

Abstract
Emerging evidence suggest that the heterogeneity of cancer limits the efficacy of immunotherapy. To search for optimal therapeutic targets for enhancing the efficacy, we used whole‐exome sequencing data of 23 early cervical tumors from Chinese women to investigate the hierarchical structure of the somatic mutations and the neo‐epitopes. The putative neo‐epitopes were predicted based on the mutant peptides’ strong binding with major histocompatibility complex class I molecules. We found that each tumor carried an average of 117 mutations and 61 putative neo‐epitopes. Each patient displayed a unique phylogenetic tree in which almost all subclones harbored neo‐epitopes, highlighting the importance of individual neo‐epitope tree in determination of immunotherapeutic targets. The alterations in FBXW7 and PIK3CA, or other members of the significantly altered ubiquitin‐mediated proteolysis and extracellular matrix receptor interaction related pathways, were proposed as the earliest changes triggering the malignant progression. The neo‐epitopes involved in these pathways, and located at the top of the hierarchy tree, might become the optimal candidates for therapeutic targets, possessing the potential to mediate T‐cell killing of the descendant cells. These findings expanded our understanding in early stage of cervical carcinogenesis and offered an important approach to assist optimizing the immunotherapeutic target selection.
Keywords: Cervical cancer, immunotherapeutic target, neo‐epitope, phylogenetic tree
Introduction
Immunotherapy is emerging as the most promising type of cancer treatment, as evidenced by recent clinical trials in which durable remission, and even cure, have been demonstrated in some patients. However, its success is limited because only a small proportion of patients respond to the therapies, whereas most remain resistant and are either unresponsive or responsive only transiently 1, 2. A growing body of evidence suggests that cancer heterogeneity is the bottleneck that limits the efficacy of immunotherapy 3. Although derived from a single initiated cell, almost all cancers comprise multiple subclones and evolve constantly, driven by mechanisms such as genome instability and Darwinian selection. Some recent cancer genome studies have revealed that subclones of a cancer comprise different compositions of genomic alterations 4, 5. It is conceivable that some of these alterations have become the determinants of whether the subclone responds to or resists current immunotherapies, including immune checkpoint inhibitors, tumor infiltration lymphocytes, chimeric antigen receptor‐modified T cells, and bispecific antibodies. Each of these immunotherapy strategies targets only one or a few subpopulations and allows the others, especially the metastatic ones, to thrive continuously 6. It is also conceivable that these genetic variabilities have become the bottleneck that limits clinical efficacy; many patients either have no response or have an incomplete response in which the cancer shrinks or even disappears but eventually recurs despite continuing treatment.
It has become apparent that the development of technology to target all of a tumor's subclones is necessary to remove the bottleneck and bring cancer immunotherapy to a new level. We recently proposed the construction of a “cancer epitope tree” to achieve this goal 3 because, although the cancer genome is highly variable both spatially and temporally, the technology is available to determine the subclonal hierarchical structure, the so‐called phylogenetic tree, to outline the temporal relationship among the genomic alterations 7, even at the single‐cell level 8. In combination with the technique to predict the neo‐epitopes created by these genomic alterations, a “cancer neo‐epitope tree” can be constructed to guide the systematic search for the optimal therapeutic targets located at the “trunk” or “major branch” that possess the potential to mediate T‐cell killing of all or most of the cancer cells 3. With few exceptions 9, most cancer phylogenetic trees were constructed with driver mutations at a cohort level. Passenger mutations outnumber driver mutations by up to 2000 times 4, 10, 11; many of them are target candidates and could play an important role in causing cancer cell death by cytotoxic T lymphocytes and antibody‐dependent cell‐mediated cytotoxicity. Therefore, for purposes of immunotherapy, it is important to include neo‐mutated epitopes derived from both driver and passenger mutations. Furthermore, an accumulating body of evidence suggests that each cancer is unique in its composition of genetic alterations and hierarchical structure of subclones 7, 9. Therefore, a “cancer epitope tree” of an individual cancer will be more useful than the one at cohort level for determination of immunotherapeutic targets.
In this article, we report the results from the first attempt to construct the individual “cancer epitope tree” of 23 early cervical cancers in Chinese women by exploring their exome sequencing data. Cervical cancer is the most lethal cancer in women worldwide, with an estimated 528,000 new cases and 266,000 deaths in 2012 12. Persistent infection with high‐risk human papillomavirus (HPV) subtypes, such as HPV 16 and 18, has been found in a majority of patients with cervical cancer. Although multiple studies have characterized the mutation landscape of cervical cancer 13, 14, the molecular events responsible for malignant transformation remain elusive. Because the 23 cervical cancers in this study were at very early stages (17 stage I ([T1, N0, M0]: the cancer has grown into (invaded) the cervix, but it is not growing outside the uterus. The cancer has not spread to nearby lymph nodes (N0) or distant sites (M0)) and 6 stage II ([T2, N0, M0]: the cancer has grown beyond the cervix and uterus, but has not spread to the walls of the pelvis or the lower part of the vagina), according to the International Federation of Gynecology and Obstetrics [FIGO] staging system 15), they allowed us to study the early events in cervical carcinogenesis while defining the optimal immunotherapeutic targets. We found that the samples from 73.9% of our patients with early‐stage cervical cancer carried a few integrated HPV sequences. The ubiquitin proteolysis and extracellular matrix (ECM) receptor pathways were significantly altered; 69.6% of cancers carried alterations in these two pathways. We propose that alterations in genes FBXW7 and PIK3CA are more likely to serve as the early genomic mutations that cause the progression of HPV‐induced precancerous cells toward invasive malignancy. Furthermore, we used the identified somatic mutations to predict the neo‐epitopes on the basis of their mutated peptides’ binding affinities with major histocompatibility complex class I molecules (MHC‐I). Using this information, we constructed the phylogenetic tree and the “cancer epitope tree” for individual tumors. We found that the mutations of individual tumors displayed a unique path of evolution, highlighting its importance in the search for therapeutic targets. HPV proteins might serve as immunotherapy targets in tumors that carry the integrated virus genome without active HPV infection. However, for tumors that do not express these proteins, our approach will suggest desirable therapeutic target candidates. The results of this study expanded our understanding of the early stages of cervical carcinogenesis and, more importantly, offered a useful systematic strategy with which to search for the optimal immunotherapeutic targets, which has important implications for cancer diagnosis, prevention, and therapy.
Materials and Methods
Sample collection and preparation
Twenty‐three pairs of cervical cancer tumors and matched normal tissues were obtained from the Southwest Hospital of Chongqing Autonomous Municipality in China. The study protocol was approved by the Institutional Review Board of Southwest Hospital, and all experiments were performed in accordance with the guidelines and regulations. Informed consent was obtained from each subject. Tumors and peripheral blood samples were collected from patients S1‐S20, who each underwent surgical resection. For patients S21, S22, and S23, adjacent tissues were used as the control samples. The surgically resected tumors were snap frozen in liquid nitrogen and stored at −80°C. The blood samples were stored at −20°C. DNA was extracted from the frozen tissues and peripheral blood lymphocytes using commercial kits (TIANamp Blood DNA Kits and Genomic DNA Kits, Tiangen Biotech) and following the manufacturer′s instructions. HPV genotyping was performed using the polymerase chain reaction (PCR)‐based mass spectrometry system 16.
