No template control was included in each run to rule out the possibility of contamination for each primer-probe set. The reaction was amplified at 95°C for 10 min followed by 45 cycles of 10 s at 95°C, 10 s at 60°C, and 10 s at 72°C, followed by a melting cycle. Each gene was accessed in duplicates. Absolute quantification was performed based on second-derivative maximum comparisons to standard curves of plasmid DNA (luciferase).
To detect AAV genome copy number in different tissues, the animals were sacrificed and the selected organs were harvested at week 2 after iv injection of rAAV. After DNA isolation using the DNeasy Blood & Tissue kit (QIAGEN, CA), real-time PCR was performed on each sample for both the luciferase gene and the mouse Mus musculus Lamin B2 gene. The primers used for mouse Mus musculus Lamin B2 gene were: 5′- GGACCCAAGGACTACCTCAAGGG-3′(forward) and 5′- AGGGCACCTCCATCTCGGAAAC -3′(reverse). The copy of genome was analyzed by Lightcycler software v.4.5 (Roche Diagnostics Cooperation, Indianapolis, IN) based on those of pTR-CBA-Luciferase plasmid used in initial transduction and the endogenous gene.
AAV binding assay
5×1010 particles of AAV/luc vector were incubated with clinical grade human serum albumin at 4 °C for different durations. Then, pre-chilled 5×105 Huh cells were added to AAV vector for 30 min at 4°C. Cells were washed four times with cold PBS and transferred to a new tube. DNA from cells was extracted and applied for Q-PCR to determine AAV genome copy number per cell using luc specific primers.
Neutralizing antibody analysis
Nab assay was carried out as described in our previous study with slight modifications(23). Briefly, 1×108 particles of AAV8/Luc vector were incubated with 100 fold dilution of 25% HSA at 4° C for 2 hours, then IVIG at different dilution was added for another 2 hours. The mixture was applied to infect Huh7 cells. 48 hours later, luciferase activity was analyzed from cell lysate and neutralizing antibody titer was calculated.
Human factor IX antigen and activity assays
The human factor IX antigen one-stage human factor IX activity assay was performed as previously described(6). The specific activity of factor IX, expressed as units of factor IX activity per milligram of protein (U/mg), was calculated by dividing the factor IX activity (U/ml) by the concentration of the factor IX protein (factor IX antigen) (mg/ml).
In vivo bleeding model
In vivo bleeding was analyzed as previously described by Meeks, et al with slightly modification(55). After anesthesia, 3mm of the distal tail was transected and the proximal tail was placed into the pre-warmed and pre-weighed tube. Forty minutes after the tail clip or before death due to bleeding, blood loss per gram body weight was calculated.
Statistical analysis
Quantitative data were presented as means ± SD. The Student t test was used to perform all statistical analyses. P values less than 0.05 were considered a statistically significant difference.
Supplementary Material
Acknowledgments
We are grateful to Xiaojing Chen and Karen Hogan for their excellent technical assistance. The authors acknowledge the UNC Biomedical Research Imaging Center (BRIC) Small Animal Imaging (SAI) facility for assistance of mouse imaging. This work was supported by National Institutes of Health Grants R01DK084033 and R01AI117408 (to C.L. and R.J.S.), R01HL125749 (To C.L.), P01HL112761, R01AI072176 (to R.J.S.), P30-CA016086-35-37, U54-CA151652-01-04 (to the BRIC SAI facility) and a research grant from Asklepios BioPharmaceutical (to C.L.). This research is based in part upon work conducted using the UNC Michael Hooker Proteomics Center, which is supported in part by the NIH-NCI Grant No. CA016086 to the Lineberger Comprehensive Cancer Center.
Footnotes
Disclosure of Conflict of Interest
R. Jude Samulski is the founder and a shareholder at Asklepios BioPharmaceutical. He receives research support through the University of North Carolina from Asklepios BioPharmaceutical. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking.
