

Summary
Here we report proteomic analyses of 129 human cortical tissues to define changes associated with asymptomatic and symptomatic stages of Alzheimer’s Disease (AD). Network analysis revealed 16 modules of co-expressed proteins, 10 of which correlated with AD phenotypes. A subset of modules overlapped with RNA co-expression networks, including those associated with neurons and astroglial cell types, showing altered expression in AD, even in asymptomatic stages. Overlap of RNA and protein networks was otherwise modest, with many modules specific to the proteome, including those linked to microtubule function and inflammation. Proteomic modules were validated in an independent cohort, demonstrating some module expression changes unique to AD and several observed in other neurodegenerative diseases. AD genetic risk loci were concentrated in glial-related modules in the proteome and transcriptome consistent with their causal role in AD. This multi-network analysis reveals protein- and disease-specific pathways involved in the etiology, initiation, and progression of AD.
eTOC Blurb
Using label-free ‘single shot’ proteomics, we define changes in the proteome of human brain linked to preclinical and clinical stages of Alzheimer’s Disease (AD). These data reveal modules of co-expressed proteins that correlate with AD phenotypes, are distinct from modules identified from gene co-expression data, and highlight non-neuronal drivers of disease.
Introduction
The neuropathological changes of Alzheimer’s disease (AD) begin two decades or more before signs of cognitive impairment (Sperling et al., 2011). Currently, our understanding of the pathological events and the molecular transition from the asymptomatic phase (AsymAD) (Driscoll et al., 2006) to clinically evident dementia is limited. Although amyloid-beta (Aβ) deposition in the brain is hypothesized to be a central force driving AD pathogenesis (Selkoe and Hardy, 2016), individuals remain cognitively normal for many years despite accumulating aggregates of Aβ plaques and tau neurofibrillary tangles (Sperling et al., 2011). Large-scale analysis of molecular alterations in human brain provides an unbiased, data-driven approach to identify the many complicated processes involved in AD pathogenesis and to prioritize their links to relevant clinical and neuropathological traits, including changes in the asymptomatic phase of disease.
Systems-level analyses of large data sets have emerged as essential tools for identifying key molecular pathways and potential new drug targets. Algorithms such as weighted gene co-expression network analysis (WGCNA) classify the transcriptome into biologically meaningful modules of co-expressed genes linked to specific cell types, organelles, and biological pathways (Miller et al., 2008; Oldham, 2014). Co-expression modules also link to disease processes in which the most centrally connected genes are highly enriched for key drivers that play prominent roles in disease pathogenesis (Cerami et al., 2010; Huan et al., 2013; Tran et al., 2011). However, there are marked spatial, temporal, and quantitative differences between mRNA and protein expression (Abreu et al., 2009). In human tissues only about one-third of mRNA-protein pairs show significant correlation in expression levels, with marked variation depending on their functions (Zhang et al., 2014a). This relationship is not well understood in complex tissues such as brain; mRNA-protein correlation coefficients reach no higher than 0.47 even in acutely isolated brain cell types (Sharma et al., 2015). While transcriptome networks in AD brain have been examined (Miller et al., 2008; Miller et al., 2013; Zhang et al., 2013), network changes in the AD brain proteome, including those associated with early asymptomatic stages of disease, have not been explored.
In this study, we coupled label-free mass spectrometry based proteomics and systems biology to define networks of highly correlated proteins associated with neuropathology and cognitive decline in the brains of healthy controls, AsymAD, and AD. Similar to RNA-based networks, the brain proteome is organized in biologically meaningful networks related to distinct functions and cell types (i.e., neurons, oligodendrocyte, astrocyte, and microglia). Downregulation of modules associated with neurons and synapses and up-regulation of astroglial modules were strongly associated with amyloid plaque and neurofibrillary tangle pathologies, consistent with RNA-based networks previously reported for late stage AD. However, comparison of RNA and protein networks shows that more than half of the protein co-expression modules are not well represented at the RNA level. These include modules associated with microtubule function, RNA/DNA binding, post-translational modification, and inflammation that were also strongly associated with AD phenotypes. Moreover, several of these were linked to AsymAD, progressively changing with cognitive status, and disease specific (i.e., not altered in other neurodegenerative diseases). Finally, common AD risk loci, identified by the IGAP consortium genome wide association study (GWAS) were concentrated in glial-related modules in both the proteome and transcriptome consistent with their causal role in AD. Our findings highlight the use of large-scale proteomics and integrated systems biology to unravel the molecular etiology promoting initiation and progression of AD.
Results
Proteomic analysis of human brain tissues
We collected post-mortem brain tissue from 50 individuals representing 15 controls, 15 AsymAD and 20 AD cases from the Baltimore Longitudinal Study of Aging (BLSA) (O’Brien et al., 2009). For 47 cases, we analyzed tissue from both the dorsolateral prefrontal cortex (FC, Brodmann Area 9) and precuneus (PC, Brodmann Area 7). Both regions are affected in AD, and PC is a site of early amyloid deposition and glucose hypometabolism (Rabinovici et al., 2010). All case metadata including neuropathological criteria for amyloid (CERAD) and tau pathology (Braak) are provided in Table S1.
Samples were uniformly homogenized and analyzed by SDS-PAGE prior to proteolytic digestion with trypsin for subsequent proteomic analysis (Fig. S1A). To account for technical variation, each peptide digest was spiked with an isotopically labeled internal reference standard before analysis by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) on an Orbitrap Q-Exactive mass spectrometer (Fig. S1B–C). Protein abundance was determined by peptide ion-intensity measurements across LC-MS runs using the label-free quantification (LFQ) algorithm in MaxQuant (Cox et al., 2014) (Fig. S1D). In total, 64,938 peptides mapping to 5,130 protein groups were identified. One limitation of “shotgun” label free quantitative proteomics is missing data (i.e. missing protein identifications or abundance values), especially for low abundance proteins (Karpievitch et al., 2012). Thus, only those proteins quantified in at least 90% of samples for each brain region were included in the analysis, resulting in the final quantification of 2,735 protein groups mapping to 2,678 unique gene symbols across the 97 samples (n=47 FC and n=50 PC) (Table S2). To quantify Aβ levels in the brain samples, the ion intensities for two amyloid precursor protein (APP) peptides, corresponding to residues 6–16 and residues 17–28 of the Aβ sequence, were summed for each individual case (Fig. S2). It is important to note that although these two peptides are derived from APP, they do not discriminate between the full-length APP and cleaved Aβ fragments. However, APP peptides mapping outside of the Aβ sequence did not show a significant increase with pathology or clinical symptoms, whereas the two peptides mapping to Aβ regions strongly correlate with CERAD and disease status across brain regions (Fig. 1A–C and Fig. S2). Thus, these two APP peptides serve as measures of Aβ levels in brain. Based on these findings, the LFQ intensities for these two Aβ peptides were calculated in each individual sample and manually included as a separate protein measurement (assigned symbol ABETA) for subsequent differential and co-expression analyses.
Fig. 1. Differential abundance of Aβ and other proteins observed in AsymAD and AD brain.
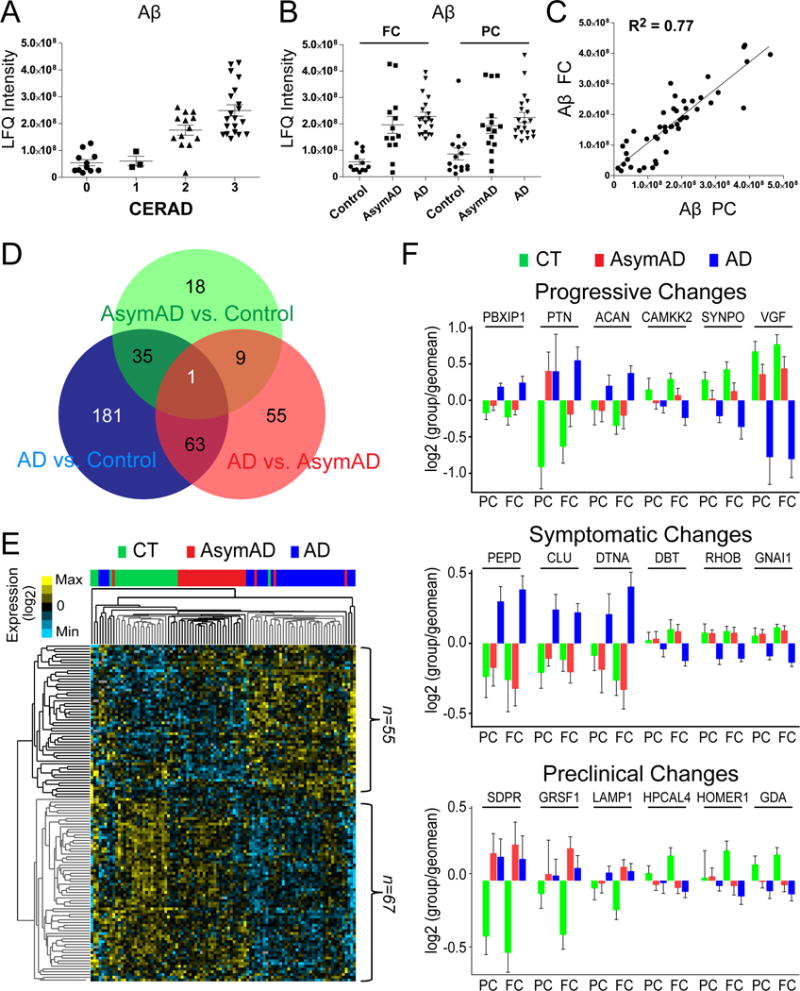
(A and B) Aβ measurements measured by LFQ ion intensity correlate with CERAD and disease status in precuneus (PC) and frontal cortex (FC). (C) Correlation for Aβ levels across 47 paired samples in the PC and FC. (D) Venn diagram showing a total of 362 unique proteins in both brain regions (FC and PC) that were determined to be significantly altered (decreased or increased) by ANOVA followed by Tukey’s post-hoc test (p <0.01) in the three comparisons. (E) Supervised hierarchical clustering of 123 significant proteins altered in FC and PC by criteria described in methods. (F) Several significant proteins in AD displayed a progressive change across comparisons of control, AsymAD and AD groups (top panel), whereas other proteins were significantly changed only during the symptomatic phase of AD (middle panel). Protein markers which trended as changing in both the preclinical and symptomatic stage of AD (bottom panel) compared to control group(s).
