

Abstract
Serum paraoxonase 1 (PON1) is a native lactonase capable of promiscuously hydrolyzing a broad range of substrates, including organophosphates, esters, and carbonates. Structurally, PON1 is a six-bladed β-propeller with a flexible loop (residues 70–81) covering the active site. This loop contains a functionally critical Tyr at position 71. We have performed detailed experimental and computational analyses of the role of selected Y71 variants in the active site stability and catalytic activity in order to probe the role of Y71 in PON1’s lactonase and organophosphatase activities. We demonstrate that the impact of Y71 substitutions on PON1’s lactonase activity is minimal, whereas the kcat for the paraoxonase activity is negatively perturbed by up to 100-fold, suggesting greater mutational robustness of the native activity. Additionally, while these substitutions modulate PON1’s active site shape, volume, and loop flexibility, their largest effect is in altering the solvent accessibility of the active site by expanding the active site volume, allowing additional water molecules to enter. This effect is markedly more pronounced in the organophosphatase activity than the lactonase activity. Finally, a detailed comparison of PON1 to other organophosphatases demonstrates that either a similar “gating loop” or a highly buried solvent-excluding active site is a common feature of these enzymes. We therefore posit that modulating the active site hydrophobicity is a key element in facilitating the evolution of organophosphatase activity. This provides a concrete feature that can be utilized in the rational design of next-generation organophosphate hydrolases that are capable of selecting a specific reaction from a pool of viable substrates.
Introduction
The chemical reactions that make life possible are dependent on enzymes for their regulation. Therefore, to understand how enzyme function evolves is to understand how life evolves at the most fundamental molecular level. It has become increasingly clear that the catalytic promiscuity of many enzymes allows them to acquire new functions on time scales much faster than would be expected from the typical time scales for divergence of new enzyme functions by natural evolution.1 That is, the promiscuous side activities provide an evolutionary starting point for these enzymes, as they allow new activities to evolve without the selective pressure that is being placed on the native reaction.1−3 This in turn acts as an evolutionary “trampoline” that allows a more rapid response to environmental change. In the present work, we have used the catalytically promiscuous organophosphatase serum paraoxonase 1 (PON1)4−8 as a model system to understand the structural origins of the selectivity and promiscuity of this enzyme and have then performed comparisons to other structurally diverse organophosphatases in order to find the chemical commonalities that facilitate their organophosphatase activities. In particular, as many organophosphatases have evolved from enzymes that were originally lactonases and/or arylesterases (and retain these activities to varying degrees),4,9−12 we have focused on the particular features necessary to facilitate lactone and ester hydrolysis that are common among these enzymes.
PON1 is a calcium-dependent hydrolytic enzyme that is found in all mammalian species.13 In vivo, this enzyme forms complexes with the membrane-like surface of high-density lipoprotein (HDL)14−17 and contributes to HDL’s antioxidant properties.18 As with many other organophosphatases, structure–activity studies suggest that the native activity of PON1 is its lactonase activity,4,19 with a particular preference for γ- and δ-lactones with long alkyl side chains as its primary substrates. The enzyme is broadly promiscuous, however, and can catalyze the hydrolysis of multiple substrates, including not just lactones and thiolactones but also esters, organophosphates,7 and carbonates.20 PON1 has been extensively studied from a biological and biomedical point of view, primarily because of its role in atherosclerosis.21,22 In addition to its direct biomedical relevance, PON1 also shows great promise as a biotherapeutic because of its ability to hydrolyze a broad range of organophosphates, including pesticides and nerve agents such as sarin, soman, and VX.23,24
Structurally, PON1 is six-bladed β-propeller with a central tunnel containing two calcium ions approximately 7.4 Å apart (Figure 1A).16 Of these calcium ions, the calcium ion buried further into the tunnel is structural and essential for PON1’s conformational stability, whereas the second solvent-exposed calcium ion residing at the bottom of the active site cavity is catalytic, playing a role both in substrate positioning and in activating the P(C)=O ester bonds of the substrate25,26 (Figure 1B). At the top of the active site are three helices, highlighted in salmon in Figure 1A, of which the first two (helices H1 and H2) are believed to play a role in mediating PON1–HDL interactions on the basis of both structural analysis and selective mutagenesis.16,25 Finally, an active site loop comprising residues 70–81, which is absent in all apo crystal structures of PON1 because of its high flexibility, closes over the active site upon binding of a ligand (2-hydroxyquinoline, Figure 1A).
Figure 1.

(A) Overall structure of RePON1 in complex with the inhibitor 2-hydroxyquinoline (2HQ) obtained at pH 6.5 (PDB ID 3SRG).7 Here the flexible loop (residues 70–81) is highlighted in red, the three surface helices are highlighted in salmon, the Ca2+ ions are highlighted in green, and 2HQ is highlighted in orange. (B) Close-up of the active site tunnel leading from the catalytic Ca2+ to the surface helix H2. Key residues involved in this network as well as the corresponding hydrogen-bonding distances in the 3SRG crystal structure are shown.
Despite great progress in this area,27−36 our knowledge of how interfacial activation of enzymes by protein–lipid interactions regulates enzyme stability, efficiency, and specificity still remains limited. As PON1 shares many structural features with other interfacially activated enzymes, including in particular secreted phospholipase 2 (sPLA2),37−40 it provides a promising model system with which to understand the molecular details of such interfacial activation and, following from this, also a further probe for the role of enzyme dynamics in catalysis. Structural considerations have identified a key hydrogen-bonding network that spans from the protein–membrane interface to the catalytic calcium center, interacting in particular with N168, which interacts with the substrate and plays a role in fixing the position of the catalytic calcium, as well as Y71, which lies on the active site loop and interacts with residues in the central tunnel (Figure 1B).25 The importance of this hydrogen-bonding network can be in part highlighted by the fact that disturbing even its peripheries by removal of HDL or detergents or mutation of key second- or third-shell residues such as D183 causes a complete loss in catalytic activity.6 Our recent combined biochemical, crystallographic, and computational study suggested that interaction with HDL rigidifies this hydrogen-bonding network, keeping the key catalytic residues in place and thus leading to the observed stimulation of PON1’s lactonase activity.25 This also touches on the “dark side” of excess enzyme dynamics and the need to sometimes reduce floppiness to enhance activity (a phenomenon also seen in directed evolution of several de novo-designed enzymes41−43).
A key remaining question is what the role of the active site loop, and in particular Y71, is in facilitating PON1’s activity, specificity, and promiscuity and how it compares to other organophosphatases with similar active site loops. Our recent computational work, which probed the effect of distorting the H2 helix on the catalytic activity, showed only minor displacement of Y71 in response to the repositioning of H2 when PON1 was in complex with the chromogenic lactone thiobutyl-γ-butyric lactone (TBBL).25 Taken together, this would suggest that Y71 does not play an active role in PON1’s lactonase activity in general, and specifically not on the HDL-induced simulation. However, as can be seen from Figure 1B, Y71 is a key participant in the hydrogen-bonding network connecting the catalytic calcium center to the membrane interface of PON1. Additionally, when the disordered loop is closed over the active site, Y71 points directly into the active site, which makes it much harder to fit larger and bulkier organophosphate substrates such as the pesticide paraoxon into the active site, even though kinetic characterization shows that PON1 is able to hydrolyze paraoxon quite efficiently, with kcat/KM = 2149 ± 393 M–1 s–1.4,7 Both docking7 and molecular dynamics (MD)25 simulations suggest that the only way paraoxon can be accommodated in the PON1 active site is by pushing Y71 out of the active site, causing the active site loop to take on a partially open conformation. This raises key questions about the role of Y71 and, following from this, about the role of active site plasticity in facilitating a catalytically favorable active site architecture in this highly promiscuous interfacially activated enzyme.