Whole‐exome sequencing
DNA from matched tumor and control samples were fragmented with an ultrasonicator UCD‐200 (Diagenode). These fragments were purified and size selected with Ampure Beads (Beckman, Beverly, MA) following three enzymatic steps (end repairing, the addition of an “A” base, and adapter ligation) according to Illumina's instructions. NimbleGen EZ 64M human exome array probes (SeqCap EZ Human Exome Library v3.0) were used in hybridization. Each captured library was then pair‐end sequenced in 100‐bp lengths with an Illumina HiSeq 2000 following the manufacturer's instructions. The raw data are available from the corresponding authors upon reasonable request and with permission of Beijing Genomics Institute.
Read mapping and somatic mutation detection
Raw whole‐exome sequencing reads were aligned to the reference human genome (hg19) using a BWA aligner (v 0.7.10) 17 with default parameters. Alignments were sorted and converted into BAM format. Picard (v1.119) (http://picard.sourceforge.net/) was used to mark possible PCR duplicates in the BAM file, and the Genome Analysis Toolkit (v3.2.2) 18 was used to improve alignment accuracy. Somatic point mutations were detected with MuTect (v1.1.4) 19. Variants from the 1000 Genome Project (Phase 3) 20, the NHLBI GO Exome Sequencing Project (version 2) 21, which represented variants from more than 200,000 individuals, and the Exome Aggregation Consortium (version 0.2) 22, which spanned variants from 60,706 unrelated individuals (with a minor allele frequency threshold of 0.1), were removed from the somatic mutations. Variants were annotated for effects on transcripts using the variant effector predictor tool 23.
Validation of somatic mutations
We validated a subset of recurrent mutations together with some randomly selected mutations by either mass spectrum or Sanger sequencing. Specific primers were designed for PCR amplification and base extension that covered the mutation sites. Genotyping assay and base calling procedures were performed on the MassArray platform of Sequenom by determining their genotypes in the tumors and matched samples. The PCR amplification products were sequenced with a 3730xl DNA Analyzer (Applied Biosystems). All sequences were analyzed with Sequencing Analysis Software Version 5.2 (Applied Biosystems, Foster City, CA).
HPV genome alignment
The reads that could not be mapped to the human reference genome were extracted and realigned to a database of multiple HPV reference genomes. HPV reference genomes were obtained from the Human Papilloma Virus Episteme (pave.niaid.nih.gov) 24. With the paired‐end read information, we determined whether the HPV genome could integrate into the human genome by screening pairs of reads with one end mapped to the human genome and the other end mapped to the HPV genome.
Pathway analysis
The KEGG pathways were obtained from the Molecular Signatures Database (MSigDB) 25, and the gene set was downloaded from http://www.broadinstitute.org/gsea/downloads.jsp (accessed 19 June 2015). The mutated genes in each tumor were compared with the KEGG pathway to determine whether the tumor had altered pathways.
For each pathway, we randomly sampled the same number of genes from all genes in the human genome without replacement. We then counted the number of tumors that harbored mutation in this random gene set. We performed 10,000 such random samplings for each pathway and calculated the P‐value as the proportion of random samples in which more patients carried mutations than the number of tumors that used the original pathway. The false discovery rate was then calculated for each pathway using the Benjamini and Hochberg method. The significantly enriched pathway was considered if the adjusted P < 0.1.
Phylogenetic inference
The evolutionary history of each of the 23 tumors was constructed on the basis of the somatic mutations’ reads count using PhyloSub 26. This approach made use of Bayesian inference and Markov chain Monte Carlo sampling (with 2500 samplings) to estimate the number of clonal lineages and their ancestry. We only considered trees with the highest likelihood.
We downloaded 194 cervical cancers’ mutation data and the RNA‐sequencing data from the International Cancer Genome Consortium (ICGC) data portal (https://dcc.icgc.org). For these mutations, we did one same filtration using the variants from the 1000 Genome Project, the NHLBI GO Exome Sequencing Project, and the Exome Aggregation Consortium. We then merged all the mutations into one matrix of genes versus tumor samples with 0/1 entries indicating the absence/presence status of a mutation in a gene for each sample. Based on this matrix, we used the BML 27 tool to infer the sequence of gene mutations for the 194 cervical cancer data.
Immunogenic variants prediction
For each somatic missense mutation, we obtained the corresponding mutated amino acid and one peptide centred on the mutated residue, flanked on each side by eight amino acids from the protein sequence. We also obtained the corresponding normal 17 amino acid peptide. We then used the NETMHC‐3.4 algorithm 28 to predict the binding affinity for the peptide with MHC‐I. The variant showed immunogenicity only if the mutated peptide showed strong binding affinity with MHC‐I (affinity < 50) and the normal peptide had no binding affinity (affinity > 500) at the same peptide position.
Gene expression analysis
The normalized read counts from the ICGC donors’ RNA‐sequencing data from the 194 cervical cancer tissues, with the log transformation, were calculated as the gene expression values. Gene set enrichment analysis (GSEA) 25, 29 using the KEGG pathway, the canonical pathway, and the biological process gene set was performed separately to identify pathways that have significantly altered expression levels in carriers of the FBXW7 or PIK3CA mutants. The significantly enriched pathway was considered if the q < 0.05.
The difference in FBXW7 and PIK3CA was tested using two‐sided Student's t test. Pearson correlation was used to assess the correlation between FBXW7 and PIK3CA gene expression. All analyses were performed with the R Version 3.1.1 statistical software (R Core Team, Vienna, Austria). A two‐sided P < 0.05 was considered to indicate statistical significance.
Results
General data
Our study included specimens of 23 patients who received surgical treatment upon diagnosis of early‐stage cervical cancer (17 stage I and 6 stage II, according to the FIGO staging system; Table 1) in the Southwest Hospital of Chongqing Autonomous Municipality in China. We performed whole‐exome sequencing of 242,232 exons, with a length of 63.8 megabases, at an average coverage of 181X (Fig. S1). Peripheral blood samples from most patients were used as germline controls, with the exception of patients S21, S22, and S23, for whom adjacent tissues were used instead. We detected HPV sequences in all but two tumor samples and found only a small number of integrated sequences in 17 exome‐captured sequencing datasets (Table S1). We used MuTect and Indelocator 19, 30 to call each case's somatic mutations by filtering out germline events from the corresponding normal sample. We also filtered out variants in the 1000 Genome Project (Phase 3) 20, the NHLBI GO Exome Sequencing Project (version 2) 21, and the Exome Aggregation Consortium (version 0.2) 22 by applying a minor allele frequency threshold of 0.1 to all three databases. The final cleaned dataset includes 2691 somatic mutations, including 730 synonymous substitutions, 1934 nonsynonymous substitutions, 18 deletions, and 9 insertions across 23 sample pairs. A subset of 59 somatic mutations was selected for validation, and 57 variants (96.6%) were validated using mass spectrum or Sanger sequencing (Table S2). The number of nonsynonymous mutations showed no correlation with the patients’ age or clinical stage (Fig. S2).