Classification of AD and AsymAD cases by proteomic measures
A total of 362 unique proteins in both brain regions (FC and PC) showed significantly altered levels of expression (ANOVA followed by Tukey’s comparison post-hoc test, p <0.01) across three comparisons: (i) controls vs. AsymAD, (ii) controls vs. AD, and (iii) AsymAD vs. AD (Fig. 1D). The number of significant proteins compared to controls was proportional to disease severity in a cross-sectional analysis of AsymAD (n=63), and AD (n=280) indicating that differences in the proteome correlate with neuropathological burden and cognitive dysfunction. The differential proteome was consistently more complex (i.e., greater n of significantly altered proteins) in the FC compared to the PC (Supplemental Figure S3).
To determine whether differentially expressed proteins could be used as classifiers of disease status, we performed a supervised cluster analysis using Cluster 3.0 across control, AsymAD, and AD cases (Fig. 1E). We restricted the analysis to those proteins significantly altered in at least two of the aforementioned comparisons across both FC and PC (n=123; Fig. S3A and Table S2). As expected, the resulting dendrogram segregated the majority of control (green) and AD (blue) cases. In addition, AsymAD (red) cases, with rare exceptions, clustered between the control and AD cases. Thus, these select differentially expressed proteins provide a molecular signature distinguishing control, AsymAD and AD brains.
Gene ontology (GO) enrichment analysis of classifier proteins that are significantly decreased (n=67) in AD indicated overrepresentation of ‘protein folding capacity’ and ‘microtubule cytoskeleton’ proteins, whereas those related to ‘response to protein stimulus’ and ‘response to biotic stimulus’, were over-represented among proteins increased in AD (n=55) (Fig. S3). Several proteins displayed progressive increases or decreases in abundance across control, AsymAD, and AD cases (Fig. 1F, and Table S2). These included progressive increases in chondroitin sulfate proteoglycan (CSPG), aggrecan (ACAN), and CSPG binding protein pleitrophin (PTN) in AsymAD and AD. In contrast, proteins involved in synaptic function and synaptogenesis, including SYNPO1, VGF, and the kinases CAMKK2 and CAMK4 displayed a progressive decrease across AsymAD and AD. Several proteins were altered exclusively in the symptomatic phase of disease, including increases in clusterin (CLU) and peptidase D (PEPD), and decreases in proteins involved in GTPase signaling including RHOB and G protein subunit alpha i protein (GNAI1). Some changes in the proteome were of maximum magnitude and significance in AsymAD cases, including decreases in the synaptic protein HOMER1 and guanine deamidase GDA, and increases in lysosomal marker LAMP1 and G-rich RNA binding protein GRSF1 (Fig. 1F). Of note, these latter 3 proteins are in a brain tissue-specific genetic interaction neighborhood (Greene et al., 2015) that is highly significantly (FDR-corrected p<1×10−7) enriched for the lysosome KEGG pathway. Endosomal-lysosomal dysfunction is strongly implicated in AD (Hu et al., 2015). Considering other proteins significantly changed in AsymAD, like TMPO and HNRNPF (Table S2), the genetic interaction network landscape had edges that were consistently supported by changes found in a microarray study which identified transcripts altered downstream of an age-associated overexpressed circulating and brain-resident microRNA, mIR34-a (Chang et al., 2007; Li et al., 2011). Overall, these results highlight the potential utility of differentially expressed proteins for classifying AD and identifying disease-related pathways including those altered in the preclinical stage of AD.
Protein co-expression network analysis of AD brain
Co-expression analysis of the brain transcriptome effectively organizes RNA into networks related to molecular pathways, organelles, and cell types impacted by AD (Miller et al., 2008; Miller et al., 2013). Correlation between RNA and protein in any given brain cell type is generally weak (Sharma et al., 2015), and it is unclear whether similar networks manifest at the protein level in AD brain. Thus, we applied WGCNA (Langfelder and Horvath, 2008) to define trends in protein co-expression across FC and PC in BLSA brains (n=97 samples). We corrected for age, sex, and PMI-mediated covariance of protein measurements, and then normalized baseline protein expression differences between regions, setting a small minimum WGCNA module size of 17 proteins with a low propensity to merge modules (merge height of 0.07). These parameters identified 16 modules of strongly co-expressed groups of proteins, and ranked by size, ranging from BLSA M1, BM-1 (largest, 396 proteins) to BM-16 (smallest, 28 proteins). Reducing the minimal module size in WGCNA for proteomic datasets facilitates module identification, although final modules were somewhat larger owing to final module member reassignment by eigenprotein correlation p value (reassignThresh parameter was set to p=0.05 instead of the default p=10−6). It should also be noted that a label-free protein expression matrix differs from a microarray or even RNA-Seq expression matrix because of the missing quantitative data for some proteins in a sample due to the stochastic nature of LC-MS/MS. Therefore, choices for the threshold of missing data allowed into the matrix, and also for imputation of those values, if any, becomes an important consideration for protein co-expression analysis. Networks were also analyzed for prefrontal and precuneus cortical regions, and because they showed significant overlap (data not shown), the expression data were combined as described above to maximize power.
Many of these modules were significantly enriched for brain-specific gene ontologies as well as established cellular functions and/or organelles (Fig. 2A and Table S3). The three largest modules were associated with categories of ‘synaptic transmission’ (B-M1), ‘myelin sheath’ (B-M2), and ‘mitochondrion’ (B-M3), whereas B-M5 represented ‘extracellular matrix’ and B-M6, ‘inflammatory response’. Other modules included those with GO terms linked to synaptic membrane and dendrites (B-M4), protein folding (B-M7 and B-M11), oxidoreductase activity (B-M9), DNA/RNA binding (B-M10), microtubules (B-M12), regulation of apoptosis (B-M13), hydrolase activity (B-M14), regulation of microtubule polymerization (B-M15) and ribosome (B-M16).
Fig. 2. Protein co-expression classifies the proteome into modules associated with specific gene ontologies and brain cell types.
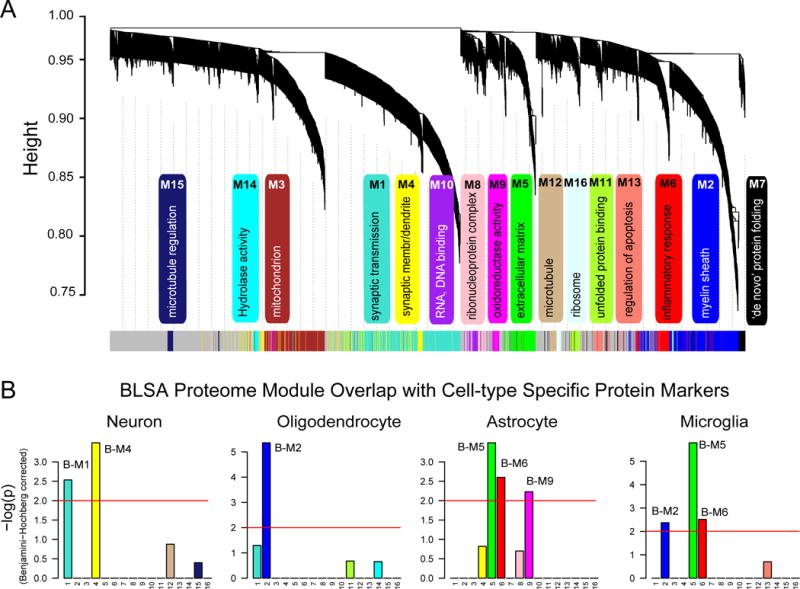
(A) WGCNA cluster dendrogram groups proteins (n=2,735) measured across FC and PC into distinct protein modules (M1–16) defined by dendrogram branch cutting. These modules were significantly enriched for gene ontologies linked to discrete cellular functions and/or organelles in the brain. (B) Cell type enrichment was assessed by cross-referencing module proteins (via matching gene symbols) using the one-tailed Fisher’s exact test against lists of proteins determined as enriched in neurons, oligodendrocytes, astrocytes and microglia (Table S4). The FDR was corrected for multiple comparisons by the Benjamini-Hochberg (BH) method, bars extending above the line represent BH p<0.01.