While the most direct way to probe the importance of the conformation of Y71 and the active site loop on catalysis would be through crystal structures of PON1 in both its substrate-free form and in complex with different inhibitors, enzyme–ligand complexes of PON1 are notoriously difficult to crystallize,7,16 and therefore, we have explored the role of Y71 and active site plasticity indirectly through a combination of site-saturation mutagenesis of Y71 (using a small library that contains the variation of Y71 to all other 19 amino acids) and screening for both lactonase activity toward TBBL and paraoxonase activity. This has then been complemented with computational studies of selected Y71 mutants. Doing so has allowed us to explore how different residues at this position in the active site loop would affect the configuration of PON1’s binding site and in turn both its native and promiscuous activities. Our data show that while the effect of mutation of Y71 on the observed catalytic activity is not radical, as expected, it affects both the loop flexibility and the rigidity of the hydrogen-bonding network connecting the active site to the surface helices. The observed changes in active site architecture upon substitution of Y71 can also to some extent help rationalize the kinetic observations from previous studies with other PON1 variants.5,44,45
Following from this, the resulting structural reshaping of the active site also affects substrate positioning, which can be directly linked to the observed changes in catalytic activity. More significantly, however, the Y71···D183 hydrogen-bonding interaction appears to allow the active site loop to act as a hydrophobic gate, allowing movement of the substrate and product in and out of the active site and, more critically, helping to exclude solvent from the active site, thus providing a hydrophobic cage in which the reaction can take place. Mutations of Y71 cause changes in the active site volume that allow an increasing number of solvent molecules access to the active site, although this is more pronounced when the organophosphate is bound to the active site compared with TBBL, which can in turn be directly linked to the observed differential effects of the same mutation on the lactonase and paraoxonase activities. This influx of water molecules will modulate the electrostatic environment of the active site, thus also impacting the corresponding catalytic efficiency of the enzyme. A comparison to other organophosphatases demonstrates that these enzymes often either have an active site loop that appears to play a similar role or have found other solutions to create a hydrophobic cage for the substrate (e.g., through a deeply buried active site pocket). We posit that this regulation of the electrostatic environment of the active site through solvent exclusion, which is clearly advantageous for the binding of hydrophobic molecules such as lactones and aryl esters, is key to the fortuitous ability of these enzymes to also hydrolyze uncharged organophosphate nerve agents and pesticides. This creates a major difference with other promiscuous phosphatases such as members of the alkaline phosphatase superfamily,3,46,47 where active site polarity and charge discrimination between different anionic substrates apparently play key roles in facilitating the observed selectivity and promiscuity.
Materials and Methods
Site-Saturation Mutagenesis and Screening of Y71 Mutants
The site-saturation library in position 71 was generated using the “inverse polymerase chain reaction” method48 with the G2E6 variant of PON1 as the template, as previously described.6 The G2E6 variant is a chimeric form of PON1 (by directed evolution) showing functional and structural properties similar to those of human PON1.49 However, unlike the latter, the G2E6 variant is highly soluble and capable of bacterial expression, which thus enabled detailed analyses of PON1’s structure and function.16,49 For simplicity, in the text we refer to the G2E6 wild-type-like variant16,49 as RePON1 throughout. The tyrosine codons in position 71 were replaced with NNS codons (where N is a mixture of all four nucleotides and S is a mixture of C and G) encoding all amino acids. The library was transformed into Origami cells, and colonies were grown on agar plates. Randomly picked colonies were individually grown in 96-deep-well plates, and protein production was induced with IPTG as previously described.6,8 Following cell lysis, clarified lysates of the Y71 variants were screened for phosphotriesterase (paraoxon; 1 mM) and lactonase (TBBL; 0.2 mM) activities. Mutants with interesting activity phenotypes were subjected to DNA sequencing and further characterization.
Purification and Kinetics of Y71 Mutants
The expression and purification of RePON1 and its Y71 mutants were performed as previously described.7 Kinetic measurements with TBBL and paraoxon were determined at substrate concentrations in a range from 0.3 × KM up to (2–3) × KM, and kcat, KM, and kcat/KM were obtained by fitting the data to the Michaelis–Menten model with PRISM (GraphPad Software).7 All of the data presented are the means of at least two independent experiments, and the error ranges represent the standard deviations from the mean.
Molecular Dynamics Simulations of PON1
The starting point for all of the PON1 simulations in this work was a 2.2 Å crystal structure of RePON1 in complex with the inhibitor 2-hydroxyquinoline (2HQ) and obtained at pH 6.5 (PDB ID 3SRG).7 We chose this structure in particular as it is one of the only crystal structures of PON1 in the Protein Data Bank in which the active site loop is sufficiently stabilized by the inhibitor to be observed in the crystal structure. For comparison, we have simulated both the substrate-free enzyme and the enzyme–substrate complexes of RePON1–TBBL and RePON1–paraoxon in both the wild-type (WT) and mutant forms of the enzyme. Because of the lack of crystal structures, the relevant Y71 mutant forms of RePON1 were generated in silico either by manual truncation of Y71 to Phe, Ala, or Gly or by choosing the most probable rotamer of Met and Trp using the Dunbrack50 rotamer library as implemented in Chimera.51 In all cases, and as in our previous work, the relevant substrate and the nucleophilic water molecule were manually placed into the active site so as to optimize the alignment of the nucleophilic water molecule and the reactive C/P center, the alignment of the nucleophilic water molecule and the relevant general base, and the alignment of the substrate with amino acid side chains in the active site. In both cases, the conformation of 2HQ in the 3SRG crystal structure was used as a guiding point for where to place the substrate into the active site. Additionally, both the structural and catalytic calcium ions were modeled using an octahedral dummy model52,53 overlaid on the position of the relevant metal ion in the crystal structure and the same parameters as in our previous study25 (see Table S1). This had the advantage of allowing for more stable metal coordination and avoided the local structural instabilities we observed when using a standard Lennard-Jones sphere as a calcium model, which were also independently alluded to in ref (54).
To solvate the system, all of the crystallographic water molecules were removed from the crystal structure, except for those directly coordinated to the Ca2+ ions, and the system was solvated in a 24 Å-radius sphere of TIP3P water molecules centered on the catalytic Ca2+ ion. This was then subjected to a 10 kcal mol–1 Å–2 surface restraint on all atoms in the outer 15% of the sphere to keep it stable, as per the surface-constrained all-atom solvent model (SCAAS).55 All atoms outside this sphere were retained in the simulation but restrained to their crystallographic positions using a 200 kcal mol–1 Å–2 harmonic positional restraint, and all atoms within the inner 85% of the sphere were allowed to move freely (also see our previous work25). In order to keep the charge of the sphere stable, all ionizable groups within 17 Å of the catalytic Ca2+ were kept in their relevant protonation states at physiological pH, and all other ionizable groups outside and close to the boundary of the sphere were kept in their neutral forms, resulting in an overall system charge of −1 (Table S2). Once the system had been solvated and correct ionization states had been assigned, each structure was gradually heated from 0 to 300 K over the course of 240 ps of simulation time, starting with a 200 kcal mol–1 Å–2 positional restraint on all heavy atoms and reducing this restraint to 0.5 kcal mol–1 Å–2 on all atoms of the substrate and nucleophilic water molecule (when present in the simulation) over the course of the heating procedure. Once the system had reached 300 K, we performed a further 30 ns of MD simulations to monitor the behavior of the loop and equilibrate the system for subsequent empirical valence bond (EVB) simulations of the chemical step of catalysis. All simulations were performed in three replicas with different initial velocities using the Q simulation package56 and the OPLS-AA force field57 with a 1 fs time step and the Berendsen thermostat with a 1 fs bath relaxation time, leading to a total of 1.62 μs of (productive) simulation time over all systems.
We note as an aside here that, as outlined in the Introduction, PON1 is a membrane-associated enzyme, and extensive delipidation has been shown to lead to complete loss of activity.58,59 As in our previous study,25 we did not explicitly include the membrane in our simulations both because of the excessive computational cost this creates when running large numbers of simulations of the enzyme-catalyzed hydrolysis of multiple substrates by both wild-type and mutant forms of RePON1 and because of the lack of any direct structural information on how PON1, or in fact any other enzyme, interacts with HDL. However, we demonstrated in our previous study that even without explicitly including the membrane in our simulations, they still provided meaningful information because we are working with a multilayer system in which the outer peripheries of RePON1 were kept restrained to their crystallographic coordinates. This restraint approach, which is standard practice in QM/MM simulations of enzyme reactivity (see, e.g., refs (60) and (61)), mimics to some extent the stabilizing effect of the membrane or detergent and prevents the protein from artificially unfolding in the absence of the membrane while keeping the active site structure stable. Following from this, we demonstrated in our previous study that our model allowed us to reproduce both relevant activation free energies and other key experimental observables with high fidelity, increasing our confidence in the reliability of our simulations (see the discussion in ref (25)), and once again, in the present work we were able to reproduce the relevant energy changes upon mutation of Y71 using our model (see the Results and Discussion).
Empirical Valence Bond Simulations of RePON1-Catalyzed Hydrolyses of Paraoxon and TBBL
Following from our initial molecular dynamics simulations of wild-type and mutant forms of RePON1, we have modeled the effect of mutating Y71 on the energetics of TBBL and paraoxon hydrolysis using the EVB approach, which is an empirically based hybrid VB/MM approach that uses classical force fields to model chemical reactivity within a quantum-mechanical framework.62 The full theoretical details of this approach have been reviewed at great length elsewhere,62−64 but in brief, the two main advantages of this approach in the context of the present work are (1) its lower computational cost as a classical approach, which allowed us to perform the extensive conformational sampling necessary to obtain convergent free energies for chemical processes in biological systems, and (2) the fact that a well-parametrized force field carries a tremendous amount of chemical information, allowing us to model bond breaking and formation processes in a chemically meaningful way (in the EVB approach, this is achieved using Morse rather than harmonic potentials to describe bonds that are changing).