Table 1.
Clinical stage and human papillomavirus (HPV) infection status of 23 patients with cervical cancer
| Tumor sample code | Age (years) | Clinical stage | HPV genotyping |
|---|---|---|---|
| S1 | 54 | Ib1 | HPV16 |
| S2 | 46 | Ib1 | HPV16 |
| S3 | 44 | Ib2 | HPV16 |
| S4 | 49 | Ib1 | HPV16 |
| S5 | 43 | Ib1 | HPV16 |
| S6 | 39 | Ib1 | HPV16 |
| S7 | 42 | Ib1 | HPV16 |
| S8 | 38 | Ib1 | HPV33 |
| S9 | 48 | Ib1 | HPV16 |
| S10 | 50 | IIa1 | HPV16 |
| S11 | 46 | Ib1 | HPV16 |
| S12 | 44 | Ib1 | HPV16 |
| S13 | 41 | Ib1 | HPV16 |
| S14 | 56 | Ib1 | HPV18 |
| S15 | 48 | Ib1 | HPV16 |
| S16 | 44 | Ib1 | HPV18 |
| S17 | 44 | IIa1 | HPV18 |
| S18 | 37 | Ib1 | HPV16 |
| S19 | 35 | Ib1 | HPV16 |
| S20 | 49 | IIa1 | HPV16 |
| S21 | 59 | IIa1 | HPV16 |
| S22 | 46 | IIb | HPV16 |
| S23 | 63 | IIa1 | HPV16 |
Frequency of mutations in cervical cancer
We first estimated the distribution of the somatic mutations and their nucleotide substitutions. C/T and G/A substitutions were the most frequent among the 23 patients (Fig. 1B), with mean frequencies of 21.8% and 21%, respectively. This observation, especially the C/T substitution pattern, agrees with findings from a previous study of 115 Norwegian and Mexican cervical cancer samples 13. We found that PIK3CA (17.4%), SYNE1 (17.4%), FBXW7 (17.4%), and MUC16 (21.7%) were among the most frequently mutated genes (Fig. 1A), which again agrees with the findings of previous studies: the Norwegian and Mexican study 13 showed that EP300 (16%), FBXW7 (15%), and PIK3CA(14%) harbored recurrent mutations; a study 31 in 80 cervical cancer samples from Boston showed that PIK3CA (31.3%), KRAS (8.8%), and EGFR (3.8%) had the highest mutation rates; and another study in 15 cervical cancer patients from Hong Kong revealed frequent alteration of FAT1 (33.3%), ARID1A (33.3%), ERBB2 (26.7%), and PIK3CA (53.3%) 32. We obtained 194 cervical cancer whole‐exome sequencing mutation datasets from ICGC. After applying the same filtering, we also noticed PIK3CA(27.3%) and FBXW7(10.3%) (Fig. S3). Despite the ethnic and geographic differences, alterations in PIK3CA, followed by FBXW7, were the most common mutations in the various cervical cancer studies.
Figure 1.
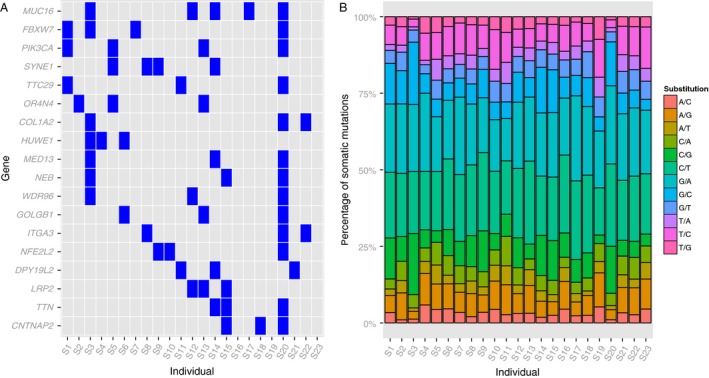
Distribution of mutated genes and base substitution patterns in the 23 patients with cervical cancer. (A) Distribution of mutated genes in at least three patients (mutation frequency > 13%). Each column represents one individual, and each row is a gene. (B) Distribution of base substitution patterns for all somatic mutations.
Prediction of neo‐epitopes
The mutated peptides derived from those mutations, if presented on MHC‐I, could potentially be immunogenic as the adaptive immune system should recognize them as “non‐self” neo‐antigens. Recent neo‐antigen prediction approach by estimating the binding affinity between the mutated peptide and MHC‐I can be used to identify therapeutic targets for immunotherapy 33, 34, 35, 36. Using the same approach (see Methods), of the 1934 nonsynonymous substitutions, we found 1405 missense mutations’ peptides exhibiting strong binding affinity with MHC‐I (Fig. 2), which suggests that tumor progression could generate antigens that may recruit immunologic cells to attack the tumor cells.
Figure 2.
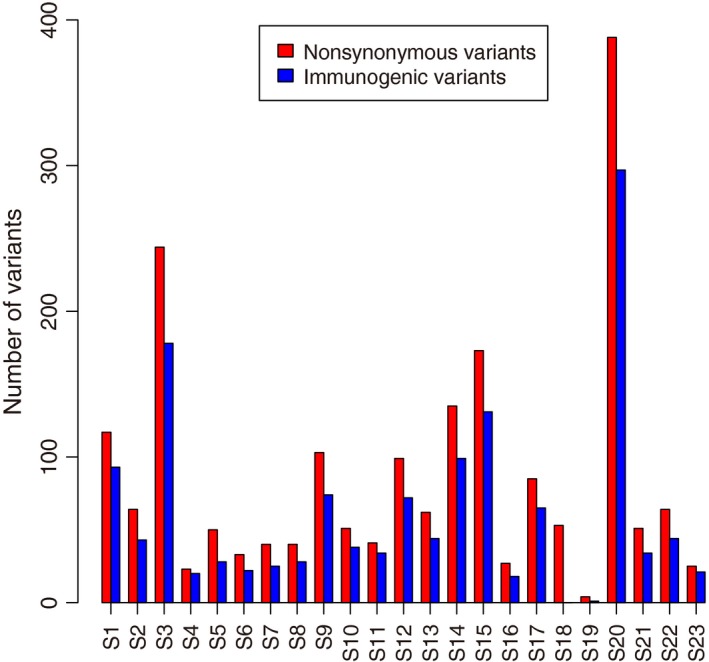
Number of immunogenic variants in 23 patients. The numbers of total nonsynonymous mutations (red) and immunogenic variants (blue) are shown for each patient. The immunogenic variants were predicted only if the mutated peptide showed strong binding affinity with MHC‐I (affinity < 50) and the normal peptide had no binding affinity (affinity > 500) at the same peptide position.