Transcript-based network analyses have shown that modules represent major sources of biological variance across the cortex and that some modules are enriched in genes expressed by different cell types (Oldham et al., 2008a). To determine if a similar relationship exists with protein-based networks, we evaluated the overlap of proteins in each module with brain cell-type-specific proteomes (Fig. 2B and Table S4) generated previously from acutely isolated neurons, oligodendrocytes, astrocytes, and microglia from mouse brain (Sharma et al., 2015). The largest module, B-M1, was enriched with neuron-specific proteins, including neuritic (CAMK2A and CAMKK2) and synaptic (SYNGAP1, RTN1, L1CAM) proteins. The B-M4 module was also enriched for neuronal protein markers including GBRA1, GBRA2, OLFM1, and NRXN3, associated with ‘synaptic membrane and dendrites’. Oligodendrocyte-specific proteins were over-represented as hub proteins in B-M2 related to myelination, including CNP, MAG, MOG, PLP1, and MBP gene products. Astrocyte proteins were predominantly enriched in B-M6 (e.g. GFAP, PBXIP1, and GJA1) and B-M9 (e.g. SLCA11, ALDOC, and GCSH). Finally, microglial protein markers were over-represented in the B-M5 module (e.g. ANXA1, ANXA4, CTSZ and CLIC), B-M6 (e.g. MSN, LCP1, LSP1, and C1QC) and, to a lesser extent, B-M2 (e.g. CRYL1, NPC2, and CNDP2). Thus, as seen in transcriptome level network analyses (Miller et al., 2008; Oldham et al., 2008a), certain modules of co-expressed proteins enrich with markers of specific brain cell types, and likely reflect changes in the abundance, activation, and/or changing phenotype(s) of these cells in AD.
Correlation of protein networks with AD neuropathology and cognitive status
We assessed the module correlations to the hallmark neuropathological traits of amyloid plaques (CERAD) and neurofibrillary tangles (Braak) across control, AsymAD, and AD cases to identify protein networks associated with both neuropathological features and clinical symptoms (Fig. 3A). Five BLSA modules had positive correlation with CERAD and/or Braak, and included those associated with ‘inflammatory response’ (B-M6), ‘ribonucleoprotein function’ (B-M8), and ‘apoptosis’ (B-M13). The eigenproteins for these modules also increased between control and AsymAD and between AsymAD and AD (Fig. 3B), indicating that protein expression changes begin early in the asymptomatic phase of disease. In WGCNA, a module eigengene, or in our case, an eigenprotein, is defined as the first principal component of a given module and serves as a representative, weighted expression profile for the module. Module B-M15 with ontologies associated with ‘microtubule regulation’ showed significant correlation exclusively with CERAD, whereas B-M5 harbored extracellular matrix proteins enriched in astrocyte/microglial markers, and showed correlation exclusively with Braak stages (Fig. 3A and B).
Fig. 3. BLSA protein modules correlate to cognitive status and AD neuropathological burden.
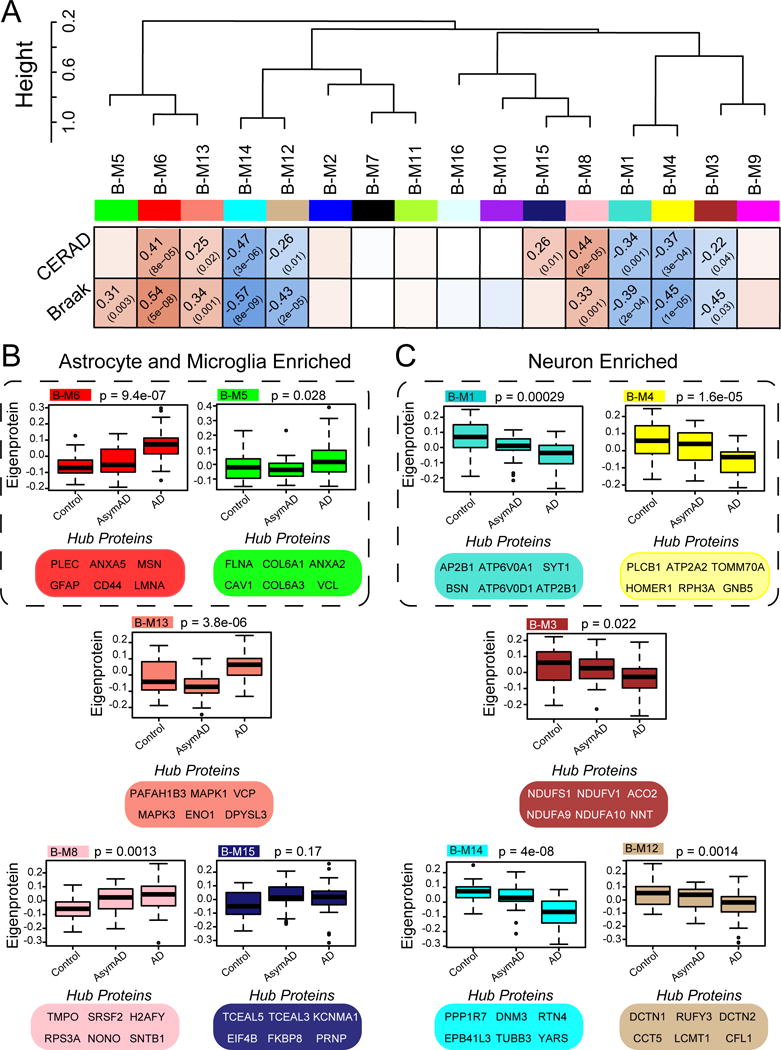
(A) BLSA protein modules were clustered to assess module relatedness based on correlation of protein co-expression eigenproteins. Pearson correlation and P value between module eigenprotein expression and CERAD (top) and Braak (bottom). (B) Module expression profiles and key hub proteins that are positively correlated with CERAD and/or Braak. (C) Module expression profiles and key hub proteins that are negatively correlated with CERAD and/or Braak. Box plots with error bars are displayed for each of the three groups of case samples (control, AsymAD and AD). Significance was measured using one-way nonparametric ANOVA, Kruskal-Wallis p-values. Cell type associated modules are encircled (dashed line).
Several module expression profiles were significantly decreased in AD and negatively correlated with AD neuropathology (Fig. 3A and C). Namely, B-M1 and B-M4, both enriched with neuronal markers (Fig. 2B) and associated with related ontologies, were negatively correlated with CERAD and Braak scores, consistent with loss of synaptic proteins in AD (Terry et al., 1991) and observed in RNA co-expression modules (Miller et al., 2008). Module B-M3, enriched with mitochondrial proteins, was also decreased across AD cases, consistent with a loss of mitochondrial function and hypometabolic phenotype observed in patients with AD (Alexander et al., 2002).
Two modules enriched with tubulin binding proteins, B-M12 and B-M14, were negatively correlated with CERAD score and Braak stage, consistent with a known loss of microtubule function with tau aggregation in AD (Feinstein and Wilson, 2005). Notably, B-M14, the module with the strongest negative correlation to Braak (cor=−0.57, p=8e–9) and CERAD (cor=−0.47, p=3e–6), was enriched with proteins associated with ‘hydrolase activity’, represented by the hub protein PPP1R7, a subunit of protein phosphatase 1, and deubiquitinase OTUB1 as well as several microtubule proteins (DNM3, RTN4, EPB41L3, TUBB3) involved in neurite formation and/or transport. All three isoforms of tau (MAPT) correlated negatively with these phosphatase-enriched modules (Table S2), consistent with the role of tau hyperphosphorylation in neurofibrillary tangle pathology.
Since little is known about the preclinical molecular changes of AD, one goal of this study was to examine AsymAD cases to identify protein networks affected early in AD progression. Decreased expression of the B-M1 and B-M4 modules in AsymAD indicates loss of synaptic proteins prior to the onset of cognitive impairment. B-M8 was particularly elevated among AsymAD and AD samples, implicating perturbations in RNA metabolism. We and others have previously identified RNA splicing deficits in AD brain which could have profound impact on transcription of many genes, including tau, APP, and presenilins (Bai et al., 2013; Tollervey et al., 2011). The correlation of module B-M8 with both AsymAD and AD suggests that changes in RNA metabolism occur in preclinical stages of disease. B-M6, linked to ‘inflammatory response’, is also increased in AsymAD, which may reflect phenotypic activation and/or proliferation of astrocytes and microglia prior to cognitive decline. Of note, B-M13 was suppressed and more tightly correlated across AsymAD samples compared to control samples (Fig. 3B). Given that this module increases in AD compared to AsymAD, proteins within this module may reflect a transient, concerted downregulation of a protein network prior to upregulation with symptom onset—potentially implicating B-M13 as important for cognitive decline. Key hub proteins in B-M13 included MAPK1 and MAPK3, stress-activated kinases previously observed as increased in AD transcriptome studies of hippocampus (Miller et al., 2013). Module B-M5, enriched with microglia/astrocyte markers, increased only in the symptomatic phase of AD, suggesting that neuroinflammation proteins in this module may be important mediators of cognitive decline or disease progression. Thus, co-expression analysis effectively organizes the brain proteome into protein modules that are strongly linked to neuropathological features of AD and presence or absence of clinical symptoms.