References
- 1.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nature medicine. 2006;12(3):342–7. doi: 10.1038/nm1358. Epub 2006/02/14. [DOI] [PubMed] [Google Scholar]
- 2.Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. The New England journal of medicine. 2011;365(25):2357–65. doi: 10.1056/NEJMoa1108046. Epub 2011/12/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. The New England journal of medicine. 2014;371(21):1994–2004. doi: 10.1056/NEJMoa1407309. Epub 2014/11/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107(7):2653–61. doi: 10.1182/blood-2005-10-4035. Epub 2005/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16(2):280–9. doi: 10.1038/sj.mt.6300355. Epub 2007/12/07. [DOI] [PubMed] [Google Scholar]
- 6.Monahan PE, Sun J, Gui T, Hu G, Hannah WB, Wichlan DG, et al. Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Human gene therapy. 2015;26(2):69–81. doi: 10.1089/hum.2014.106. Epub 2014/11/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene therapy. 2003;10(26):2112–8. doi: 10.1038/sj.gt.3302134. Epub 2003/11/20. [DOI] [PubMed] [Google Scholar]
- 8.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene therapy. 2001;8(16):1248–54. doi: 10.1038/sj.gt.3301514. Epub 2001/08/18. [DOI] [PubMed] [Google Scholar]
- 9.Shen S, Horowitz ED, Troupes AN, Brown SM, Pulicherla N, Samulski RJ, et al. Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. The Journal of biological chemistry. 2013;288(40):28814–23. doi: 10.1074/jbc.M113.482380. Epub 2013/08/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, et al. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. Journal of virology. 2012;86(15):7752–9. doi: 10.1128/JVI.00675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(22):7827–32. doi: 10.1073/pnas.0802866105. Epub 2008/05/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell AM, Li C, Samulski RJ. Arsenic trioxide stabilizes accumulations of adeno-associated virus virions at the perinuclear region, increasing transduction in vitro and in vivo. Journal of virology. 2013;87(8):4571–83. doi: 10.1128/JVI.03443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell AM, Samulski RJ. Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. Journal of virology. 2013;87(23):13035–41. doi: 10.1128/JVI.01826-13. Epub 2013/09/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douar AM, Poulard K, Stockholm D, Danos O. Intracellular trafficking of adeno-associated virus vectors: routing to the late endosomal compartment and proteasome degradation. Journal of virology. 2001;75(4):1824–33. doi: 10.1128/JVI.75.4.1824-1833.2001. Epub 2001/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. The Journal of clinical investigation. 2000;105(11):1573–87. doi: 10.1172/JCI8317. Epub 2000/06/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. Journal of virology. 1996;70(5):3227–34. doi: 10.1128/jvi.70.5.3227-3234.1996. Epub 1996/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson JS, Samulski RJ. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. Journal of virology. 2009;83(6):2632–44. doi: 10.1128/JVI.02309-08. Epub 2008/12/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monahan PE, Lothrop CD, Sun J, Hirsch ML, Kafri T, Kantor B, et al. Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18(11):1907–16. doi: 10.1038/mt.2010.170. Epub 2010/08/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ju XD, Lou SQ, Wang WG, Peng JQ, Tian H. Effect of hydroxyurea and etoposide on transduction of human bone marrow mesenchymal stem and progenitor cell by adeno-associated virus vectors. Acta pharmacologica Sinica. 2004;25(2):196–202. Epub 2004/02/11. [PubMed] [Google Scholar]
- 20.Prasad KM, Xu Y, Yang Z, Toufektsian MC, Berr SS, French BA. Topoisomerase inhibition accelerates gene expression after adeno-associated virus-mediated gene transfer to the mammalian heart. Molecular therapy : the journal of the American Society of Gene Therapy. 2007;15(4):764–71. doi: 10.1038/sj.mt.6300071. Epub 2007/02/15. [DOI] [PubMed] [Google Scholar]