Alteration in ubiquitin‐mediated proteolysis and ECM receptor interaction pathways
To determine whether any biological functions were significantly altered, we integrated all of the nonsynonymous mutations from the 23 patients and determined the pathways that were enriched. In permutation tests of 10,000 samples among the 177 mutated pathways, the ubiquitin‐mediated proteolysis and ECM receptor interaction pathways were the most significantly altered, with false discovery rates of <0.1 (Table 2). All of the mutated genes involved in these two pathways are shown in Figure S4.
Table 2.
Top 10 altered pathways
| KEGG Pathway | Number of patients altered | P value | FDR |
|---|---|---|---|
| UBIQUITIN‐MEDIATED_PROTEOLYSIS | 15 | 0.0014 | 0.072 |
| ECM_RECEPTOR_INTERACTION | 11 | 0.0016 | 0.072 |
| INSULIN_SIGNALING_PATHWAY | 14 | 0.0028 | 0.1008 |
| HYPERTROPHIC_CARDIOMYOPATHY_HCM | 9 | 0.0036 | 0.108 |
| FOCAL_ADHESION | 14 | 0.0044 | 0.1131 |
| DILATED_CARDIOMYOPATHY | 9 | 0.0065 | 0.14625 |
| ALDOSTERONE_REGULATED_SODIUM_REABSORPTION | 7 | 0.0075 | 0.15 |
| MTOR_SIGNALING_PATHWAY | 8 | 0.0124 | 0.2094 |
| SMALL‐CELL_LUNG_CANCER | 10 | 0.0128 | 0.2094 |
| INOSITOL_PHOSPHATE_METABOLISM | 9 | 0.0146 | 0.219 |
The ubiquitin‐mediated proteolysis pathway mediates protein degradation via the ubiquitin conjugation and proteasome system. It is reported to be the most frequently altered pathway in clear cell renal cell carcinomas and a contributor to the tumorigenesis 37. In our cervical cancer data, the mutated genes involved in this pathway included FBXW7 (altered in 17%), HUWE1 (altered in 13%), and BIRC6 (altered in 9%). It has been suggested that mutations in FBXW7 cause increased genetic instability because several prominent oncogenes (Notch, c‐Myc, JunB, and mTOR) are its substrates 38, 39. In cervical cancer, the ubiquitin‐mediated proteolysis pathway can be best characterized by high‐risk HPV‐16 E6 binding activity to the tumor‐suppressor protein p53 to induce ubiquitylation and proteasomal degradation 40, 41, and the abrogation of p53 allows the accumulation of genetic mutations that would normally have been repaired. The HPV‐18 E7 oncoprotein also targets the tumor‐suppressor Rb proteins for proteasomal degradation via the ubiquitin‐dependent pathway 42. Although the mTOR signaling pathway is not the most significant, it is among the top 10 altered pathways (with PIK3CA altered in 17% of patients). Thus, the alterations in genes involved in the ubiquitin‐mediated proteolysis pathway may trigger a cascade of reactions that lead to malignancy.
We found that, in the ECM receptor interaction pathway, genetic alterations mainly occurred in COL1A2 (altered in 13%) and ITGA3 (altered in 13%). These alterations may disrupt the signaling transfer function during interactions with extracellular proteins, leading to malfunction in cellular activities such as adhesion, migration, differentiation, proliferation, and apoptosis. This pathway was also involved in the focal adhesion pathway comprising mutations in PIK3CA (altered in 17%), COL1A2 (13%), and ITGA3 (13%). HPV‐positive cells have been found to express high levels of focal adhesion kinase, which regulates the interaction between the signal transduction of ECM and integrins 43. The virus oncoprotein HPV‐16 E6 also binds to the ECM protein, leading to cytoskeletal reorganization and formation of focal adhesions 44. This interaction, in combination with deregulation of focal adhesion kinase, promotes resistance to anoikis and allows the HPV‐infected cells to proliferate in the absence of adherence to the ECM, that is, anchorage‐independent growth 45. Thus, the altered genes in these pathways may allow cells to escape anoikis and play a role in transformation and tumor invasion.
The cancer neo‐epitope trees
Tumors usually contain multiple genetically diverse clones or subclones that have constantly evolved from an earlier population through expansion and selection 4, 46, 47. Outlining the evolutionary history of these mutations will aid in understanding the cancer development and guide design of therapy targets 48. We therefore constructed phylogenetic trees for each tumor using nonsynonymous substitutions (Fig. 3, Table S3) and named the clones in chronical order as the ancestor, descendant, and later subclones. Consistent with their early stage of malignancy, the subclonal hierarchy structures of all tumors were simple. Five tumors (S4, S9, S11, S12, and S21) harbored only one ancestor subclone, and no descendants were observed. The evolutionary paths in the other tumors showed either linear (S3, S7, S8, S19, and S20) or branching (the remaining 13) patterns. Five of the 13 tumors with branching paths (S6, S13, S15, S16, S23) had two ancestor subclones (S6 and S15 derived one descendant subclone from one of the two ancestors), and the other eight carried only one ancestor subclone with multiple descendants or later subclones (Fig. 3).
Figure 3.
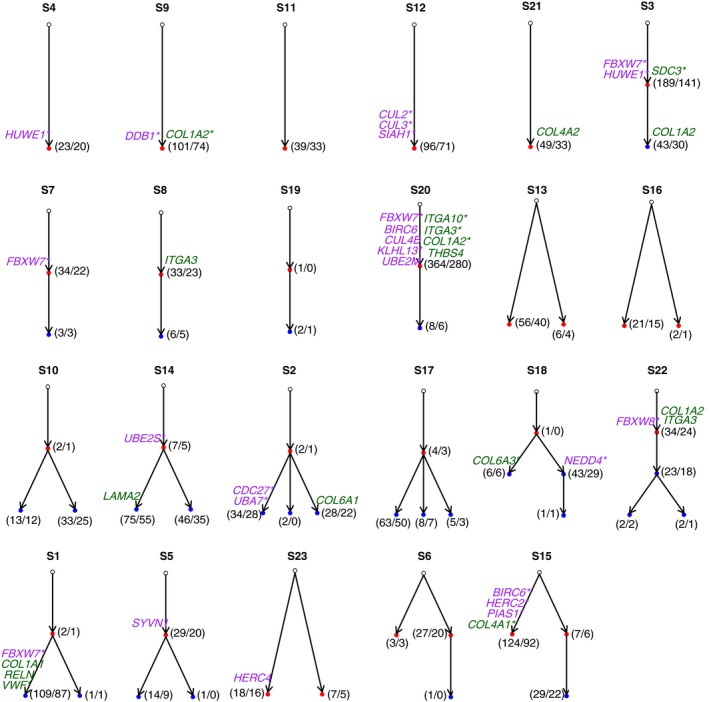
The clonal structures and phylogenic relationships for all 23 patients. In each patient, a phylogenic tree was constructed using somatic mutations. Each node represents one clone. Each clone harbors multiple mutations, and only the genes involved in the ubiquitin‐mediated proteolysis (purple) and ECM receptor interaction (dark green) pathways are labeled on the corresponding node. Normal clones (non‐pathogenic) are in open circles, early clones are shown in red, and later clones are shown in blue. Arrows point from the parent node to the child node (i.e., the descendant clone derived from the ancestor clone). Asterisks indicate that the gene harbors putative neo‐epitopes. The numbers in parentheses indicate the number of total mutated genes (before the slash) and the number of genes that harbored putative neo‐epitopes in the clone.