Validation of protein expression changes in an independent cohort of brain tissues
To assess the reproducibility of our findings in the BLSA cases, and the specificity of the changes for AD versus other neurodegenerative diseases, we performed a second proteomic analysis of brain homogenates prepared from the dorsolateral prefrontal cortex of clinically and pathologically characterized control, AD, Parkinson’s Disease (PD), and amyotrophic lateral sclerosis (ALS) cases (n=8 per group; Table S5) collected from the Emory Alzheimer’s Disease Research Center. In total, 53,313 peptides mapping to 4,356 proteins groups were identified. As with the BLSA cases, only those proteins quantified in at least 90% of cases were included in the analysis, resulting in the final use of 2,491 quantified protein groups mapping to 2,433 unique gene symbols. A total of 48 differentially expressed proteins in the Emory AD cases overlapped with significant protein changes in the BLSA AD cases (Fig. S4). Of these, 47 were consistent in their direction of change in the BLSA and Emory AD cases. As seen in the BLSA cases, Aβ (ABETA) represented the most increased protein in the Emory AD cases, followed by the small heat shock protein, HSP27 (HSPB1), which is significantly and selectively elevated only in AD compared to control, PD, and ALS (Fig. S4). Thus, the combination of Emory and BLSA proteomic datasets confirmed the reproducibility of differentially expressed proteins, and allowed us to identify those changes that occur during the asymptomatic phase as well as those which are AD-specific or shared among neurodegenerative diseases.
We next compared the protein co-expression networks between the BLSA and Emory cases. A total of 23 modules (E-M1 to E-M23) were generated from the Emory brain proteome using WGCNA (see experimental procedures). The increased number of modules in the Emory network (Table S6) compared to the BLSA network can be attributed to the additional diversity of neurodegenerative disease cases (AD, PD, and ALS). Importantly, 13 of the 16 modules in the BLSA network (Fig. 4A) were preserved in the Emory network, with Zsummary scores above 2 (p<0.05) (Fig. 4A) (Langfelder et al., 2011). Module overlap across networks was also assessed using a hypergeometic Fisher’s exact test (Fig. 5A, left panel). Modules showed either significant positive correlation (red), anti-correlation (blue), or no correlation (white) in protein membership (via matched gene symbols). The vast majority of BLSA modules (14/16) showed at least one cognate module within Emory proteome, and conversely, 17/23 modules in the Emory brain network overlapped in the BLSA proteome. Thus, protein co-expression networks are conserved across both BLSA and Emory brains, supporting the robustness of WGCNA in defining consistent patterns of protein co-expression representing shared biology (Parikshak et al., 2015).
Fig. 4. BLSA network changes are preserved in the Emory proteome.
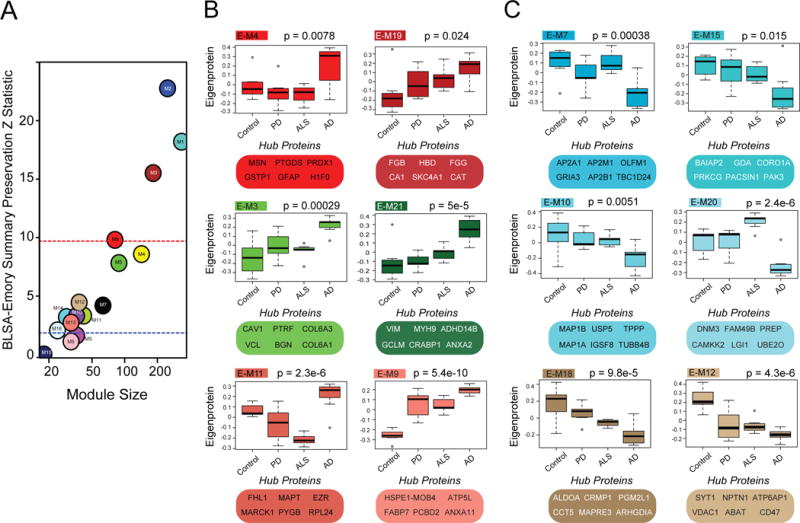
(A) In the BLSA proteomic network, 13 of the 16 modules were highly preserved in the Emory proteomic network, with Zsummary scores above 2 (p<0.05). Larger modules were some of the most preserved (above red line, p<0.01). (B) Emory protein modules and top hub proteins that increased in AD selectively or across all neurodegenerative diseases. (C) Emory protein modules and top hub proteins that decreased in AD selectively or across all neurodegenerative diseases. Significance was measured using one-way nonparametric ANOVA, Kruskal-Wallis p-values.
Fig. 5. Overlap between RNA and protein co-expression networks in AD.
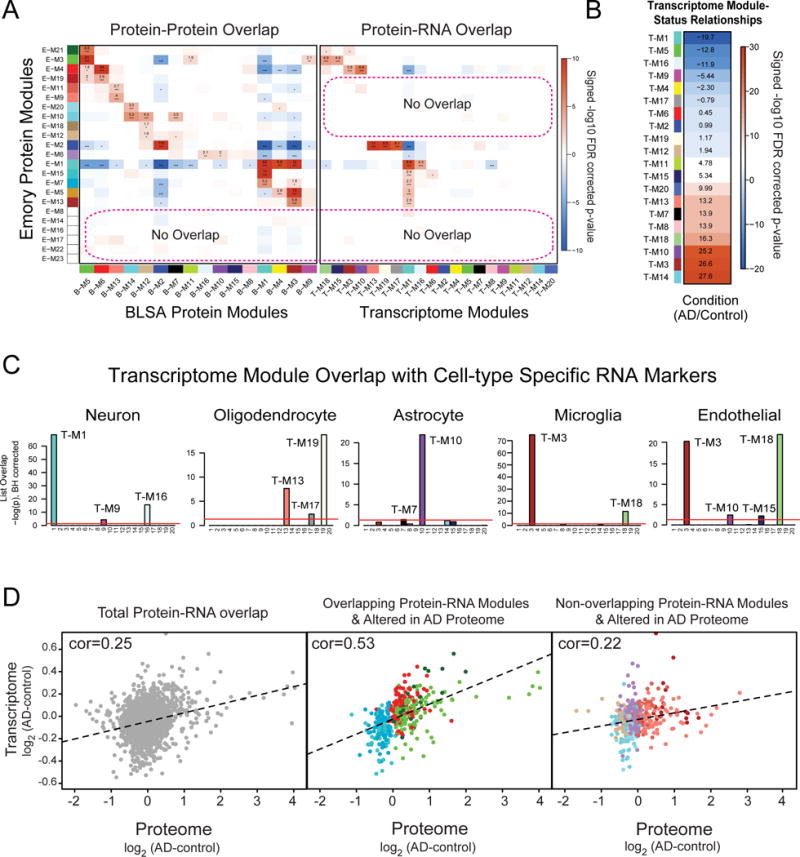
(A) A hypergeometric two-tailed Fisher’s exact test was used to determine which modules shared significant overlap or depletion of module members between the Emory and BLSA proteome networks (left panel) and Emory proteome and RNA networks (right panel). The 23 modules in the Emory case network (x-axis), clustered by module eigenprotein relatedness, were aligned to the 16 modules in the BLSA network (y-axis). Module gene symbol lists showed either significant overlap (red), depletion (blue) or no significant under- or over-representation (white) in protein membership. Numbers are positive signed −log10(FDR-corrected overrepresentation p values) representing degree of significance of overlap; asterisks also represent degree of significance for either positive or depleted comparisons: *, p<0.05; **, p<0.01, ***, p<0.005. (B) The degree of significance related to AD with correlation sign [signed −log10(FDR corrected p value)] is provided for each transcriptome module. Module eigengenes were correlated to AD status and multiple comparisons were accounted for by FDR correction (Benjamini-Hochberg) across modules with significance. (C) RNA modules were found enriched for specific cell type markers (Table S4) including neuronal, oligodendrocyte, astrocyte, microglia and endothelial cells following one-way Fisher’s exact test overlap with cell-type specific transcriptomes. (D) Pearson correlation analysis between all overlapping protein-RNA targets (n=2,406, left panel). Pearson correlation analysis between Protein-RNA targets in Emory modules that overlap with the transcriptome and change in the AD proteome (n=411, middle panel). This included protein modules increased in AD (E-M4, E-M21 and E-M3) that are enriched in astrocyte/microglia/endothelial markers and modules decreased in AD (E-M15 and E-M7) that are enriched with neuronal markers. Pearson correlation analysis between Protein-RNA targets in Emory modules that did not overlap with modules in the transcriptome (n=411, right panel), yet were increased (E-M19, E-M11 and E-M9) or decreased (E-M20, E-M10, E-M6, E-M18 and E-M12) in AD. Genes and cognate proteins were grouped and colored by their Emory protein module membership.
AD-specific network changes compared to other neurodegenerative diseases
The Emory proteome allowed us to identify networks that were specific to AD as well as those shared across AD, PD, and ALS, the latter potentially representing common mechanisms of neurodegeneration. For example, four Emory modules (E-M3, E-M4, E-M11 and E-M21), were each significantly increased in AD compared to PD, ALS, and control cases (Fig. 4B and Fig S5). Three of these modules (E-M3, E-M4 and E-M21) showed strong overlap with BLSA modules B-M5 and B-M6. Notably, E-M21 included humoral immune response proteins C4 and LTA4H, whereas E-M11, showed a high degree of overlap with BLSA module B-M13 that associated with apoptosis (Fig. 5A). In contrast, two modules, E-M9 and E-M19 in the Emory network (overlapping with B-M13, and B-M5 plus B-M6, respectively, in the BLSA network) (Fig. 4B and Fig. 5A) were increased across AD, PD, and ALS cases. E-M9 was represented by GO terms including “defense response”, whereas E-M19 was represented by “extracellular region” and “immune response,” indicating that common mechanisms in neurodegeneration also involve these ontologies. Other Emory proteome modules, especially E-M3, E-M4, and E-M11 (overlapping with B-M5, B-M6 and B-M13 respectively in the BLSA) were specifically altered in AD (Fig. 4B). Representative module gene ontologies for the Emory network are in Table S7.