On the basis of previous experimental and computational studies,25,65−69 here we describe the hydrolyses of paraoxon and TBBL as one- and two-step reactions, respectively (Figure 2), using the valence bond states illustrated in Figure S1. In our recent EVB study,25 we considered only the nucleophilic attack step to form a tetrahedral intermediate for TBBL hydrolysis, as this step is most likely to be rate-limiting. However, in the present study, for completeness we have also considered the subsequent ring-opening step. Nucleophile activation has been modeled as proceeding through general base catalysis by the side chains of H115 and D269 for TBBL and paraoxon hydrolysis, respectively, on the basis of extensive mutagenesis data4,6−8 (also see our previous work25), and in the case of TBBL hydrolysis, we modeled the ring-opening step with proton transfer back from the protonated H115 side chain, which now acts as a general acid catalyst instead, as this is the most likely proton source.
Figure 2.

Proposed mechanisms of (A) paraoxon and (B) TBBL hydrolysis by serum paraoxonase 1. For further details, see refs (7) and (25).
All of the EVB simulations for each system were performed by taking the end point of one of our replicas from the initial MD simulations for each system and preparing 10 new starting structures by running another 500 ps of MD simulations with a new set of initial velocities for each starting structure. The subsequent EVB simulations were then performed from these new starting structures using the standard EVB free energy perturbation umbrella sampling (FEP/US) approach outlined in, e.g., refs (62−64), and all of the EVB simulations for each system were performed in 51 mapping frames of 200 ps/frame, for simulation times of 10.2 ns for each individual system for paraoxon hydrolysis and 20.4 ns for TBBL hydrolysis, as the latter was modeled as a two-step process (306 and 612 ns per system, respectively, over 30 independent EVB trajectories, considering three independent initial MD simulations as starting points and then 10 EVB simulations initiated from each individual equilibrated system). This leads to a total simulation time of 5.51 μs over all systems (5.69 μs when including also the initial 500 ps MD simulations). The EVB simulations were performed using the same setup as our initial MD simulations and applying a weak 0.5 kcal mol–1 Å–2 harmonic constraint on all free reacting fragments during the simulation to prevent them from drifting out of the simulation sphere. In each case, our reference state was the uncatalyzed reaction in aqueous solution, the EVB parameters for which were calibrated to fit activation and reaction free energies of ΔG1⧧ = 20.7 kcal mol–1, ΔGint = 14.4 kcal mol–1, and ΔG2 = 17.5 kcal/mol for TBBL hydrolysis (where ΔG1⧧ and ΔG2 are the free energies of activation for nucleophilic attack and ring opening, respectively, and ΔGint is the free energy of the corresponding tetrahedral intermediate) and ΔG⧧ = 23.9 kcal mol–1 and ΔG0 = −10.5 kcal mol–1 for paraoxon hydrolysis. This calibration was based on extensive consideration of existing experimental and computational data25,65−69 and is described in detail in the Supporting Information of ref (25). The EVB simulations of the background reaction in aqueous solution were performed using the same setup as for the wild-type enzyme, with ethylimidazole and propionic acid as models for H115 and D269, respectively. The only differences in simulation setup between the reactions in water and the enzyme active site were that we used only 10 ns of equilibration time for the water reaction and that a slightly larger harmonic restraint of 1 kcal mol–1 Å–2 (compared with 0.5 kcal mol–1 Å–2 for the corresponding enzyme-catalyzed reactions) was placed on the reacting atoms to keep them at the center of the simulation sphere. Once each background reaction had been calibrated to reproduce the relevant energetics in aqueous solution, the same parameter set was then used unchanged in order to model the reactivities of the wild-type and mutant forms of RePON1 with each substrate. The resulting calculated activation free energies were obtained using the QCalc module of Q, and unless otherwise specified, all other external analysis was performed using the CPPTRAJ71 module as implemented in AMBER 16.72 All of the EVB parameters necessary to reproduce our work are provided in the Supporting Information.
Results and Discussion
Effect of Mutations at Position Y71 on the Stability of RePON1’s Mobile Loop
An examination of the structure of RePON1 (Figure 1) strongly suggests that the active site loop of RePON1 (defined here as residues 70–81, following ref (7)) and related enzymes acts as a lid, facilitating both entrance of the substrate to the active site and subsequent product release.7,73,74 However, in addition to the overall structural role of this loop, previous work has shown that Y71 in particular is an important residue that plays a key role in PON1’s function. This includes, for example, modulation of substrate selectivity and catalytic stimulation by HDL.7,25,75 The side chain of Y71 is highly mobile and has been shown to adopt various conformational substates7 that have been suggested to differentially affect PON1’s native and promiscuous functions.7,76 Therefore, in order to further probe the role of residue Y71, we have generated a saturation library at position 71 (i.e., substitution of Y71 with all 20 amino acids) and used it for library screening of both the chromogenic lactone TBBL and the organophosphate pesticide paraoxon. In general, substitutions at position 71 mainly affected the paraoxonase activity. Only 16% of the tested library mutants (see Materials and Methods) exhibited paraoxonase activity ≥50% compared with Y71, whereas the lactonase activity was retained in 52% of the library mutants. The mutants showing the most diverse functional properties in the library screens (i.e., Y71M, Y71W, Y71F, Y71A, and Y71G), such as differential activity between RePON1’s native (lactonase) and promiscuous (paraoxonase) activities, were purified, and we experimentally measured the rates of hydrolysis of both TBBL and paraoxon by each RePON1 variant (Table 1). These mutations potentially introduce significant structural perturbations to the active site of PON1, which consequently affect its function. Thus, further analysis of these mutants may allow us to examine both the structural impact of Y71 mutations on the active site configuration, especially with regard to the flexible loop, and their corresponding effect on the activity and selectivity of RePON1 toward its native (lactonase) and promiscuous (paraoxonase) substrates.
Table 1. Comparison of Computational and Experimental Data for the Hydrolyses of TBBL and Paraoxon by Wild-Type RePON1 and a Range of Y71 Mutantsa.
| variant | kcat | KM | kcat/KM | ΔGexp⧧ | ΔGcalc⧧ |
|---|---|---|---|---|---|
| TBBLc | |||||
| WT | 189 ± 16 | 1.1 ± 0.1 | 169104 ± 22915 | 14.4 | 13.5 ± 0.4 |
| Y71G | 50 ± 4 | 1.5 ± 0.13 | 31338 ± 5207 | 15.1 | 11.9 ± 0.5 |
| Y71A | 41 ± 6 | 4.9 ± 1.2 | 8411 ± 2045 | 15.3 | 11.6 ± 1.5 |
| Y71F | 24 ± 4 | 1.6 ± 0.1 | 15291 ± 3903 | 15.6 | 11.3 ± 1.0 |
| Y71M | NDb | >5 | 6762 ± 170 | NDb | 12.5 ± 1.3 |
| Y71W | NDb | >5 | 26514 ± 2629 | NDb | 12.6 ± 3.3 |
| Paraoxon | |||||
| WT | 5 ± 0.6 | 2.3 ± 0.3 | 2149 ± 393 | 16.5 | 16.3 ± 1.1 |
| Y71M | 3.4 ± 0.01 | 1.5 ± 0.03 | 2253 ± 47 | 16.7 | 17.0 ± 0.5 |
| Y71F | 0.85 ± 0.07 | 2.3 ± 0.3 | 327 ± 53 | 17.5 | 18.3 ± 0.8 |
| Y71A | 0.24 ± 0.01 | 1.9 ± 0.1 | 124 ± 9 | 18.3 | 18.9 ± 1.3 |
| Y71G | 0.05 ± 0.01 | 2.7 ± 0.4 | 18 ± 3 | 19.2 | 19.5 ± 2.4 |
| Y71W | NDb | >5 | 85 ± 4 | NDb | 16.8 ± 1.2 |
Kinetic parameters were measured at 25 °C and pH 8, and kcat, KM, and kcat/KM are shown in s–1, mM, and M–1 s–1, respectively.
Because of limited substrate solubility, the reaction rates did not show saturation; thus, kcat could not be determined, and only an upper limit for the KM value based on the maximal substrate concentration used is provided. The catalytic efficiency was obtained by linear fit of the initial velocity to the substrate concentration. ΔGexp⧧ denotes the experimental activation free energy obtained as an upper limit from kcat using transition state theory, and ΔGcalc denotes the corresponding calculated activation free energy obtained from our empirical valence bond (EVB) simulations using the EVB parameters shown in the Supporting Information. The ΔGcalc⧧ values shown in this table are averages and standard deviations over 30 independent trajectories as described in Materials and Methods. All of the activation energies are shown in kcal mol–1. For a detailed description of the derivation of the energetics of the background reaction in aqueous solution and the corresponding EVB calibration used to obtain the calculated values shown in this table, we refer the reader to Materials and Methods and the Supporting Information of ref (25).