Each tumor displayed accumulation of different mutations and evolutionary paths over time, suggesting heterogeneity between patients. Thus, for therapeutic considerations, the individual phylogenetic tree should offer clues for the selection of therapy targets. The number of altered genes in each subclone and the number of genes that harbored neo‐epitopes are shown in Figure 3. Each tumor carried an average of 117 mutations and 67 antigenic targets. All tumors but S19, which only harbored three mutations, had subclones that harbored neo‐epitopes, which makes immunotherapy a feasible approach. An individualized “cancer epitope tree” could be constructed using neo‐epitopes. Selection of targets in the ancestor subclones would inhibit the majority of the tumor cells because the descendants are derived from the ancestor subclone. We defined the ancestor subclones as the “trunk” and the descendant subclones as the “major branches” in the phylogenic tree. Among the many neo‐epitopes in the trunk and major branches, one possibility to choose functional mutations or to scale down the mutation is to choose the genes involved in important pathways. We therefore list in Figure 3 the 34 altered genes involved in the ubiquitin‐mediated proteolysis and ECM receptor interaction pathways in these trees, together with the number of neo‐epitopes.
Alteration of FBXW7 and PIK3CA
Mutations of both passenger and driver genes occur during a lesion's transition from precancerous to malignant. Among the approximately 20,000 protein‐coding genes in the human genome, only 138 genes were reported in a previous study as driver genes 49, which play a significant role in tumorigenesis. We found that 24 of the 138 proposed driver genes were mutated in our 23 tumors (Fig. 4). In addition to FBXW7 (17%) and PIK3CA (17%), which were the most frequent, NFE2L2 (13%) and CREBBP (9%) were also frequently mutated. NFE2L2 participates in protein processing and amino acid metabolism and was recently identified in a recent cervical cancer study 13. Interestingly, in tumor S2, two driver genes, GATA2 and STK11, were located in the ancestor subclone, and STK11 also harbors neo‐epitope (Table S3). It is possible that the two ancestral mutated driver genes granted a selective growth advantage to allow the cancer cells to derive more descendants in S2, as we observed. In the S2‐specific “epitope tree,” STK11, which is part of the mTOR signaling pathway, may be a “trunk” target candidate.
Figure 4.
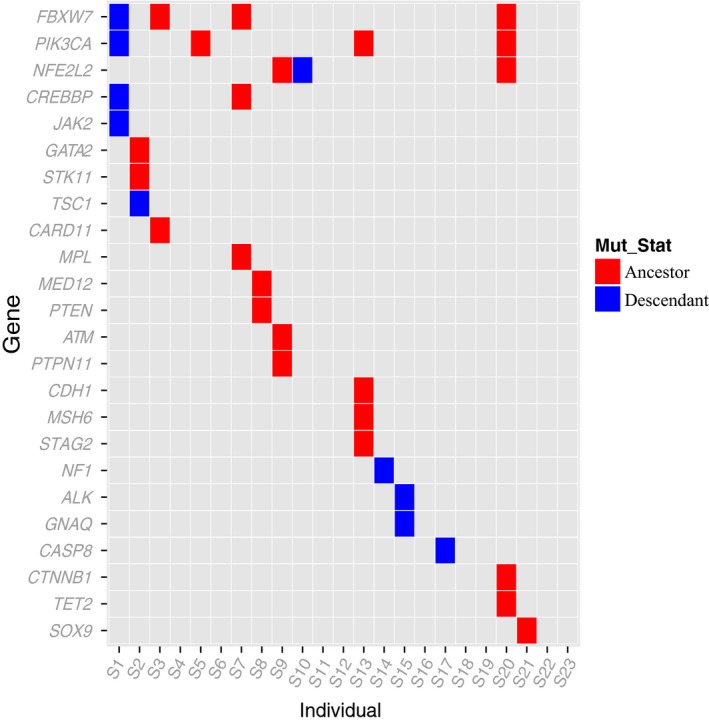
Distribution of the reported driver genes in our 23 patients. Driver genes were obtained from the literature 49. The ancestral (red) or descendant (blue) status for each gene mutation in each patient was labeled.
Overall, FBXW7 and PIK3CA seem to play more important roles in these early‐stage tumors. Both showed mutations in four patients. In the individual phylogenetic trees, both genes were located on the ancestor subclones in three tumors, which suggests they were likely early events during tumorigenesis. Due to the limited number of samples available in our study, we made one extension study on the mutations of 194 ICGC cervical cancer donors which also represented the cervical cancer's pathogenesis. We inferred the likely sequence of mutation for these patients. The results supported the symptom that FBXW7 and PIK3CA mutations were more likely to be the second mutation event starting at the normal status, with probability 0.86 and 0.97 separately (Fig. S5). We propose that alterations in FBXW7 and PIK3CA are likely the early changes that trigger the progression of the HPV‐induced precancerous cells toward invasive malignancy.
Using the genome‐wide gene expression from ICGC donors’ RNA‐sequencing data, we performed GSEA between the mutant and wild‐type for these two genes. No pathway or biological process was found to significantly differ in carriers of the mutants (Tables S4, S5). We further compared the expression levels of FBXW7 and PIK3CA between carriers of the mutants and the wild‐types. We found the mutated FBXW7′s expression was significantly increased (Student's t test, P: 0.006231; Fig. S6A) while PIK3CA generally showed no changes (Student's t test, P: 0.1293; Fig. S6B). When comparing PIK3CA expression in FBXW7 mutant carriers and wild‐type, we found FBXW7 mutants were marginally associated with PIK3CA expression levels (Student's t test, P: 0.02241; Fig. S6C), and FBXW7′s expression displayed no changes in PIK3CA mutant and wild‐type cases (Student's t test, P: 0.4807; Fig. S6D). The expression of these two genes was significantly correlated (Fig. S7). Further investigations with a larger sample size and in relevant tissue and cell lines are needed to reach a convincing conclusion.