Decreases in expression in neuronal and synaptic protein enriched modules E-M7 and E-M15, overlapping with B-M1 in the BLSA proteome, were selectively observed in AD (Fig. 4C). Two modules enriched with microtubule/cytoskeletal proteins (E-M10 and E-M20), with shared biology common to B-M14 in the BLSA proteomic network, were also selectively decreased in AD. In contrast, E-M12 and E-M18, related to ontologies associated with ‘centrosome’ and ‘cytoskeleton’ were decreased across all neurodegenerative disease groups. Thus, loss of certain cytoskeletal proteins appears to be a common mechanism in AD, PD, and ALS, whereas AD-specific modules are linked to a loss of a select group of microtubule and related proteins represented respectively by hubs MAP1B (E-M10) and DNM3 (E-M20) (Gray et al., 2003; Kawakami et al., 2003). Overall, the direction of change of overlapping modules in the Emory AD cases compared to non-AD neurodegenerative diseases is highly consistent with the network analysis from the BLSA cohort.
Overlap between RNA and protein co-expression networks in AD reveals both shared and protein-specific network changes
Well-established differences between expression of mRNA and their respective translated proteins prompted us to investigate the degree of conservation in RNA vs protein-based co-expression networks. We first generated an RNA co-expression network from previously reported microarray measurements of 18,204 genes across AD (n=308) and control (n=157) cases from dorsolateral prefrontal cortex (Narayanan et al., 2014). A total of 20 distinct transcriptome modules were identified (Fig. 5A, right panel; and Table S8) that closely correspond to the originally reported network (Narayanan et al., 2014). The relationships of these modules to AD vs control status is shown in Fig. 5B with full list of modules gene ontologies provided in Table S9. As expected, we identified a series of RNA modules that were enriched for specific cell-type markers (Zhang et al., 2014b), including neuronal (T-M1, T-M9, TM-16), oligodendrocyte (T-M13, T-M17, T-M19), astrocyte (T-M10, T-M7), microglia (T-M3, T-M18), and endothelial cells (T-M3, T-M18, TM-10, T-M15). Moreover, consistent with our findings in the protein co-expression networks, transcriptome-based modules enriched with microglia and astrocytes genes were among the most significantly increased in AD, whereas modules enriched with neuronal genes were among the most decreased (Fig. 5B and 5C). Protein and transcriptome modules with the highest degree of overlap were those associated with shared cell-type markers; these proteins showed strong correlation (cor=0.53) with their cognate RNA compared to all overlapping proteins mapped in the transcriptome (cor=0.25) (Fig. 5C and D). Despite the presence of shared cell-type specific modules in the protein and RNA networks, only 39% (9/23) of the Emory protein modules overlapped with RNA modules. This stands in contrast to the 74% (17/23) module overlap observed across the Emory and BLSA brain protein networks (Fig. 5A).
A key finding of this study is that several Emory protein modules that significantly increased (E-M9, E-M11, and E-M19) or decreased (E-M6, E-M10, E-M12, E-M18, and E-M20) in AD, did not overlap with transcriptome modules, consistent with a weak overall RNA-protein correlation (cor= 0.22) for these genes and cognate proteins (Fig. 5D, right panel). For example, E-M10 and E-M20 (and the highly related B-M14 module in the BLSA proteome) do not have a cognate transcriptome module. Thus, despite manifold deeper coverage, the transcriptome does not capture all key changes occurring in the AD brain, including reduced levels of protein phosphatases and microtubule-binding proteins that are enriched in these modules.
Another protein-based module that shows weak overlap in the RNA networks in brain is E-M19, which harbors several acute phase reactants (e.g. A2M and CP), immunoglobulins (IGHG1 and IGHA1) as well as blood fibrinogens involved in coagulation (Mosesson, 2005). As most of these genes are not generally well expressed in brain (Zhang et al., 2014b), protein members of this module could potentially be blood-derived and deposited in brain following disruption of the blood-brain barrier, which is exacerbated with aging and neurodegenerative disease (Ryu and McLarnon, 2009; Zlokovic, 2008). For example, insoluble fibrinogen or fibrin can be deposited in brain and drive adaptive immune responses as well as microglial inflammation (Ryu et al., 2015). Other notable protein modules that did not overlap in the transcriptome included E-M6, which was decreased across all neurodegenerative disease (Fig. S5) and enriched with DNA/RNA-associated proteins including splicing factors. Discordant RNA-protein correlation between splicing factors have previously been reported (Zhang et al., 2014a). Thus, our findings reveal shared modules between the transcriptome and proteome networks that mainly associate with cell-type, as well as an AD associated group of divergent or ‘protein-only’ networks associated with microtubule binding, inflammation, and DNA/RNA binding functions.
AD GWAS candidates are enriched in convergent protein and RNA networks preferentially associated with microglia and oligodendrocytes
Integration of genetic risks and gene expression networks has proven fruitful for identifying potential causal mechanisms (Voineagu et al., 2011). We used a similar approach to assess for enrichment of the proteins encoded by AD risk loci in the BLSA and Emory protein networks. Summary data from GWAS of AD, schizophrenia, and autism spectrum disorder (ASD) were used as input for MAGMA, which controls for confounders including gene length (de Leeuw et al., 2015), to generate a single p-value for each protein coding risk loci (Table S10). In the BLSA network, translated protein products from AD candidate genes defined with a MAGMA p-value of <0.05 (−log p value >1.3) were over-represented in the oligodendrocyte (B-M2) and astrocyte/microglia (B-M5) extracellular matrix protein modules. A total of 24 candidate genes mapped to B-M2 including BIN1 and PICALM, whereas 11 candidate genes mapped to B-M5 module including APOE, CLU, and FERMT2 (Fig. 6A and Fig. 2B). Enrichment of candidate genes in B-M5 was not solely driven by the presence of APOE, as B-M5 was still significant after omitting APOE from the analysis (data not shown). In contrast, candidate genes for ASD and schizophrenia with MAGMA p-values of <0.05 were significantly over-represented in the B-M1 and B-M4 neuron/synaptic modules, which is consistent with a previous finding of enrichment of ASD risk genes in neuronal modules (Voineagu et al., 2011). Similar results were also observed for the Emory network (Fig. S6). These findings in the proteome were corroborated at the mRNA level, as AD candidate risk loci were enriched in T-M3, associated with microglial/endothelial cell markers (Fig. 6B and Fig. 5C). A total of 33 GWAS candidates including HMHA1, MS4A6A, CD2AP, MS4A4A, HLA-DRA, and FERMT2 were enriched in T-M3. Also as seen in the protein networks, T-M13, associated with oligodendrocyte genes, was also enriched with 27 AD GWAS candidates including BIN1, whereas ASD and schizophrenia GWAS candidates were mainly over-represented in neuronal modules (T-M1 and T-M9) as well as module T-M13 in ASD. Thus, we conclude that genetic risk loci for late onset AD overlap in convergent RNA and protein networks preferentially enriched in microglia and oligodendrocytes, further providing network-driven evidence implicating glial dysfunction in the etiology of late onset AD.
Fig. 6. AD GWAS candidates are over-represented in protein and RNA networks associated glial cell types.
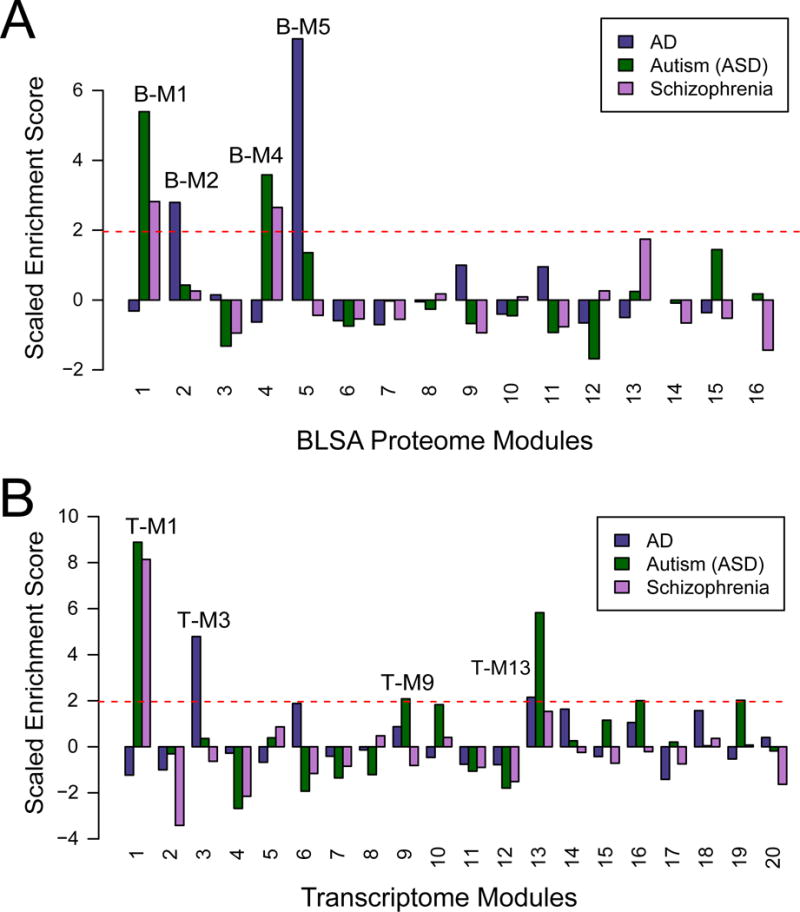
(A) AD GWAS candidate genes (darkslateblue) in the BLSA network were found significantly over-represented in oligodendrocyte module (B-M2), and in the astrocyte/microglia (B-M5) module. GWAS risk candidate genes for both ASD (darkgreen) and schizophrenia (magenta) were significantly over-represented in the B-M1 and B-M4 neuron/synaptic modules (B) AD GWAS candidate genes in the transcriptome were overrepresented microglial/endothelial modules T-M3 and T-M18 and an oligodendrocyte associated module (T-M13). Similar to the proteome, ASD and schizophrenia GWAS targets were significantly over-represented in neuronal transcriptome modules (T-M1 and T-M9). ASD was also found in the oligodendrocyte enriched T-M13 module. Random sampling (10,000 times) of the MAGMA gene list was used to assess the significance of the module enrichment score (* p value <0.05).