- 21.Russell DW, Alexander IE, Miller AD. DNA synthesis and topoisomerase inhibitors increase transduction by adeno-associated virus vectors. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(12):5719–23. doi: 10.1073/pnas.92.12.5719. Epub 1995/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan Z, Zak R, Luxton GW, Ritchie TC, Bantel-Schaal U, Engelhardt JF. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. Journal of virology. 2002;76(5):2043–53. doi: 10.1128/jvi.76.5.2043-2053.2002. Epub 2002/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Crosby A, Hastie E, Samulski JJ, McPhee S, Joshua G, et al. Prediction of adeno-associated virus neutralizing antibody activity for clinical application. Gene therapy. 2015;22(12):984–92. doi: 10.1038/gt.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonntag F, Kother K, Schmidt K, Weghofer M, Raupp C, Nieto K, et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. Journal of virology. 2011;85(23):12686–97. doi: 10.1128/JVI.05359-11. Epub 2011/09/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao PJ, Li C, Neumann A, Samulski RJ. Quantitative 3D tracing of gene-delivery viral vectors in human cells and animal tissues. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20(2):317–28. doi: 10.1038/mt.2011.250. Epub 2011/11/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machida A, Kishimoto S, Ohnuma H, Miyamoto H, Baba K, Oda K, et al. A hepatitis B surface antigen polypeptide (P31) with the receptor for polymerized human as well as chimpanzee albumins. Gastroenterology. 1983;85(2):268–74. Epub 1983/08/01. [PubMed] [Google Scholar]
- 27.Pontisso P, Petit MA, Bankowski MJ, Peeples ME. Human liver plasma membranes contain receptors for the hepatitis B virus pre-S1 region and, via polymerized human serum albumin, for the pre-S2 region. Journal of virology. 1989;63(5):1981–8. doi: 10.1128/jvi.63.5.1981-1988.1989. Epub 1989/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehdi H, Kaplan MJ, Anlar FY, Yang X, Bayer R, Sutherland K, et al. Hepatitis B virus surface antigen binds to apolipoprotein H. Journal of virology. 1994;68(4):2415–24. doi: 10.1128/jvi.68.4.2415-2424.1994. Epub 1994/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andre P, Perlemuter G, Budkowska A, Brechot C, Lotteau V. Hepatitis C virus particles and lipoprotein metabolism. Seminars in liver disease. 2005;25(1):93–104. doi: 10.1055/s-2005-864785. Epub 2005/02/26. [DOI] [PubMed] [Google Scholar]
- 30.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr., et al. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(14):5848–53. doi: 10.1073/pnas.0700760104. Epub 2007/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. Journal of virology. 2007;81(24):13783–93. doi: 10.1128/JVI.01091-07. Epub 2007/10/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker AL, Waddington SN, Nicol CG, Shayakhmetov DM, Buckley SM, Denby L, et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood. 2006;108(8):2554–61. doi: 10.1182/blood-2006-04-008532. Epub 2006/06/22. [DOI] [PubMed] [Google Scholar]
- 33.Jiang H, Wang Z, Serra D, Frank MM, Amalfitano A. Recombinant adenovirus vectors activate the alternative complement pathway, leading to the binding of human complement protein C3 independent of anti-ad antibodies. Molecular therapy : the journal of the American Society of Gene Therapy. 2004;10(6):1140–2. doi: 10.1016/j.ymthe.2004.08.015. Epub 2004/11/27. [DOI] [PubMed] [Google Scholar]
- 34.Zinn KR, Szalai AJ, Stargel A, Krasnykh V, Chaudhuri TR. Bioluminescence imaging reveals a significant role for complement in liver transduction following intravenous delivery of adenovirus. Gene therapy. 2004;11(19):1482–6. doi: 10.1038/sj.gt.3302331. Epub 2004/08/06. [DOI] [PubMed] [Google Scholar]
- 35.Denard J, Marolleau B, Jenny C, Rao TN, Fehling HJ, Voit T, et al. C-reactive protein (CRP) is essential for efficient systemic transduction of recombinant adeno-associated virus vector 1 (rAAV-1) and rAAV-6 in mice. Journal of virology. 2013;87(19):10784–91. doi: 10.1128/JVI.01813-13. Epub 2013/08/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denard J, Beley C, Kotin R, Lai-Kuen R, Blot S, Leh H, et al. Human galectin 3 binding protein interacts with recombinant adeno-associated virus type 6. Journal of virology. 2012;86(12):6620–31. doi: 10.1128/JVI.00297-12. Epub 2012/04/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaiss AK, Cotter MJ, White LR, Clark SA, Wong NC, Holers VM, et al. Complement is an essential component of the immune response to adeno-associated virus vectors. Journal of virology. 2008;82(6):2727–40. doi: 10.1128/JVI.01990-07. Epub 2008/01/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pulicherla N, Shen S, Yadav S, Debbink K, Govindasamy L, Agbandje-McKenna M, et al. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;19(6):1070–8. doi: 10.1038/mt.2011.22. Epub 2011/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nature biotechnology. 2010;28(1):79–82. doi: 10.1038/nbt.1599. Epub 2009/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sleep D. Albumin and its application in drug delivery. Expert opinion on drug delivery. 2015;12(5):793–812. doi: 10.1517/17425247.2015.993313. Epub 2014/12/19. [DOI] [PubMed] [Google Scholar]