In the case of TBBL hydrolysis, the calculated activation free energies shown here correspond to the rate-limiting step for a two-step process (Figure 2). For a breakdown of the calculated energies for each individual reaction step, see Table S5. Representative structures of the corresponding stationary points for each reaction step for the reactions catalyzed by the wild-type enzyme are shown in Figures S5 and S6.
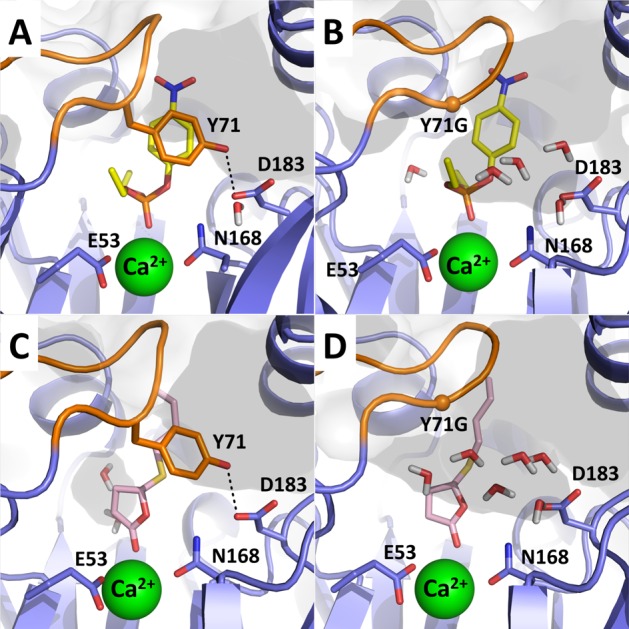
As can be seen from Figure 1, in the wild-type enzyme Y71 forms a hydrogen-bonding interaction with the side chain of D183, which is itself part of a hydrogen-bonding network that leads from N168 on the catalytic Ca2+ ion to the outer periphery of the protein. D183 is central to this hydrogen-bonding network, as in addition to the hydrogen bond it forms with the side chain of Y71, it also forms hydrogen-bonding interactions with the side chains of S166, N168, and H184, the backbone carbonyl group of S166, and an active site water molecule. Therefore, D183 acts as an “anchor”, keeping this hydrogen-bonding network together. The importance of the hydrogen-bonding network in general and residue D183 in particular is further supported by the fact that mutations at position 183 have been shown to be detrimental to both PON1’s lactonase and paraoxonase activities,6,8,77 even though this residue is 7 Å away from the catalytic Ca2+ and makes no obvious contacts with the bound substrate in the active site.
Our starting point, therefore, was to examine the structural implications of the Y71 substitutions and to see the extent to which breaking the hydrogen bond between Y71 and D183 affects the dynamics of the active site loop and the hydrogen-bonding network along the central tunnel. The most direct way to examine this would be to generate crystal structures of the relevant mutant forms of the enzyme; however, the active site loop, including Y71, is disordered and absent from almost all of the few existing PON1 structures in the Protein Data Bank. Recently, we were able to make significant progress in this direction by using an inhibitor, 2HQ, to obtain a structure of RePON1 at pH 6.5 with a closed loop conformation and a visible side chain at position 71.7 However, as was previously shown,7 mutations in the flexible loop, especially at positions 71 and 74 (both make direct contact with 2HQ), abolish the binding of 2HQ. Therefore, crystallization of the Y71 mutants, even in the presence of 2HQ, is likely to produce crystal structures with a disordered active site loop, including the side chain at position 71. Because of this, we are dependent on simulation in order to elucidate the effect of mutations on the positioning and dynamics of the active site loop.
Therefore, we began by performing MD simulations of RePON1 and each of the Y71 mutants mentioned above in either the substrate-free form of the enzyme or in complex with either paraoxon or TBBL (three 30 ns trajectories for each system), as described in Materials and Methods. The root-mean-square deviations (RMSDs) of all backbone Cα atoms during our MD simulations (shown in Figure S2 and Table S3) demonstrate the stability of our system during our simulations and that the system adequately equilibrated during the time scale of the simulations. Similarly, a comparison of the metal–ligand distances between the catalytic Ca2+ ion and its surrounding ligands (Table S4) shows that the metal center remained stable during our simulations, and that while the metal–ligand distances were slightly perturbed by the Y71 mutations, they were highly stable throughout our simulations without the need for any restraints or artificial bonds to the ligands.
We have also examined the root-mean-square fluctuations (RMSFs) of the backbone Cα atoms during the course of our MD simulations (Figure S3) and observed that in all of the simulations, the regions of the protein with the largest fluctuations are the active site loop (residues 70–81), a second loop (residues 46–55) that is close to the metal-bound E53, and a shorter loop segment (residues 133–140) that is adjacent to H134, which forms part of the H115–H134 catalytic dyad in the active site.6 As would be expected, the simulations of RePON1 in complex with either TBBL (Figure S3B) or paraoxon (Figure S3C) show the flexibility of the active site loop either to be similar to that in the wild-type enzyme or to slightly increase upon substitution of Y71 (as a result of breaking of the Y71–D183 hydrogen bond). Interestingly, however, binding of paraoxon to the active site appears to greatly reduce the flexibility of the active site loop, even though it increases the active site volume compared with those of the corresponding RePON1–TBBL complexes, as shall be discussed below. Additionally, comparing the wild-type and mutant forms of RePON1 in both the substrate-free forms and in complex with paraoxon and TBBL (Figure S4) illustrates that despite these changes in flexibility, the active site loop is still very floppy and can take multiple conformations. Therefore, the loop itself appears to be moving back and forth in the simulations, and there is not a direct correlation between either loop position and mutation or loop position and which substrate is bound to the active site, except that substrate binding appears to significantly increase the overall mobility of the enzyme compared with the substrate-free form (Figure S3).
Differential Effect of Mutating Y71 on the Native and Promiscuous Activities of PON1
As mentioned in the previous section, the Y71 substitution library overall seems to affect more drastically the promiscuous paraoxonase activity compared with the native lactonase activity (16% vs 52% of screened variants showing ≥50% paraoxonase and lactonase activity, respectively, compared with the Y71 WT). The kinetic parameters determined for the individual Y71 mutants also largely agree with this observation (Table 1). That is, it can be seen, for instance, that kcat/KM is reduced by up to 25-fold for TBBL hydrolysis and by up to 120-fold for paraoxon hydrolysis, with the Y71M and Y71G mutants showing the largest decrease, respectively. For most of the mutants, however, KM is minimally affected by the mutation, and thus, the changes in activity primarily arise from changes in kcat. An exception is the Y71W mutant, which shows higher KM values for both substrates, presumably due to structural hindrances induced by the bulkier side chain of Trp, which might affect substrate binding. More curiously, the same mutation apparently impacts the activities of RePON1 toward the two substrates differently. That is, in the case of TBBL hydrolysis, the most detrimental effect on kcat is observed in the conservative mutation of Tyr to Phe (which involves only loss of a hydrogen bond), whereas much smaller effects are observed upon replacement of Y71 with Ala or even Gly, although none of the observed effects are particularly large for any of these mutations in free energy terms. In contrast, in the case of paraoxon hydrolysis, the trend is reversed, with only a small effect from mutation to Met, a slightly larger effect from mutation to Phe, and then a significant effect of truncation to either Gly or Ala, resulting in a 100-fold reduction in kcat (corresponding to a 2.8 kcal/mol change in activation free energy).
Overall, our experimental results clearly show a differential effect of the Y71 mutations on RePON1’s native and promiscuous functions. Specifically, the Y71 mutations seem to have a much larger effect on paraoxon hydrolysis (promiscuous function) than on TBBL hydrolysis (native function). This observation is interesting, as the shape of the active site wall is complementary to the native substrate, even though Y71 has no direct contacts with the lactone (it should be noted, however, that binding of the promiscuous organophosphate substrate is driven by nonspecific hydrophobicity4,78). This is therefore in agreement with the suggestion that native functions are more robust and resistant to mutations compared with promiscuous functions1,79−81 (although recent work suggests that this may not be valid for all systems82). That is, it has been argued that mutations that have a neutral or near-neutral effect on the native function might remarkably affect promiscuous function(s) and have thus been suggested to serve as a driving force for the evolvability of promiscuous functions.79 Similarly to Y71, these mutations are often located in the flexible regions of the binding-site walls, such as loops. Thus, in the present case, it is plausible that variations in the composition of the flexible loop, especially at position 71, have contributed greatly to the evolvability of PON1’s paraoxonase activity.