Discussion
Our proteomic studies offer new insights into changes in individual proteins and protein networks linked to AD, including changes related to poorly-understood preclinical stages of AD. Protein co-expression modules linked to astrocytic/microglial and neuronal functions in the BLSA cases revealed strong positive and negative correlations, respectively, with AD neuropathological burden and clinical symptoms. These BLSA networks were highly preserved in an independent cohort of Emory cases, which also revealed modules specific for AD, as well as networks of co-expressed proteins shared with other neurodegenerative diseases. We also observed relatively strong preservation of select modules derived from proteomics and previously reported transcriptomic studies of AD, especially those enriched in cell type-specific markers (neurons, oligodendrocytes, astrocytes, and microglia). However, most modules altered in the AD proteome were not well represented in the transcriptome, including those associated with microtubule function, RNA/DNA binding, and inflammation. These discrepancies may be due to well-known differences between the regulation, localization, and functions of mRNA and proteins. For example, a poor correlation between expression levels of microtubule-associated mRNA and protein in AD may in part be due to a spatial mismatch between neuronal cell bodies that express the mRNA and the degenerating axons where the proteins accumulate. Another example is the deposition of plasma proteins and peripheral immune cells that are often deposited in brain following disruption of the blood brain barrier (Zlokovic, 2008). The observed differences between protein- and RNA-based modules highlights the value of incorporating both transcriptomic and proteomic data in efforts to advance our understanding of AD pathogenesis and identifying novel therapeutic targets.
Despite the divergence in RNA and protein networks, one important similarity across the brain transcriptome and proteome was the consistent mapping of AD GWAS candidates to networks enriched in oligodendrocytes and microglia markers. This observation strongly implies that these modules reflect causal pathogenic processes involved in AD. Protein and RNA networks enriched in microglial proteins were significantly increased in AD and strongly correlated with both neurofibrillary tangle pathology and cognitive decline. These results echo recent large transcriptomic and AD GWAS network studies that show the involvement of microglial gene products and GWAS candidates in AD (Jones et al., 2010; Zhang et al., 2013). Thus, while significant synaptic and neuronal loss can occur prior to symptom onset in the asymptomatic phase of AD, our findings in the proteome suggest that clinical symptoms are associated with the increase in protein networks linked to inflammation, in particular those enriched with microglia.
One limitation of our study was the relative lack of proteome depth (~4,500 proteins per sample) compared to transcriptomics (~18,000). Each individual case proteome was generated from a ‘single shot’ LC-MS/MS analysis (120-minute gradient) without prior fractionation at the protein or peptide level. However, the trade-off of proteome depth was the increased number of individual samples analyzed (n=129), which bolsters network-based approaches requiring a high number of biological replicates to reach consensus (Oldham, 2014). Of note, future studies incorporating multiplex isobaric peptide mass tagging approaches and fractionation will enable both deeper proteome coverage with fewer missing values across individual samples (McAlister et al., 2012). Direct comparison of the transcriptome and proteome in the same cases will also be possible in the future with the Accelerating Medicine Partnership for AD projects using postmortem brains from several thousand cases to identify novel therapeutic targets and biomarkers (Hodes and Buckholtz, 2016). Nevertheless, our studies reveal an impressive integrated view of protein co-expression that validate across two independent groups of brains highlighting the robustness of our approach. Furthermore, our application of multi-network driven approaches (RNA and protein) offer new insights into the pathways underlying preclinical changes in AD brain and provide further evidence that non-neuronal cells mediate the genetic risk of developing AD.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the lead contact corresponding author, Dr. Nicholas T. Seyfried (nseyfri@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All brain tissue used in this analysis was derived from the autopsy collection of the Baltimore Longitudinal Study of Aging (BLSA) or the Emory Alzheimer’s Disease Research Center (ADRC) Brain Bank. Human postmortem tissues were acquired under proper Institutional Review Board (IRB) protocols with consent from family. Postmortem neuropathological evaluation of amyloid plaque distribution was performed according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria (Mirra et al., 1991), while extent of spread of neurofibrillary tangle pathology was assessed in accordance with the Braak staging system (Braak and Braak, 1991). Diagnoses were also made in accordance with established criteria and guidelines for Parkinson’s Disease (PD) (Gelb et al., 1999) and Amyotrophic Lateral Sclerosis (ALS) (Ince et al., 1998). All case metadata including, disease status, neuropathological criteria, age, sex, post-mortem interval and APOE genotype are provided in Table S1 and Table S5.
METHOD DETAILS
Brain tissue homogenization and protein digestion
Each piece of tissue was individually weighed (~0.1 g) and homogenized in 500 uL of urea lysis buffer (8M urea, 100 mM NaHPO4 buffer system, pH 8.5), including 5 μL (100× stock) HALT protease and phosphatase inhibitor cocktail (ThermoFisher, Cat# 78440). All homogenization was performed using a Bullet Blender (Next Advance) according to manufacturer protocols. Briefly, each tissue piece was added to Urea lysis buffer in a 1.5 mL Rino tube (Next Advance) harboring 750 mg stainless steel beads (0.9–2 mm in diameter) and blended twice for 5 minute intervals in the cold room (4°C). Protein supernatants were transferred to 1.5 mL Eppendorf tubes and sonicated (Sonic Dismembrator, Fisher Scientific) 3 times for 5 s with 15 s intervals of rest at 30% amplitude to disrupt nucleic acids and subsequently vortexed. Protein concentration was determined by the bicinchoninic acid (BCA) method, and samples were frozen in aliquots at −80°C. Each brain homogenate was analyzed by SDS-PAGE to assess protein integrity (Fig. S1). Protein homogenates (150 ug) were diluted with 50 mM NH4HCO3 to a final concentration of less than 2M urea and then treated with 1 mM dithiothreitol (DTT) at 25°C for 30 minutes, followed by 5 mM iodoacetamide (IAA) at 25°C for 30 minutes in the dark. Protein was digested with 1:100 (w/w) lysyl endopeptidase (Wako) at 25°C for 2 hours and further digested overnight with 1:50 (w/w) trypsin (Promega) at 25°C. Resulting peptides were desalted with a Sep-Pak C18 column (Waters) and dried under vacuum.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS)
Brain derived tryptic peptides (2 μg) were resuspended in peptide loading buffer (0.1% formic acid, 0.03% trifluoroacetic acid, 1% acetonitrile) containing 0.2 pmol of isotopically labeled peptide calibrants (ThermoFisher, #88321). Peptide mixtures were separated on a self-packed C18 (1.9 um Dr. Maisch, Germany) fused silica column (25 cm × 75 μM internal diameter; New Objective, Woburn, MA) by a NanoAcquity UHPLC (Waters, Milford, FA) and monitored on a Q-Exactive Plus mass spectrometer (ThermoFisher Scientific, San Jose, CA). Elution was performed over a 120 minute gradient at a rate of 400 nL/min with buffer B ranging from 3% to 80% (buffer A: 0.1% formic acid and 5% DMSO in water, buffer B: 0.1 % formic and 5% DMSO in acetonitrile). The mass spectrometer cycle was programmed to collect one full MS scan followed by 10 data dependent MS/MS scans. The MS scans (300–1800 m/z range, 1,000,000 AGC, 150 ms maximum ion time) were collected at a resolution of 70,000 at m/z 200 in profile mode and the MS/MS spectra (2 m/z isolation width, 25% collision energy, 100,000 AGC target, 50 ms maximum ion time) were acquired at a resolution of 17,500 at m/z 200. Dynamic exclusion was set to exclude previous sequenced precursor ions for 30 seconds within a 10 ppm window. Precursor ions with +1, and +6 or higher charge states were excluded from sequencing.