Discussion
Cervical cancer is among the few malignancies that allows convenient study from morphologic, cytological, and molecular events during the formation of precancerous lesions and their transition to invasive cancers. Consequently, timely diagnosis and treatment of early‐stage cervical cancer is possible. The whole‐exome sequencing data in this study were obtained from 23 patients with early‐stage cervical cancer (FIGO stage I or II) to allow identification of early genomic events without the complication of late‐stage genomic alterations. Infection of high‐risk HPV is a prerequisite for cervical cancer, and integration of the viral genome occurs throughout the course of carcinogenesis. Twenty of the 23 patients had infection with high‐risk HPV, and integrated HPV sequences were detected in 17 cases, albeit at relatively low sequences covered. A total of 2691 genomic alterations, mostly single nucleotide substitutions, were identified, of which 1405 were predicted to encode neo‐epitopes on the basis of their strong binding affinity with MHC‐I. Each cancer carried an average of 117 nonsynonymous somatic mutations and 61 predictive neo‐epitopes. To outline the phylogenetic relationship among these somatic mutations, we constructed the subclonal hierarchical structures of individual tumors and named the identified cancer cell populations in temporal order as ancestor, descendant, and late subclones. We found that five patients carried only the ancestor subclone, 16 carried an additional descendant, and only two had all three subclones. Furthermore, we found that 17% of the tumors had mutations in PIK3CA and FBXW7 without mutation of typical driver genes, such as KRAS, TP53, and EGFR, as reported in other cervical cancer genome studies 13, 31; these were not found in our study, which suggests that they may be the later‐stage events.
It has been well documented that HPV viral oncoproteins E6 and E7 can induce precancerous lesions and that additional genetic alterations are required for malignant transformation 50. Through our exome sequencing data, although we observed multiple HPV‐related sequences, we detected only a small number of HPV integrated sequences, while in some patients there was no observation. Currently, we do not have enough evidence that either whether the whole HPV genome is integrated with the host cell or a part of them is integrated by the exome capture technique. The low HPV integrated sequences may confirm that most of the cervical cancers in this study were at a very early stage of malignancy. Therefore, the somatic mutations in our samples provide more information that they must have been capable of triggering the transition from benign to invasive lesions. We here highlighted the early somatic mutations for critical targets selection. Based on our analyses, we propose that mutation of FBXW7 and PIK3CA and other members in these two pathways were among the earliest alterations that triggered malignant transformation. This hypothesis is consistent with earlier studies in a large number of cancer types in which mutation of FBXW7 and PIK3CA was a frequent event, including cancers of the colon, brain, gastrointestinal system 51, 52, cervix 32, head and neck 53, and breast 54. This hypothesis is further supported by recent evidence that mutated PIK3CA initiates breast cancer by triggering multiple key events during the cancer initiation stage 55.
Our findings will be very useful in guiding cancer immunotherapy. A growing body of evidence suggests that cancer heterogeneity is the bottleneck that limits the efficacy of cancer immunotherapy 6, 56. Most of the current immunotherapeutic technologies, including tumor infiltration lymphocytes, immune checkpoint inhibitors, chimeric antigen receptor‐modified T cells, and bispecific antibodies, kill only one or a few subclones in a cancer and allow the others to continue to grow. In our study, we found that each of the 23 cervical tumors had a unique subclonal hierarchical structure that comprised a different composition of genetic alterations and predicted neo‐epitopes. Therefore, the “cancer neo‐epitope tree” of each tumor is critical to help determine the optimal targets at the trunk or major branch shared by all descendant cells that have the potential to lead to a cure.
Another important observation is that a large number of passenger mutations encoded neo‐epitopes that were potential target candidates. Many earlier studies also demonstrated that each cancer encodes a unique set of genetic alterations, but they focused on driver mutations and demonstrated the phylogenic tree at the cohort level without indicating their temporal relationship; thus, they are very useful in outlining cancer signal pathways, but not in determining the most suitable therapeutic target. Passenger mutations greatly outnumber driver mutations, so that they may play an important role in cancer immunotherapy 57, 58. Conceivably, the “cancer neo‐epitope tree” strategy as established in this study will help to determine optimal therapeutic targets and result in a great increase in clinical efficacy or even cure, especially when a cocktail of targets is used to reduce the chances of escape due to sporadic loss of the targets.
In this study, we constructed the “cancer neo‐epitope tree” using genomic data derived from a single DNA sample from each tumor. It should be noted that this approach is limited by many factors, such as the heterogeneous composition of the tumor's cell population, the exome capture efficiency, the genomic sequencing and assembly technique, and the tumor cell collection method. Therefore, our technique is able to draw out a “cancer neo‐epitope tree” that comprises only the major subpopulations. Even so, integrity is achievable only when the tumor is small and DNA is fully representative. For a large tumor, however, DNA from well‐designed multiple samples 4, 11 would be more appropriate. Currently, we predict the immunogenic mutations based on their mutated peptides’ binding affinity with the MHC‐I. We should note that further immunogenic experiments are warranted to validate the real immunogenicity of those putative neo‐epitopes so that we can better test our idea.
In summary, our results show that each tumor carried a unique set of genetic alterations and associated putative epitopes and that the construction of individual “cancer epitope trees,” together with the earliest genomic events, such as alterations in FBXW7, PIK3CA, and other members in the pathways, could assist in the understanding of the early genetic events involved in cervical carcinogenesis and, more importantly, the systematic search for optimal immunotherapeutic targets at the trunk or major branches.
Conflict of Interest
The authors declare no competing financial interests.
Supporting information
Figure S1. Distribution of alignment rate and coverage. Bar plot shows the alignment rate (A) and coverage (B) in each tumor (red) and control (blue) sample for each patient.
Figure S2. Correlation of mutations with patients’ age and stage. (A) Distribution of patients’ nonsynonymous mutations number with the age. The correlation coefficient and the significance P‐value are shown. (B) Distribution of patients’ nonsynonymous mutations number with the disease stage of the patients. P value was calculated by two‐sided Student's t test (mean ± S.D.; n = 23 subjects).
Figure S3. Distribution of mutated genes among 194 donors from ICGC. Distribution of mutated genes in 194 cervical cancer patients (mutation frequency > 8.2%). Each column represents one individual, and each row is a gene.
Figure S4. Mutated genes involved in the two significantly altered pathways. The heat map shows the mutated genes in the 23 patients involved in the two significantly altered pathways: the ubiquitin‐mediated proteolysis (red) and ECM receptor interaction pathways (blue).
Figure S5. The temporal order of mutations in the 194 ICGC cervical cancer donors. Beginning with the normal circle, figure shows all possible sequence of somatic mutated genes after leaving out low‐probability (the probability from the parent circle to the child circle below 0.4) events. The arrow points to possible order among mutated genes. Color for each circle is scaled according to the relative probability from the parent circle to the child circle (the color scale probability labeled under each panel).
Figure S6. Relationship between FBXW7 and PIK3CA mRNA expression and mutation statuses. Distribution of the gene expression values of FBXW7 (A) and PIK3CA (B) in patients harboring mutant and the wild‐type, separately. PIK3CA or FBXW7 mRNA expression levels were also compared between samples with FBXW7 (C) or PIK3CA (D) mutant and wild‐type forms. The normalized read counts from the ICGC donors’ RNA‐sequencing data, with the log transformation, were calculated as the gene expression values. P value was calculated by two‐sided Student's t test (mean ± S.D.; n = 194 subjects). Mut, mutant; Wld, wild‐type.