To further probe the origin of these differential changes in PON1’s native and promiscuous activities, we have performed EVB simulations of the RePON1-catalyzed hydrolysis of TBBL and paraoxon as described in Materials and Methods, and our data are summarized in Figure 3 and Table 1. On the basis of extensive experimental studies of PON1-catalyzed TBBL hydrolysis,6−8 we have modeled TBBL hydrolysis as proceeding through general base catalysis by H115 and paraoxon hydrolysis as proceeding through general base catalysis by the metal-coordinated D269, in line with our previous work.25 We note that the geometries of nucleophilic attack are different for TBBL and paraoxon hydrolysis, with the scissile bond of paraoxon needing to be in-line with the attacking water molecule,83 whereas in the case of TBBL the nucleophile attacks from a Bürgi–Dunitz angle of 102°.84 However, the fact that PON1 appears to use different general bases for nucleophile activation for the two substrates resolves this issue, as the substrates occupy different positions in PON1’s binding pocket, placing them in the correct position relative to the nucleophile in each case (because of steric constraints, paraoxon cannot bind in a conformation that would allow for in-line attack by a water molecule that is deprotonated by H115) (Figure 1B). This causes the two substrates to use different subsets of active site residues to catalyze their respective hydrolysis reactions, which has been suggested to be one of the reasons for PON1’s selectivity and promiscuity.7,25 In addition, even though the overall substrate positions are different, in these two cases the C=O and P=O ester bonds overlay almost perfectly (Figure 1B), allowing for activation of the ester bond by the catalytic calcium ion (also see the discussion in refs (10) and (25)).
Figure 3.
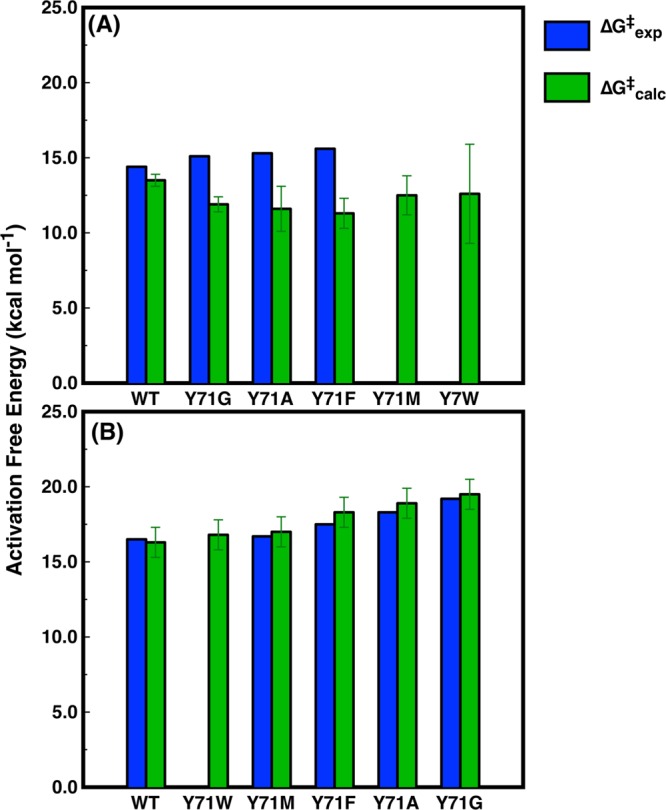
Comparison of calculated (green) and experimental (blue) activation free energies for the hydrolysis of (A) TBBL and (B) paraoxon by wild-type RePON1 and the Y71 variants of interest in this work. All energies are in kcal mol–1 and correspond to the values shown in Table 1. Error bars correspond to the standard deviations of over 30 individual EVB trajectories, which were generated as described in Materials and Methods. In the case of TBBL hydrolysis, the calculated activation free energies correspond to the rate-limiting step for a two-step process (Figure 2). For a breakdown of the calculated energies for each individual reaction step, see Table S5. Representative structures of the corresponding stationary points for each reaction step for the reactions catalyzed by the wild-type enzyme are shown in Figures S5 and S6.
Using this model, we are able to reproduce the effect of mutations of Y71 on the energetics of paraoxon hydrolysis with high accuracy and to obtain excellent quantitative agreement with the experimental data (Figure 3 and Table 1). In addition, we can clearly reproduce the increase in activation free energy observed for paraoxon hydrolysis upon perturbation of Y71, with a steady increase in the calculated activation free energy that is in line with the experimental observations.
In the case of TBBL hydrolysis, our calculated activation free energies appear to be slightly too low compared with the corresponding experimental values, and in addition, the Y71W substitution appears to disrupt the stability of the active site, considering the large standard deviation we obtain in the calculated activation free energy. Following from this, they also do not appear to capture the correct trend in reactivity. The lower activation free energies could be due to the calibration of the corresponding reference reaction in aqueous solution, to which we fit our EVB parabola, as this value is not known experimentally for TBBL hydrolysis and we were therefore forced to fit to quantum-chemical calculations of hydroxide attack on analogous lactone substrates.68 If our parametrization of the energetics of imidazole-catalyzed TBBL hydrolysis in aqueous solution underestimates the activation barrier for this process, this will cause the corresponding enzyme-catalyzed reactions to have artificially low activation free energies. In addition, correlating the calculated experimental activation free energies for PON1-catalyzed TBBL hydrolysis is more complex than for paraoxon hydrolysis for two reasons: (1) the actual experimental energy differences in the case of TBBL hydrolysis are extremely small (up to only a 1.2 kcal mol–1 increase relative to the wild type across the series of mutants; see Table 1) and thus very hard to reproduce computationally, and (2) the measured kcat actually corresponds to breakdown of the final product to release a thiol moiety, which is detected by using Ellman’s reagent (see ref (4)), and thus only provides us with an upper limit for the activation free energy for the lactone hydrolysis by each variant. However, our calculated values are never larger than the experimental values, and also, they never vary by more than about 2 kcal mol–1 across the series of all six RePON1 variants studied here, which, when the standard deviations of the calculated values are also taken into account, shows much less sensitivity to the Y71 substitutes than is observed in the case of paraoxon hydrolysis. This increases our confidence in the ability of our model to rationalize the origin of these differential effects of the Y71 substitutions on the two substrates.
From our MD simulations of the Michaelis complex, it was clear that although the loop dynamics is affected by mutation of Y71, there is no direct trend between the behavior of the loop and the specific mutation (i.e., the loop flops back and forth), and thus, the position of the active site loop alone is not the origin for the changes in paraoxonase activity upon mutation of Y71. However, there are significant differences in the position of the side chain of residue 71 upon mutation, with concomitant effects on the overall organization of the active site residues (Figures S4 and S7). The most conservative substitution is that of Y71 into Phe, where the hydrogen-bonding interaction with D183 is lost because of the absence of the −OH group. The model structures from our simulations reveal significant changes in the orientation of the F71 side chain compared with that of the wild-type Y71. For instance, in the absence of substrate, the F71 side chain is oriented into a hydrophobic region located below the flexible loop and formed by residues L69, I74, and V346. This in turn perturbs the configuration of this hydrophobic region in order to accommodate the bulkiness of the Phe side chain (Figure S7). In the presence of either paraoxon or TBBL, however, the F71 side chain is pushed toward outside the active site, which results in the opening of the flexible loop as well as changes in the configuration of H134. The substitution of Y71 into Trp leads to very similar observations (Figure S7).
Interestingly, the residues forming the hydrophobic region (i.e., L69, I74, and V346; Figure S7) were shown to play a critical role in PON1’s function and specificity, especially toward paraoxonase activity. For instance, it was shown that truncation of I74 into Ala reduced the paraoxonase activity by >25-fold, while the lactonase activity was not affected.7 Moreover, mutations of both L69 and V346 were shown by several studies to increase the organophosphatase activity of PON1 (e.g., mutations L69S/G and V346A).5,44,45 This could be due to changes in the configuration of the hydrophobic region upon mutation of these residues, which would affect the accommodation of bulky organophosphate side chains such as the O-ethyl side chains of paraoxon.
Substitution of Y71 with either Ala or Gly also seems to induce structural changes not only to the loop but also to the residues of the hydrophobic region. The absence of the bulkiness of Y71 seems to affect specifically the orientations of residues L69, I74, and V346 (Figure S7). In contrast, as a result of the flexible nature of the Y71M side chain, it occupies a position very similar to that of Y71, with minimal perturbations to the active site. This side chain is also relatively bulky and flexible and can accommodate multiple configurations, and therefore, one would not expect a significant perturbation upon substitution with Met. Overall, this analysis suggests that mutation at position 71 might also induce an indirect effect on the active site configuration, especially by changes in the orientations of residues L69, I74, and V346A, which in turn would be expected to have a functional impact on PON1 activity.