Western blotting
Total brain homogenates in urea (50 μg) were mixed with Laemmli sample buffer and resolved by SDS-PAGE before an overnight wet transfer to nitrocellulose membranes (BioRad) as previously reported (Diner et al., 2014). Membranes were blocked with casein blocking buffer (Sigma B6429) and probed with primary polyclonal antibodies for Anti-Hsp27 (HSPB1) antibody [G3.1] (Abcam #ab2790) and TDP-43 (Proteintech, Cat#) overnight at 4 °C. Membranes were incubated with secondary antibodies conjugated to Alexa Fluor 680 (Invitrogen) or IRDye800 (Rockland) fluorophores for one hour at room temperature. Images were captured using an Odyssey Infrared Imaging System (Li-Cor Biosciences).
Supervised Clustering
Individual BLSA cases were clustered using Cluster 3.0 (de Hoon et al., 2004) on a scaled log2 (case-specific protein measurement/all-sample geometric mean) range of values, with the range of values for each protein fixed at 5 log2 units. Proteins were clustered using uncentered correlation and samples were clustered using city-block distance, with an average linkage clustering method. Clustering results were visualized using Java TreeView 1.1.6r2 (Saldanha, 2004).
Weighted Gene Co-expression Network Analysis (WGCNA) of BLSA and Emory proteome datasets
Prior to network analysis, missing protein LFQ values were imputed using the k-nearest neighbor imputation function in R (impute.knn). Samples with low intra-region (PC or FC) network connectivity Z.K<−2.5 standard deviations (outlier samples) were identified iteratively and removed using SampleNetworks R package 1.06 essentially as described in (Oldham et al., 2008b), respectively reducing sample number to 47+42 samples (n=89 in PC+FC). Bootstrap regression of the LFQ intensity matrix using a model incorporating case status and case covariates for age, gender, and postmortem interval (PMI) was performed following principal component analysis of the PC+FC data set, where notably, none of the top five principle components correlated more than |rhoSpearman| >±0.3 with any of these three covariates. The ComBat function (Johnson et al., 2007) was then used to regress out region-specific (PC versus FC) quantitative baseline protein differences. A weighted protein co-expression network was built using the above LFQ-normalized, outlier-removed, and post-regressed protein expression values using blockwiseModules WGCNA function with the following parameters: soft threshold power beta=6.00, deepSplit=4, minimum module size of 17, merge cut height of 0.07, signed network with partitioning about medioids respecting the dendrogram, and a reassignment threshold of p=0.05. Module preservation was tested using the modulePreservation() WGCNA R package function, over 500 iterations for PC+FC vs. a similar network built using FC region- specific expression alone and confirmed that the modules were representative of FC specific networks.
WGCNA of the AD transcriptome dataset
The R package WGCNA was used to construct a co-expression network using the data downloaded from GEO (#GSE33000) (Narayanan et al., 2014). Briefly a thresholding power of 9 was chosen (as it was the smallest threshold that resulted in a scale-free R2 fit of 0.8) and the network was created by calculating the component-wise minimum values for topologic overlap (TO). Using 1 − TO (dissTOM) as the distance measure, genes were hierarchically clustered. Initial module assignments were determined by using a dynamic tree-cutting algorithm (cutreeHybrid, using default parameters except deepSplit = 4, cutHeight = 0.999, minModulesize = 100, dthresh=0.1 and pamStage = FALSE). The resulting 20 modules or groups of co-expressed genes were used to calculate module eigengenes (MEs; or the 1st principal component of the module). MEs were correlated with different biological traits indicating certain AD-specific modules. Multiple comparisons were accounted for by FDR correction across modules, and the P values for the model are reported.
QUANTIFICATION AND STATISTICAL ANALYSIS
MaxQuant for label-free proteome quantification
Data files for the 97 samples were analyzed using MaxQuant v1.5.2.8 with Thermo Foundation 2.0 for RAW file reading capability. The search engine Andromeda was used to build and search a concatenated target-decoy UniProt Knowledgebase (UniProtKB) containing both Swiss-Prot and TrEMBL human reference protein sequences (90,411 target sequences downloaded April 21, 2015), plus 245 contaminant proteins included as a parameter for Andromeda search within MaxQuant (Cox et al., 2011). Methionine oxidation (+15.9949 Da), asparagine and glutamine deamidation (+0.9840 Da), and protein N-terminal acetylation (+42.0106 Da) were variable modifications (up to 5 allowed per peptide); cysteine was assigned a fixed carbamidomethyl modification (+57.0215 Da). Only fully tryptic peptides were considered with up to 2 miscleavages in the database search. A precursor mass tolerance of ±20 ppm was applied prior to mass accuracy calibration and ±4.5 ppm after internal MaxQuant calibration. Other search settings included a maximum peptide mass of 6,000 Da, a minimum peptide length of 6 residues, 0.05 Da tolerance for high resolution MS/MS scans. The false discovery rate (FDR) for peptide spectral matches, proteins, and site decoy fraction were all set to 1 percent. Quantification settings were as follows: re-quantify with a second peak finding attempt after protein identification has completed; match full MS1 peaks between runs; a 0.7 min retention time match window was used after an alignment function was found with a 20 minute retention time search space. The label free quantitation (LFQ) algorithm in MaxQuant (Cox et al., 2014; Luber et al., 2010) was used for protein quantitation. The quantitation method only considered razor and unique peptides for protein level quantitation. The total summed protein intensity was also used to assess overall signal drift across samples prior to LFQ normalization. Retention time internal standards were identified by searching against a small database containing the isotopic peptide standard sequences, keratins, and brain-specific spectrins with fixed modification of +8.014 Da (Lys) and +10.008 Da (Arg) using Proteome Discoverer 1.4. Briefly, precursor mass tolerance was ±10 ppm, fragment ion tolerance was 0.05 Da, and FDR was enforced by Percolator (Yang et al., 2012) at 1%, enabling identification of MS/MS spectra for 14 of the 15 standard peptides. Thermo MSF files were imported as peptide searches into Skyline 3.1 (MacLean et al., 2010) with chromatograms within 2 minutes of a MS/MS match to one of the standard peptides. Identified peak boundaries were visually checked and adjusted as necessary, and precursor (M, M+1, and M+2) isotopic peak areas were exported. These areas were summed and each peak area was divided by the average area of the corresponding peak across all runs to calculate aggregate signal responsiveness within the run.
Differential Expression Analysis
Differentially expressed proteins in the BLSA and Emory cohorts were found using one-way ANOVA followed by Tukey’s comparison post-hoc test in both the PC and FC independently. Three comparisons were considered for BLSA: i) controls vs. AsymAD ii) controls vs. AD and iii) AsymAD vs. AD samples in each brain region (FC and PC). All six pair-wise comparisons were considered for the Emory dataset. Significantly altered proteins with corresponding p-value are provided in Table S2 and Table S6.
Cell Type Enrichment
Cell type enrichment was assessed by cross-referencing module genes with lists of genes known to be preferentially expressed in different cell types. Cell type-specific gene lists from RNA-Seq (Zhang et al., 2014b) and mass spectrometry-based proteomics (Sharma et al., 2015) were filtered as follows: transcript or protein log2(abundance/geometric mean abundance), (log2 value) was required to be 0.8 log2 units in measurement above the next most abundant cell type measurement if the log2 value was greater than 1.4, and was required to be greater than 1.6 log2 units higher if the log2 value for that cell type was less than 1.4. Mouse homologs of the module genes were identified using the mygene R package (Mark et al., 2014). The total list of identified protein groups was used as the background and the cell-type specific gene lists were filtered for presence in the total proteins list prior to cross-referencing. Significance of cell type enrichment was assessed using the one tailed Fisher’s exact test and corrected for multiple comparisons by the FDR (Benjamini-Hochberg) method. These lists are provided in Supplemental Table S4.
Gene Ontology (GO) Enrichment
Functional enrichment of differentially expressed proteins and within the WGCNA modules was determined using the GO-Elite (v1.2.5) python package (Zambon et al., 2012). GO-Elite Hs (human) databases were downloaded on or after June 2016. The set of all proteins identified and considered in the network was used as the background. Z score determines overrepresentation of ontologies in a module and one tailed Fisher’s exact test (Benjamini-Hochberg FDR corrected) was used to assess the significance of the Z score. Z score cut off of 1.96, P value cut off of 0.01 and a minimum of 5 genes per ontology were used as filters prior to pruning the ontologies.
Over-representation analyses for RNA and protein networks
Gene set enrichment analysis was performed using a two-sided Fisher exact test with 95% confidence intervals calculated according to the R function fisher test. We used p values from this two-sided approach to the one-sided test (which is equivalent to the hypergeometric p-value) as we did not a priori assume enrichment (Rivals et al., 2007). To reduce false positives, FDR adjusted p-values were used for multiple hyper-geometric test comparisons. The background for over-representation analysis was chosen as brain region (frontal cortex) expression levels from array data.