Figure S7. Significant correlation between FBXW7 and PIK3CA expression.
Table S1. Number of reads mapped to the HPV genomes.
Table S2. Validated mutations.
Table S3. Somatic mutations with the phylogenic relationship, immunogenicity, and altered pathways.
Table S4A. KEGG pathway enrichment of GSEA report for mutant FBXW7.
Table S5A. KEGG pathway enrichment of GSEA report for mutant PIK3CA.
Acknowledgment
This work was supported by the government funds of Shenzhen, China (SFG 2012.566 and SKC 2012.237), the National Natural Science Foundation of China (Grant number: 31501065), and the China Postdoctoral Science Foundation (Grant number: 2016M590824).
Cancer Medicine 2017, 6(1):207–219
Contributor Information
Ming Qi, Email: qiming@genomics.cn.
Zhi‐Ying Chen, Email: zy.chen1@siat.ac.cn.
References
- 1. Brahmer, J. R. , and Pardoll D. M.. 2013. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol. Res. 1:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borghaei, H. , Paz‐Ares L., Horn L., Spigel D. R., Steins M., Ready N. E., et al. 2015. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N. Engl. J. Med. 373:1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen, Z. Y. , Ma F., Huang H. L., and He C. Y.. 2015. Synthetic immunity to break down the bottleneck of cancer immunotherapy. Sci. Bulletin. 60:977–985. [Google Scholar]
- 4. Gerlinger, M. , Rowan A. J., Horswell S., Larkin J., Endesfelder D., Gronroos E., et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nik‐Zainal, S. , Van Loo P., Wedge D. C., Alexandrov L. B., Greenman C. D., Lau K. W., et al. 2012. The life history of 21 breast cancers. Cell 149:994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waclaw, B. , Bozic I., Pittman M. E., Hruban R. H., Vogelstein B., and Nowak M. A.. 2015. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 525:261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carreira, S. , Romanel A., Goodall J., Grist E., Ferraldeschi R., Miranda S., et al. 2014. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 6:254ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eirew, P. , Steif A., Khattra J., Ha G., Yap D., Farahani H., et al. 2015. Dynamics of genomic clones in breast cancer patient xenografts at single‐cell resolution. Nature 518:422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gundem, G. , Van Loo P., Kremeyer B., Alexandrov L. B., Tubio J. M., Papaemmanuil E., et al. 2015. The evolutionary history of lethal metastatic prostate cancer. Nature 520:353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Couzin‐Frankel, J. 2013. Breakthrough of the year 2013. Cancer immunotherapy. Science 342:1432–1433. [DOI] [PubMed] [Google Scholar]
- 11. Nik‐Zainal, S. , Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., Raine K., et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. GLOBOCAN 2012: Cervical Cancer Incidence, Mortality and Prevalence Worldwide in 2012 Avaliable at: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx(accessed 12 June 2015).
- 13. Ojesina, A. I. , Lichtenstein L., Freeman S. S., Pedamallu C. S., Imaz‐Rosshandler I., Pugh T. J., et al. 2014. Landscape of genomic alterations in cervical carcinomas. Nature 506:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chung, T. K. , Van Hummelen P., Chan P. K., Cheung T. H., Yim S. F., Yu M. Y., et al. 2015. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. Int. J. Cancer 137:776–783. [DOI] [PubMed] [Google Scholar]
- 15. FIGO Committee on Gynecologic Oncology . 2014. FIGO staging for carcinoma of the vulva, cervix, and corpus uteri. Int. J. Gynaecol. Obstet.. 125:97–98. [DOI] [PubMed] [Google Scholar]
- 16. Yi, X. , Li J., Yu S., Zhang A., Xu J., Yi J., et al. 2011. A new PCR‐based mass spectrometry system for high‐risk HPV, part I: methods. Am. J. Clin. Pathol. 136:913–919. [DOI] [PubMed] [Google Scholar]
- 17. Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKenna, A. , Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. 2010. The genome analysis toolkit: a mapreduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cibulskis, K. , Lawrence M. S., Carter S. L., Sivachenko A., Jaffe D., Sougnez C., et al. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Genomes Project C ; Auton, A. Brooks L. D., Brooks L. D., DePristo M. A., and Durbin R. M., et al. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Exome Variant Server . 2015. NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Avaliable at: http://evs.gs.washington.edu/EVS/ (accessed 4 January 2015).
- 22. Exome Aggregation Consortium (ExAC) . 2015. Cambridge, MA. Avaliable at: http://exac.broadinstitute.org. (accessed 4 January 2015)
- 23. McLaren, W. , Pritchard B., Rios D., Chen Y., Flicek P., and Cunningham F.. 2010. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26:2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Doorslaer, K. , Tan Q., Xirasagar S., Bandaru S., Gopalan V., Mohamoud Y., et al. 2013. The papillomavirus episteme: a central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 41:D571–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subramanian, A. , Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., et al. 2005. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc. Natl Acad. Sci. USA 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiao, W. , Vembu S., Deshwar A. G., Stein L., and Morris Q.. 2014. Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC Bioinformatics 15:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Misra, N. , Szczurek E., and Vingron M.. 2014. Inferring the paths of somatic evolution in cancer. Bioinformatics 30:2456–2463. [DOI] [PubMed] [Google Scholar]
- 28. Lundegaard, C. , Lamberth K., Harndahl M., Buus S., Lund O., and Nielsen M.. 2008. NetMHC‐3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8‐11. Nucleic Acids Res. 36:W509–W512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mootha, V. K. , Lindgren C. M., Eriksson K. F., Subramanian A., Sihag S., Lehar J., et al. 2003. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34:267–273. [DOI] [PubMed] [Google Scholar]
- 30. Banerji, S. , Cibulskis K., Rangel‐Escareno C., Brown K. K., Carter S. L., Frederick A. M., et al. 2012. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wright, A. A. , Howitt B. E., Myers A. P., Dahlberg S. E., Palescandolo E., Van Hummelen P., et al. 2013. Oncogenic mutations in cervical cancer: genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer 119:3776–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chung, T. K. , Van Hummelen P., Chan P. K., Cheung T. H., Yim S. F., Yu M. Y., et al. 2015. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. Int. J. Cancer 137:776–783. [DOI] [PubMed] [Google Scholar]
- 33. Tran, E. , Turcotte S., Gros A., Robbins P. F., Lu Y. C., Dudley M. E., et al. 2014. Cancer immunotherapy based on mutation‐specific CD4+ T cells in a patient with epithelial cancer. Science 344:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robbins, P. F. , Lu Y. C., El‐Gamil M., Li Y. F., Gross C., Gartner J., et al. 2013. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor‐reactive T cells. Nat. Med. 19:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trajanoski, Z. , Maccalli C., Mennonna D., Casorati G., Parmiani G., and Dellabona P.. 2015. Somatically mutated tumor antigens in the quest for a more efficacious patient‐oriented immunotherapy of cancer. Cancer Immunol. Immunother. 64:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castle, J. C. , Kreiter S., Diekmann J., Lower M., van de Roemer N., de Graaf J., et al. 2012. Exploiting the mutanome for tumor vaccination. Cancer Res. 72:1081–1091. [DOI] [PubMed] [Google Scholar]