Clearly these structural perturbations reshape the active site, thus impacting the catalytic activity of the enzyme. This can be further observed by considering the O–P/O–C distances between the nucleophile and substrate as well as the distances associated with deprotonation of the nucleophilic water molecule, where perturbations appear to primarily affect the position of the nucleophile relative to the substrate and relative to the general base (Tables S6 and S7). We note additionally that we cannot directly compare these calculations to previous QM cluster85,86 and QM/MM87,88 studies of paraoxon hydrolysis, as these have primarily focused on the alkaline hydrolysis of paraoxon catalyzed by enzymes with bimetallo active sites. However, despite this limitation, we have obtained similar concerted and highly associative transition states as in refs (87) and (88), which is in good agreement with the experimental data on both enzymatic and nonenzymatic paraoxon hydrolysis65−67 and quantum-chemical calculations on the uncatalyzed reaction.89
Following from this, in an examination of the average occupancies (%) of hydrogen bonds in the hydrogen-bonding network along the active site tunnel at both the Michaelis complex and the transition state for each substrate (Tables S8 and S9, respectively), it can be seen that, in particular, mutation of Y71 disrupts hydrogen-bonding interactions between N168 and the substrate as well as interactions involving S166 and D183 as well as S166 and K192. The disruption of the hydrogen bond to K192 is particularly relevant, as we demonstrated in our recent study25 that the correct positioning of this residue is crucial to the paraoxonase activity of PON1. The importance of a positive charge at this position can also be seen from the effects of PON1 192R/Q polymorphs upon interaction with HDL. That is, in human PON1 both in vitro and in vivo, the 192Q isozyme exhibits significantly lower HDL binding and lipolactonase activity than the 192R polymorph.90 In addition, recombinant PON1-192K and PON1-192R show similar stability and lipolactonase activity upon association with HDL, whereas recombinant PON1-192Q shows both reduced stability and lipolactonase stimulation.90 Finally, in our computational work,25 we demonstrated that the effect of K192 on the paraoxonase activity is much larger than that on the lactonase activity, which may explain part of the observed differential effect of the Y71 mutations, which disrupt the hydrogen-bonding network keeping this residue in place, on the two activities. However, while it is clear that mutation of Y71 both affects the positioning of the active site loop and reshapes critical interactions in the active site pocket and along the central tunnel, these structural changes do not follow a specific trend with different mutations or explain why paraoxon hydrolysis is so much more affected by mutation of Y71 than TBBL hydrolysis. What, then, is the role of Y71 and the active site loop and where do these differences come from?
Active Site Hydrophobicity and the Role of the Active Site Loop in Determining Substrate Selectivity
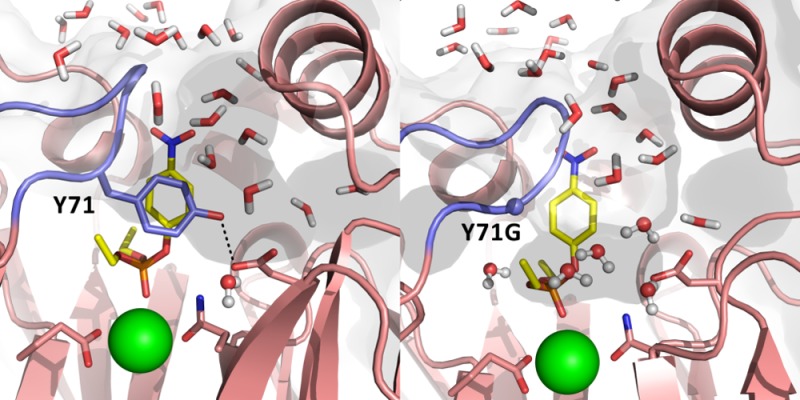
Up to this point, we have not taken into account changes in penetration of water into the active site upon Y71 modifications that lead to increased opening of the active site loop. This can be significant, as both paraoxon and TBBL are hydrophobic substrates, and, as can be seen from Figure 4, once the active site lid closes over the substrate, this creates a hydrophobic cage with only the nucleophilic water molecule in the immediate vicinity of the reacting atoms. This then alters the local electrostatic environment of the active site, which would be expected to strengthen the effect of local electrostatic interactions through lowering of the local dielectric constant. As expected, and as shown in Figure 4 for the example of the Y71G variant, substitutions of Y71 also affect the degree of solvent accessibility in PON1’s active site. To examine the extent to which this is affected by mutation of Y71 and breaking of the Y71–D183 hydrogen bond, we have calculated the average numbers of water molecules within 6 Å of the reacting parts of TBBL and paraoxon, defined as the carbonyl group and ring oxygen of TBBL and the phosphorus atom and connecting oxygen atoms of paraoxon, during our MD simulations of the Michaelis complex (Table 2). We note that the numbers of active site water molecules shown in Table 2 for simplicity do not include the nucleophilic water molecule and refer only to water molecules coordinating to the catalytic calcium ion and the mobile nonreactive water molecules in the active site. The corresponding values at the transition state and for the solvation of the nitro moiety of paraoxon are shown in Tables S10 and S11, respectively. These tables demonstrate that the Michaelis complex trends are largely retained in the transition state and that the main changes are observed in the proximity of the reacting atoms rather than the nitro group.
Figure 4.
Comparison of solvent accessibility in representative active sites of RePON1 in complex with (A, B) paraoxon and (C, D) TBBL in the Michaelis complexes of the wild-type and Y71G RePON1 variants, respectively. The solvent-accessible area is shown in shaded gray, and all water molecules that are within 6 Å of the reacting centers are shown explicitly.
Table 2. Solvent Accessibility of the Active Sites of Different RePON1 Variants in Their Substrate-Bound Forms and Its Impact on Solute–Solvent Interactions during the Hydrolysis Reactionsa.
| variant | system | WT | Y71F | Y71W | Y71M | Y71A | Y71G |
|---|---|---|---|---|---|---|---|
| number of active site water molecules | TBBL | 2.8 ± 0.5 | 2.9 ± 0.8 | 3.3 ± 1.0 | 3.3 ± 1.0 | 5.0 ± 2.2 | 5.6 ± 1.2 |
| paraoxon | 1.4 ± 0.6 | 3.1 ± 0.5 | 3.2 ± 0.6 | 3.0 ± 0.5 | 4.8 ± 1.1 | 5.9 ± 1.4 | |
| active site volume (Å3)b | TBBL | 163.2 ± 13.6 | 207.9 ± 22.9 | 199.3 ± 22.5 | 191.5 ± 22.1 | 248.4 ± 66.1 | 244.6 ± 32.1 |
| paraoxon | 192.3 ± 18.8 | 250.5 ± 19.8 | 232.0 ± 23.1 | 217.0 ± 19.3 | 258.7 ± 23.6 | 302.1 ± 37.1 | |
| λsolute–solvent (electrostatic)c | TBBL | 3.2 ± 1.5 | 3.3 ± 1.3 | 3.9 ± 1.3 | 3.5 ± 1.3 | 4.5 ± 2.6 | 6.0 ± 1.5 |
| paraoxon | 6.8 ± 0.9 | 7.7 ± 2.0 | 8.5 ± 1.6 | 7.3 ± 1.2 | 8.7 ± 2.7 | 13.4 ± 3.9 |
The number of water molecules in the active site refers to the number of water molecules within 6 Å of the reacting atoms of the substrate (defined as the carbonyl group and ring oxygen of TBBL and the phosphorus center and connecting atoms of paraoxon), calculated as averages over the last 25 ns of three molecular dynamics trajectories using Visual Molecular Dynamics.91 These numbers are not integers because they are averages over all snapshots from our molecular dynamics simulations. The corresponding values in the transition state are shown in Table S10.
The active site volume was calculated with Pocket Volume Measurer, version 2.0.1,92 using the default settings based on a sphere with an 8 Å radius located at the pocket center (−7, –21, 31). As can be seen from the data, substitution of Y71 not only increases the average active site volume but also the standard deviation of the active site volume, suggesting an increase in active site flexibility.
λsolute–solvent denotes the contribution of the interaction between the reacting atoms and solvent molecules to the total calculated reorganization energy, λ, in kcal mol–1. The data shown here are averages and standard deviations of the pocket volume over the last 25 ns of our equilibration runs for each system, averaged over three independent trajectories (i.e., a total of 75 ns of simulation time over the three trajectories).