GWAS module association
To determine if any protein products of GWAS targets were enriched in a particular module, we used the single nucleotide polymorphism (SNP) summary statistics from the International Genomics of Alzheimer’s Project (http://www.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php) (Lambert et al., 2013) to calculate the gene level association value using MAGMA (de Leeuw et al., 2015). Notably, MAGMA takes the mean of all the transformed SNP P values associated with a particular gene and uses a known approximation of the distribution to get the gene association value. MAGMA accounts for linkage disequilibrium (LD) using reference data with similar ancestry. Gene identifications in the Magma output file were converted to gene symbols using the biological Database Network (https://biodbnet-abcc.ncifcrf.gov/db/db2db.php). These gene lists were further filtered to select for genes that have a MAGMA defined gene association value > 1.3 (−log p value). APOE was added to the gene list and assigned a −log p value of 50, given its known strong association with AD (Coon et al., 2007). For each module in the protein network, the mean GWAS significance value (−log P) was calculated as the enrichment score for the module. Random sampling (10,000 times) of the MAGMA gene list was used to assess the significance of the module enrichment score. The enrichment scores were then scaled by subtracting the mean and dividing by the standard deviation of the random samplings. The P value was calculated as the proportion of samplings that have a scaled enrichment score greater than or equal to the module enrichment score. Similar analyses were performed with GWAS candidates for Schizophrenia (SCZ) and Autism Spectrum Disorders (ASD) (Sullivan, 2010). These GWAS datasets were provided and downloaded from the Psychiatric Genomics Consortium (http://www.med.unc.edu/pgc/downloads). MAGMA significance values for each candidate gene associated with AD, ASD, or SCZ used in these analyses are provided in supplemental TableS10.
Supplementary Material
Highlights.
Protein modules correlated with cognition and Alzheimer’s Disease (AD) pathology
Modules associated with brain cell types overlapped in protein and RNA networks
Many protein-based modules were distinct from those in RNA-directed networks
AD risk loci converged in glial-related modules in the proteome and transcriptome
Acknowledgments
We are grateful to participants in the Baltimore Longitudinal Study of Aging and Emory brain bank donors for their invaluable contribution. We acknowledge Dr. William Hu (Department of Neurology, Emory School of Medicine) and Drs. Giovanni Coppola (Departments of Psychiatry, UCLA) Chris Gaiteri (Rush University) and Sara Mostafavi (University of British Columbia) for helpful comments and discussion.
Funding: Support was provided by the Accelerating Medicine Partnership AD grant (U01AG046161-02), the NINDS Emory Neuroscience Core (P30NS055077), the Johns Hopkins Alzheimer’s Disease Research Center (P50AG05146), and the Emory Alzheimer’s Disease Research Center (P50AG025688). N.T.S. is supported in part by an Alzheimer’s Association (ALZ), Alzheimer’s Research UK (ARUK), The Michael J. Fox Foundation for Parkinson’s Research (MJFF), and the Weston Brain Institute Biomarkers Across Neurodegenerative Diseases grant (11060). J.C.T was also supported by a BrightFocus Foundation (A2015332S). This research was also supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
All raw proteomic data generated contributing to the described work will be deposited electronically the Synapse Web Portal (https://www.synapse.org and doi:10.7303/syn2580853) in accordance with data sharing policies established by the NIH Accelerating Medicine Partnership (AMP) AD consortium. Specific software will also be made available upon request.
Author Contributions:
Conceptualization, N.T.S, E.B.D., A.I.L, J.J.L. and D.H.G; Methodology, D.M.D., E.B.D., and N.T.S.; Investigation, D.M.D., L.Y., T.N., and C.M.H; Formal Analysis, D.N., D.M.D., Q.D., V.S., T.W. and E.B.D., Writing – Original Draft, N.T.S, E.B.D, and A.I.L.; Writing – Review & Editing, N.T.S, E.B.D, J.J.L., V.S., D.H.G. and A.I.L.; Funding Acquisition, A.I.L and N.T.S.; Resources, J.G., J.C.T, M.G. and M.T.; Supervision, D.H.G. N.T.S, and A.I.L.
References
- Abreu RDS, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Molecular bioSystems. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. American Journal of Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, et al. Proceedings of the National Academy of Sciences. 2013. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PloS one. 2010;5:e8918. doi: 10.1371/journal.pone.0008918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Molecular cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. The Journal of clinical psychiatry. 2007;68:613–618. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Molecular & Cellular Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. Journal of proteome research. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS computational biology. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner I, Hales CM, Bishof I, Rabenold L, Duong DM, Yi H, Laur O, Gearing M, Troncoso J, Thambisetty M, et al. Aggregation Properties of the Small Nuclear Ribonucleoprotein U1-70K in Alzheimer Disease. Journal of Biological Chemistry. 2014;289:35296–35313. doi: 10.1074/jbc.M114.562959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I, Resnick SM, Troncoso JC, An Y, O’Brien R, Zonderman AB. Impact of Alzheimer’s pathology on cognitive trajectories in nondemented elderly. Ann Neurol. 2006;60:688–695. doi: 10.1002/ana.21031. [DOI] [PubMed] [Google Scholar]
- Feinstein SC, Wilson L. Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2005;1739:268–279. doi: 10.1016/j.bbadis.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Gelb DJ, Oliver E, Gilman S. DIagnostic criteria for parkinson disease. Archives of Neurology. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- Gray NW, Fourgeaud L, Huang B, Chen J, Cao H, Oswald BJ, Hemar A, McNiven MA. Dynamin 3 is a component of the postsynapse, where it interacts with mGluR5 and Homer. Current biology: CB. 2003;13:510–515. doi: 10.1016/s0960-9822(03)00136-2. [DOI] [PubMed] [Google Scholar]
- Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, et al. Understanding multicellular function and disease with human tissue-specific networks. Nature genetics. 2015;47:569–576. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodes RJ, Buckholtz N. Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opinion on Therapeutic Targets. 2016;20:389–391. doi: 10.1517/14728222.2016.1135132. [DOI] [PubMed] [Google Scholar]
- Hu YB, Dammer EB, Ren RJ, Wang G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Translational neurodegeneration. 2015;4:18. doi: 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Zhang B, Wang Z, Joehanes R, Zhu J, Johnson AD, Ying S, Munson PJ, Raghavachari N, Wang R, et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1427–1434. doi: 10.1161/ATVBAHA.112.300112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince PG, Ince PG, Lowe J, Shaw PJ. Review. Neuropathology and Applied Neurobiology. 1998;24:104–117. doi: 10.1046/j.1365-2990.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R, et al. Genetic Evidence Implicates the Immune System and Cholesterol Metabolism in the Aetiology of Alzheimer’s Disease. PloS one. 2010;5:e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpievitch YV, Dabney AR, Smith RD. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinformatics. 2012;13:S5–S5. doi: 10.1186/1471-2105-13-S16-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami S, Muramoto K, Ichikawa M, Kuroda Y. Localization of microtubule-associated protein (MAP) 1B in the postsynaptic densities of the rat cerebral cortex. Cellular and molecular neurobiology. 2003;23:887–894. doi: 10.1023/B:CEMN.0000005317.79634.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature genetics. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559–559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Khanna A, Li N, Wang E. Circulatory miR34a as an RNAbased, noninvasive biomarker for brain aging. Aging (Albany NY) 2011;3:985–1002. doi: 10.18632/aging.100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, O’Keeffe M, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark A, Thompson R, Wu C. MyGene R Package 2014 [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Analytical chemistry. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Medicine. 2013;5:48–48. doi: 10.1186/gm452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mosesson MW. Fibrinogen and fibrin structure and functions. Journal of Thrombosis and Haemostasis. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- Narayanan M, Huynh JL, Wang K, Yang X, Yoo S, McElwee J, Zhang B, Zhang C, Lamb JR, Xie T, et al. Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Molecular Systems Biology. 2014;10:743–743. doi: 10.15252/msb.20145304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Resnick SM, Zonderman AB, Ferrucci L, Crain BJ, Pletnikova O, Rudow G, Iacono D, Riudavets MA, Driscoll I, et al. Neuropathologic Studies of the Baltimore Longitudinal Study of Aging (BLSA) Journal of Alzheimer’s disease: JAD. 2009;18:665–675. doi: 10.3233/JAD-2009-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC. Transcriptomics: From Differential Expression to Coexpression. In: Coppola G, editor. The OMICs: Applications in Neuroscience. New York: Oxford University Press; 2014. pp. 85–113. [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008a;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nature neuroscience. 2008b;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16:441–458. doi: 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O’Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, et al. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivals I, Personnaz L, Taing L, Potier MC. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG. A leaky blood–brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. Journal of Cellular and Molecular Medicine. 2009;13:2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, Vagena E, Bedard C, Machado MR, Coronado PER, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun. 2015;6 doi: 10.1038/ncomms9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015 doi: 10.1038/nn.4160. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF. The Psychiatric GWAS Consortium: Big Science Comes to Psychiatry. Neuron. 2010;68:182–186. doi: 10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Wang Z, Hortobagyi T, Witten JT, Zarnack K, Kayikci M, Clark TA, Schweitzer AC, Rot G, Curk T, et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome research. 2011;21:1572–1582. doi: 10.1101/gr.122226.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran LM, Zhang B, Zhang Z, Zhang C, Xie T, Lamb JR, Dai H, Schadt EE, Zhu J. Inferring causal genomic alterations in breast cancer using gene expression data. BMC systems biology. 2011;5:121. doi: 10.1186/1752-0509-5-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Ma J, Wang P, Zhu Y, Zhou BB, Yang YH. Improving X!Tandem on peptide identification from mass spectrometry by self-boosted Percolator. IEEE/ACM transactions on computational biology and bioinformatics/IEEE, ACM. 2012;9:1273–1280. doi: 10.1109/TCBB.2012.86. [DOI] [PubMed] [Google Scholar]
- Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, Conklin BR, Pico AR, Salomonis N. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics. 2012;28:2209–2210. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014a;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014b;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.