- 41.Fiume L, Manerba M, Di Stefano G. Albumin-drug conjugates in the treatment of hepatic disorders. Expert opinion on drug delivery. 2014;11(8):1203–17. doi: 10.1517/17425247.2014.913567. Epub 2014/04/30. [DOI] [PubMed] [Google Scholar]
- 42.Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. Journal of controlled release : official journal of the Controlled Release Society. 2008;132(3):171–83. doi: 10.1016/j.jconrel.2008.05.010. Epub 2008/06/28. [DOI] [PubMed] [Google Scholar]
- 43.Sand KM, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT. Unraveling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Frontiers in immunology. 2014;5:682. doi: 10.3389/fimmu.2014.00682. Epub 2015/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. Journal of controlled release : official journal of the Controlled Release Society. 2012;157(2):168–82. doi: 10.1016/j.jconrel.2011.07.031. Epub 2011/08/16. [DOI] [PubMed] [Google Scholar]
- 45.Elsadek B, Kratz F. Impact of albumin on drug delivery--new applications on the horizon. Journal of controlled release : official journal of the Controlled Release Society. 2012;157(1):4–28. doi: 10.1016/j.jconrel.2011.09.069. Epub 2011/10/01. [DOI] [PubMed] [Google Scholar]
- 46.Bern M, Sand KM, Nilsen J, Sandlie I, Andersen JT. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. Journal of controlled release : official journal of the Controlled Release Society. 2015;211:144–62. doi: 10.1016/j.jconrel.2015.06.006. Epub 2015/06/10. [DOI] [PubMed] [Google Scholar]
- 47.Moriyama T, Takei T, Itabashi M, Uchida K, Tsuchiya K, Nitta K. Caveolae may enable albumin to enter human renal glomerular endothelial cells. Journal of cellular biochemistry. 2015;116(6):1060–9. doi: 10.1002/jcb.25061. Epub 2015/02/03. [DOI] [PubMed] [Google Scholar]
- 48.Zloza A, Kim DW, Broucek J, Schenkel JM, Kaufman HL. High-dose IL-2 induces rapid albumin uptake by endothelial cells through Src-dependent caveolae-mediated endocytosis. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2014;34(11):915–9. doi: 10.1089/jir.2013.0155. Epub 2014/06/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razzak M. Glomerular disease: Albumin endocytosis is caveolin-mediated. Nature reviews Nephrology. 2014;10(5):242. doi: 10.1038/nrneph.2014.47. Epub 2014/03/20. [DOI] [PubMed] [Google Scholar]
- 50.Yumoto R, Nishikawa H, Okamoto M, Katayama H, Nagai J, Takano M. Clathrin-mediated endocytosis of FITC-albumin in alveolar type II epithelial cell line RLE-6TN. American journal of physiology Lung cellular and molecular physiology. 2006;290(5):L946–55. doi: 10.1152/ajplung.00173.2005. Epub 2005/12/20. [DOI] [PubMed] [Google Scholar]
- 51.Lambot N, Lybaert P, Boom A, Delogne-Desnoeck J, Vanbellinghen AM, Graff G, et al. Evidence for a clathrin-mediated recycling of albumin in human term placenta. Biology of reproduction. 2006;75(1):90–7. doi: 10.1095/biolreprod.105.050021. Epub 2006/02/24. [DOI] [PubMed] [Google Scholar]
- 52.Montoyo HP, Vaccaro C, Hafner M, Ober RJ, Mueller W, Ward ES. Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2788–93. doi: 10.1073/pnas.0810796106. Epub 2009/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. The Journal of experimental medicine. 2003;197(3):315–22. doi: 10.1084/jem.20021829. Epub 2003/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. Journal of virology. 1998;72(3):2224–32. doi: 10.1128/jvi.72.3.2224-2232.1998. Epub 1998/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Non-classical anti-factor VIII C2 domain antibodies are pathogenic in a murine in vivo bleeding model. Journal of thrombosis and haemostasis : JTH. 2009;7(4):658–64. doi: 10.1111/j.1538-7836.2009.03299.x. Epub 2009/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.