- 37. Guo, G. , Gui Y., Gao S., Tang A., Hu X., Huang Y., et al. 2012. Frequent mutations of genes encoding ubiquitin‐mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat. Genet. 44:17–19. [DOI] [PubMed] [Google Scholar]
- 38. Welcker, M. , and Clurman B. E.. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8:83–93. [DOI] [PubMed] [Google Scholar]
- 39. Mao, J. H. , Kim I. J., Wu D., Climent J., Kang H. C., DelRosario R., et al. 2008. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321:1499–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tommasino, M. , Accardi R., Caldeira S., Dong W., Malanchi I., Smet A., et al. 2003. The role of TP53 in Cervical carcinogenesis. Hum. Mutat. 21:307–312. [DOI] [PubMed] [Google Scholar]
- 41. Scheffner, M. , Huibregtse J. M., Vierstra R. D., and Howley P. M.. 1993. The HPV‐16 E6 and E6‐AP complex functions as a ubiquitin‐protein ligase in the ubiquitination of p53. Cell 75:495–505. [DOI] [PubMed] [Google Scholar]
- 42. Boyer, S. N. , Wazer D. E., and Band V.. 1996. E7 protein of human papilloma virus‐16 induces degradation of retinoblastoma protein through the ubiquitin‐proteasome pathway. Cancer Res. 56:4620–4624. [PubMed] [Google Scholar]
- 43. McCormack, S. J. , Brazinski S. E., Moore J. L. Jr, Werness B. A., and Goldstein D. J.. 1997. Activation of the focal adhesion kinase signal transduction pathway in cervical carcinoma cell lines and human genital epithelial cells immortalized with human papillomavirus type 18. Oncogene 15:265–274. [DOI] [PubMed] [Google Scholar]
- 44. Du, M. , Fan X., Hong E., and Chen J. J.. 2002. Interaction of oncogenic papillomavirus E6 proteins with fibulin‐1. Biochem. Biophys. Res. Commun. 296:962–969. [DOI] [PubMed] [Google Scholar]
- 45. Chiarugi, P. , and Giannoni E.. 2008. Anoikis: a necessary death program for anchorage‐dependent cells. Biochem. Pharmacol. 76:1352–1364. [DOI] [PubMed] [Google Scholar]
- 46. Nowell, P. C. 1976. The clonal evolution of tumor cell populations. Science 194:23–28. [DOI] [PubMed] [Google Scholar]
- 47. Hughes, A. E. , Magrini V., Demeter R., Miller C. A., Fulton R., Fulton L. L., et al. 2014. Clonal architecture of secondary acute myeloid leukemia defined by single‐cell sequencing. PLoS Genet. 10:e1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aparicio, S. , and Caldas C.. 2013. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 368:842–851. [DOI] [PubMed] [Google Scholar]
- 49. Vogelstein, B. , Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A. Jr, and Kinzler K. W.. 2013. Cancer genome landscapes. Science 339:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moody, C. A. , and Laimins L. A.. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10:550–560. [DOI] [PubMed] [Google Scholar]
- 51. Samuels, Y. , Wang Z., Bardelli A., Silliman N., Ptak J., Szabo S., et al. 2004. High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554. [DOI] [PubMed] [Google Scholar]
- 52. Ciriello, G. , Miller M. L., Aksoy B. A., Senbabaoglu Y., Schultz N., and Sander C.. 2013. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rusan, M. , Li Y. Y., and Hammerman P. S.. 2015. Genomic landscape of human papillomavirus‐associated cancers. Clin. Cancer Res. 21:2009–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dumont, A. G. , Dumont S. N., and Trent J. C.. 2012. The favorable impact of PIK3CA mutations on survival: an analysis of 2587 patients with breast cancer. Chin J. Cancer 31:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Keymeulen, A. , Lee M. Y., Ousset M., Brohee S., Rorive S., Giraddi R. R., et al. 2015. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525:119–123. [DOI] [PubMed] [Google Scholar]
- 56. Nguyen, L. V. , Pellacani D., Lefort S., Kannan N., Osako T., Makarem M., et al. 2015. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature 528:267–271. [DOI] [PubMed] [Google Scholar]
- 57. Schreiber, R. D. , Old L. J., and Smyth M. J.. 2011. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331:1565–1570. [DOI] [PubMed] [Google Scholar]
- 58. Coulie, P. G. , Van den Eynde B. J., van der Bruggen P., and Boon T.. 2014. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14:135–146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of alignment rate and coverage. Bar plot shows the alignment rate (A) and coverage (B) in each tumor (red) and control (blue) sample for each patient.
Figure S2. Correlation of mutations with patients’ age and stage. (A) Distribution of patients’ nonsynonymous mutations number with the age. The correlation coefficient and the significance P‐value are shown. (B) Distribution of patients’ nonsynonymous mutations number with the disease stage of the patients. P value was calculated by two‐sided Student's t test (mean ± S.D.; n = 23 subjects).
Figure S3. Distribution of mutated genes among 194 donors from ICGC. Distribution of mutated genes in 194 cervical cancer patients (mutation frequency > 8.2%). Each column represents one individual, and each row is a gene.
Figure S4. Mutated genes involved in the two significantly altered pathways. The heat map shows the mutated genes in the 23 patients involved in the two significantly altered pathways: the ubiquitin‐mediated proteolysis (red) and ECM receptor interaction pathways (blue).
Figure S5. The temporal order of mutations in the 194 ICGC cervical cancer donors. Beginning with the normal circle, figure shows all possible sequence of somatic mutated genes after leaving out low‐probability (the probability from the parent circle to the child circle below 0.4) events. The arrow points to possible order among mutated genes. Color for each circle is scaled according to the relative probability from the parent circle to the child circle (the color scale probability labeled under each panel).
Figure S6. Relationship between FBXW7 and PIK3CA mRNA expression and mutation statuses. Distribution of the gene expression values of FBXW7 (A) and PIK3CA (B) in patients harboring mutant and the wild‐type, separately. PIK3CA or FBXW7 mRNA expression levels were also compared between samples with FBXW7 (C) or PIK3CA (D) mutant and wild‐type forms. The normalized read counts from the ICGC donors’ RNA‐sequencing data, with the log transformation, were calculated as the gene expression values. P value was calculated by two‐sided Student's t test (mean ± S.D.; n = 194 subjects). Mut, mutant; Wld, wild‐type.
Figure S7. Significant correlation between FBXW7 and PIK3CA expression.
Table S1. Number of reads mapped to the HPV genomes.
Table S2. Validated mutations.
Table S3. Somatic mutations with the phylogenic relationship, immunogenicity, and altered pathways.
Table S4A. KEGG pathway enrichment of GSEA report for mutant FBXW7.
Table S5A. KEGG pathway enrichment of GSEA report for mutant PIK3CA.