As can be seen from Table 2, and as expected, the truncation of Y71 to a smaller side chain affects the active site similarly for both paraoxon and TBBL. However, while in both cases we observe an increase in the number of water molecules, for paraoxon the relative effect across the series of mutations seems slightly more significant. That is, while there are originally more water molecules near the reacting atoms when TBBL is bound to the active site, the number of water molecules within 6 Å of the TBBL ring during our simulations increases by only two additional water molecules moving closer to the substrate upon truncation of Y71 to either Ala or Gly compared with the wild-type enzyme. In contrast, in the case of paraoxon, where the bulky ethyl groups of the substrate push on the active site loop,8 the mutation of Y71 allows a steadily increasing number of water molecules to access to the active site, going from only one water molecule in the wild-type enzyme to six water molecules on average for the Y71G variant (also see Figure 4). This difference is interesting in light of the much smaller effect of mutations of Y71 on the lactonase activity than on the paraoxonase activity. In parallel, from an examination of the changes in active site volume upon substitution of Y71 (Table 2), it seems that there is a clear increase in active site volume upon substitution of Y71, which appears to follow the decrease in the paraoxonase activity of RePON1. We note that Table 2 shows not only the average volumes over the course of our MD simulations but also the corresponding standard deviations in these values. This gives an estimate of how “floppy” the active site is during the simulations (the larger the standard deviation, the more the active site volume changes). These data show not only that the active site volume increases upon substitution of Y71 but also that the active site becomes floppier. Finally, in order to quantify the contribution of the increasing number of water molecules to the calculated activation free energies, we have also applied the linear response approximation to our calculated EVB trajectories in order to extract λsolute–solvent, the contribution of solute–solvent interactions (where the “solute” is defined as the reacting atoms) to the total reorganization energy λ (Table 2). This provides a quantitative measure of the system reorganization upon moving between the different EVB states (in kcal mol–1), where a larger reorganization energy corresponds to a higher the activation free energy and vice versa (see clarifications in, e.g., refs (63) and (93)). As can be seen from these data, the change in the solute–solvent reorganization energy across the series of RePON1 variants is much larger in the case of paraoxon hydrolysis than in the case of TBBL hydrolysis, by a factor of ∼2. Taken together, these data suggest that the role of Y71 is to act as a hydrophobic gate, keeping the active site loop closed and thus restricting solvent access to the active site, in turn creating a hydrophobic cage for the hydrolysis of hydrophobic lactones and neutral organophosphates.
In view of the fact that many enzymes capable of organophosphatase activity are primarily either lactonases or have evolved from lactonases,4,9−12 this raises the intriguing question of whether active site hydrophobicity and mobile active site loops play a similar role in other organophosphatases. The most immediate point of comparison is the enzyme DFPase, which hydrolyzes diisopropylfluorophosphate (DFP) and has an active site virtually identical to that of PON1 (Figure 5). The key differences between the two active sites are the absence of a histidine at a position structurally equivalent to H115 in PON1, which presumably leads to the observed lack of lactonase activity in this enzyme94 (although other enzymes that lack a histidine at this position have shown lactonase activity), and the fact that the DFPase loop, which is the closest corresponding loop to PON1’s active site loop, is far more rigid and does not act as a lid over the active site.95 While both PON1 and DFPase share a similar six-bladed β-propeller fold with two Ca2+ ions in the central tunnel, PON1 has a unique addition of three helices covering the binding site that form the HDL-binding interface,16 which is not present in DFPase. More significantly, however, a more recently characterized lactonase with relatively high organophosphatase hydrolase activity from the phosphotriesterase-like lactonase family, SSoPox, has an active site loop that undergoes major conformational changes upon lactone binding and thus provides a hydrophobic channel to the active site.9,96 Another active site loop has been implicated in regulating the promiscuous organophosphatase activity of a lactonase from Geobacillus kaustophilus HTA426.74 Finally, several other organophosphatases, such as the bacterial phosphotriesterase97 (PTE), acetylcholine esterase,98 and organophosphate hydrolase,99 have largely hydrophobic active sites, and such active site hydrophobicity has been suggested to play a role in the multispecificity of these enzymes.16 In the case of PTE, it has also been possible to introduce lactonase activity through selective active site loop modeling, where shortening a key active site loop minimally reduces the paraoxonase activity (∼45-fold) while allowing the emergence of homoserine lactonase activity.100 It is important to note that these enzymes not only come from very different organisms but also have both very different overall secondary structures and active site architectures, and thus, creating a hydrophobic binding pocket (whether through active site gates like PON1’s active site loop or through having deeply buried active sites) is the only significant structural feature these enzymes have in common.
Figure 5.

Comparison of the tertiary structures of (A) RePON1 (PDB ID 3SRG) and (B) DFPase (PDB ID 1E1A), with the active site loops highlighted in orange. (C) Overlay of the catalytic Ca2+ and coordinating residues for the two enzymes, with RePON1 shown in blue and DFPase shown in green.
Interestingly, we recently performed a similar comparison of the structures and physicochemical properties of highly promiscuous members of the alkaline phosphatase superfamily,101 showing a correlation between the active site volume, the polar solvent-accessible surface area, and the number of characterized promiscuous activities in the literature. In the case of the alkaline phosphatases, these enzymes preferentially hydrolyze a range of mono- and dianionic phosphate and sulfate esters,3,46,47 and where paraoxonase activity has been measured for members of this superfamily, it has been extremely low.102 In contrast, for example, the bacterial phosphotriesterase, which has evolved to hydrolyze hydrophobic and neutral lactones and organophosphates, is a very poor catalyst of charged phosphodiesters.103 Therefore, the active site architectures that these enzymes have evolved to catalyze reactions involving charged substrates appear to be quite different from those evolved to catalyze neutral and hydrophobic organophosphates such as paraoxon and DFP. Active site architecture and charge discrimination are therefore clearly very important in determining the specificity and promiscuity in both sets of reactions, as not only do these bulky hydrophobic substrates prefer the solvent-excluded environment created by, for instance, closing of the active site loop of PON1, but also, in a low-dielectric environment, electrostatic interactions in the active site will be magnified,104 leading to much greater stabilization of, for instance, the oxyanion intermediate created during lactone and ester hydrolysis. Therefore, these enzymes have presumably evolved the ability to form hydrophobic cages in order to facilitate their native lactonase/esterase activities (for which the substrates are generally more hydrophobic), and the corresponding organophosphatase activities coincidentally happen to have similar chemical requirements as the native lactones/aryl esters, taking advantage of the architecture the enzyme provides for facilitating the native reaction. We note also that the importance of hydrophobic residues in facilitating organophosphate hydrolysis has also been discussed in other studies of individual organophosphatases (e.g., refs (77), (96), and (99)) and in the case of acetylcholine esterases, where the hydrophobic binding site105 shows sensitivity to both the hydrophobicity of inhibitors106 and to mutations of hydrophobic active site residues.107
Overview and Conclusions
The flexible loop in the mammalian PON family (i.e., residues 70–81 in PON1’s numbering) consists of both conserved and variable positions. The most conserved positions are 72 and 73 (Pro and Gly, respectively), which seem to have a structural role in shaping the flexible loop as a “lid” that covers the binding site. On the other hand, positions 74 and 75 are the most diverse and show different preferences for each family member (e.g., PON1-I74, PON2-L74, and PON3-M74). Position 71 also shows different preferences, such that while both PON1 and PON3 have Tyr and show paraoxonase activity, PON2 has either Phe or Cys (rarely) and shows no detectable paraoxonase activity.11,19 Similarly, the bacterial PON (PONX-OCCAL), which has Arg in position 71, also shows no detectable paraoxonase activity.11 Interestingly, the predicted mammalian ancestor of the PON family, N9,11 which shows a “generalist” activity profile, including paraoxonase activity, has a Tyr in position 71. It is therefore appealing to speculate that Y71 enables an optimized binding-site configuration for the binding and catalysis of organophosphates. Indeed, in attempts at directed evolution of PON1 toward efficient catalysis of organophosphates (including nerve agents), Y71 was substituted with various alternative side chains, but Tyr was retained in all variants with improved catalytic activities.108
In the present work, we have probed the role of the Tyr at position 71 further by performing detailed experimental and computational analyses of the role of selected Y71 variants in the active site stability and catalytic activity of PON1. We have demonstrated that while the measured kcat for the lactonase activity is perturbed only up to 8-fold upon substitution of Y71, the corresponding paraoxonase activity is perturbed 100-fold. Clearly, substitution of Y71 changes the active site by creating volume at this position, and Y71 plays a role in positioning all of the other participants in the hydrogen-bonding network along the central tunnel in the appropriate positions for substrate binding and catalysis. When this residue is removed, our simulations show that the new residue at position 71 is displaced from the original Y71 position, either through side-chain rotations in the case of the Trp/Phe substitutions or through being pushed out of the active site by the spectator ethyl groups of paraoxon. Therefore, substitutions at this position have a much larger effect on the active site architecture than would be anticipated, which in turn affects both substrate binding and the catalytic activity of the enzyme. From a structural perspective, studying the effect of substitutions at position 71 also highlights the potential roles of residue D183 in catalysis. That is, this residue, which forms a hydrogen bond to Y71 (Figure 1), has been shown to be completely immutable,6,7,25,77 despite the fact that it makes no direct contacts with either the lactone or organophosphate substrates. Previous work highlighted the significance of this residue but did not rationalize why this position is sensitive to substitution. Our computational analysis demonstrates that D183 appears to play an important role in “catching” Y71 to close the active site lid, rather than the other way around, as the loop does not fly out when this interaction is removed.
In addition, we have demonstrated that while substitution of Y71 clearly has an impact on the flexibility of the active site loop, the largest effect of substituting Y71 appears to be disrupting the position of K192 (which is a very important residue for modulating PON1 activity25,90) as well as, most critically, affecting the active site hydrophobicity. That is, substitution of Y71 expands the active site volume and allows for an increasing number of water molecules to enter the active site, an effect that is unsurprisingly most pronounced upon substitution of Y71 with either Ala or Gly. This effect is far more pronounced when paraoxon is present in the active site than when TBBL is present in the active site and thus appears to be the most likely origin of the differential effect of the Y71 substitutions on the two activities. This differential effect is also interesting in light of studies showing that enzymes’ native activities are far more robust toward mutations than their promiscuous activities,79,80 and such minor changes in the native activity and large changes in the promiscuous activity hint at the greater evolvability of promiscuous functions.
A comparison of the active site architecture of PON1 to those of other organophosphatases demonstrates that either a similar “gating” loop or a deeply buried active site that excludes solvent is a common feature of these enzymes. In addition, as with related promiscuous phosphatases such as members of the alkaline phosphatase superfamily, charge discrimination still plays a major role in determining the selectivity. That is, in the case of the alkaline phosphatase superfamily, these enzymes are promiscuous catalysts that preferentially facilitate the hydrolysis of mono- and dianionic substrates and show only poor activity toward neutral organophosphate esters such as paraoxon.3,46,102 In contrast, in the case of organophosphate hydrolases, these enzymes preferentially catalyze large hydrophobic substrates such as lipolactones or neutral phosphate esters, and where for example the diesterase activity has been measured, it has been significantly lower than the triesterase activity despite the similar compact and associative transition states expected for the hydrolysis of these two phosphate esters.83 We therefore posit that this ability to generate a hydrophobic active site cage to enclose the substrate is a key feature in facilitating the evolution of organophosphatase activity in these promiscuous enzymes, which are often primarily (in turn hydrophobic) lipolactonases or arylesterases. In addition to being of fundamental interest from a functional evolution perspective, these insights can also be utilized in order to rationally design next-generation catalysts. That is, a number of studies have explored the role of water in both enzyme engineering studies109,110 and engineered binding sites for drug design.111 We have demonstrated here as well that selective hydration of the active site allows PON1 to discriminate between its native lactonase and promiscuous organophosphatase activities. Therefore, if one can, for example, open up a space to allow more water molecules into the active site, this might be a rational way to limit one enzymatic activity without increasing or impacting the other. Therefore, active site hydration can also be used as a tool to control the artificial evolution of enzyme function, allowing the design of enzymes to select the substrate of a specific reaction from a pool of viable substrates.
Acknowledgments
The authors thank the Royal Swedish Academy of Sciences and the Knut and Alice Wallenberg Foundation for a Wallenberg Academy Fellowship to S.C.L.K. The European Research Council provided financial support under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement 306474. All computational work was performed on the Abisko Cluster at HPC2N in Umeå and on the Tintin Cluster at Uppmax in Uppsala through a generous allocation of computational time by the Swedish National Infrastructure for Computing (SNIC, 2015/16-12). We also thank Profs. Dan S. Tawfik and John Richard for valuable discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b10801.
Supplementary figures, including valence bond states used in the present study, root-mean-square deviations of Cα atoms in our MD simulations, and structures of representative stationary points; supplementary tables, including additional simulation details; and all of the EVB parameters used in the present work (PDF)
Author Present Address
§ M.B.-D.: Banting and Best Department of Medical Research, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, 160 College Street, Toronto, ON M5S 3E1, Canada.
The authors declare no competing financial interest.
Supplementary Material
References
- Khersonsky O.; Tawfik D. S. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Copley S. D. Trends Biochem. Sci. 2015, 40, 72–78. 10.1016/j.tibs.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabis A.; Duarte F.; Kamerlin S. C. L. Biochemistry 2016, 55, 3061–3081. 10.1021/acs.biochem.6b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Biochemistry 2005, 44, 6371–6382. 10.1021/bi047440d. [DOI] [PubMed] [Google Scholar]
- Amitai G.; Gaidukov L.; Adani R.; Yishay S.; Yacov G.; Kushnir M.; Teitlboim S.; Lindenbaum M.; Bel P.; Khersonsky O.; Tawfik D. S.; Meshulam H. FEBS J. 2006, 273, 1906–1919. 10.1111/j.1742-4658.2006.05198.x. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. J. Biol. Chem. 2006, 281, 7649–7656. 10.1074/jbc.M512594200. [DOI] [PubMed] [Google Scholar]
- Ben-David M.; Elias M.; Filippi J. J.; Dunach E.; Silman I.; Sussman J. L.; Tawfik D. S. J. Mol. Biol. 2012, 418, 181–196. 10.1016/j.jmb.2012.02.042. [DOI] [PubMed] [Google Scholar]
- Ben-David M.; Wieczorek G.; Elias M.; Silman I.; Sussman J. L.; Tawfik D. S. J. Mol. Biol. 2013, 425, 1028–1038. 10.1016/j.jmb.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Elias M.; Dupuy J.; Merone L.; Mandrich L.; Porzio E.; Moniot S.; Rochu D.; Lecomte C.; Rossi M.; Masson P.; Manco G.; Chabriere E. J. Mol. Biol. 2008, 379, 1017–1028. 10.1016/j.jmb.2008.04.022. [DOI] [PubMed] [Google Scholar]
- Elias M.; Tawfik D. S. J. Biol. Chem. 2012, 287, 11–20. 10.1074/jbc.R111.257329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Rogovsky H.; Hugenmatter A.; Tawfik D. S. J. Biol. Chem. 2013, 288, 23914–23927. 10.1074/jbc.M112.427922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier F.; Tokuriki N. J. Mol. Biol. 2014, 426, 2442–2456. 10.1016/j.jmb.2014.04.013. [DOI] [PubMed] [Google Scholar]
- Zech R.; Chemnitius J. M. In Paraoxonase (PON1) in Health and Disease; Costa L. G., Furlong C. E., Eds.; Springer: New York, 2002. [Google Scholar]
- Aviram M.; Rosenblat M.; Bisgaier C. L.; Newton R. S.; Primo-Parmo S. L.; La Du B. N. J. Clin. Invest. 1998, 101, 1581–1590. 10.1172/JCI1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrington P. N.; Mackness B.; Mackness M. I. Arterioscler., Thromb., Vasc. Biol. 2001, 21, 473–480. 10.1161/01.ATV.21.4.473. [DOI] [PubMed] [Google Scholar]
- Harel M.; Aharoni A.; Gaidukov L.; Brumshtein B.; Khersonsky O.; Meged R.; Dvir H.; Ravelli R. B.; McCarthy A.; Toker L.; Silman I.; Sussman J. L.; Tawfik D. S. Nat. Struct. Mol. Biol. 2004, 11, 412–419. 10.1038/nsmb767. [DOI] [PubMed] [Google Scholar]
- Gaidukov L. A.; Aharoni A.; Khersonsky O.; Tawfik D. S. FASEB J. 2008, 22 (1 Suppl.), 811.1. [Google Scholar]
- Mackness M. I.; Arrol S.; Abbott C.; Durrington P. N. Atherosclerosis 1993, 104, 129–135. 10.1016/0021-9150(93)90183-U. [DOI] [PubMed] [Google Scholar]
- Draganov D. I.; Teiber J. F.; Speelman A.; Osawa Y.; Sunahara R.; La Du B. N. J. Lipid Res. 2005, 46, 1239–1247. 10.1194/jlr.M400511-JLR200. [DOI] [PubMed] [Google Scholar]
- Billecke S.; Draganov D.; Counsell R.; Stetson P.; Watson C.; Hsu C.; La Du B. N. Drug Metab. Dispos. 2000, 28, 1335–1342. [PubMed] [Google Scholar]
- Lusis A. J. Nature 2000, 407, 233–241. 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soran H.; Younis N. N.; Charlton-Menys V.; Durrington P. Curr. Opin. Lipidol. 2009, 20, 265–274. 10.1097/MOL.0b013e32832ec141. [DOI] [PubMed] [Google Scholar]
- Rochu D.; Chabrière E.; Masson P. Toxicology 2007, 233, 47–59. 10.1016/j.tox.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Kanamori-Kataoka M.; Seto Y. Anal. Biochem. 2009, 385, 94–100. 10.1016/j.ab.2008.09.044. [DOI] [PubMed] [Google Scholar]
- Ben-David M.; Sussman J. L.; Maxwell C. I.; Szeler K.; Kamerlin S. C.; Tawfik D. S. J. Mol. Biol. 2015, 427, 1359–1374. 10.1016/j.jmb.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Kuo C.-L.; La Du B. N. Drug Metab. Dispos. 1998, 26, 653–660. [PubMed] [Google Scholar]
- Perera H. D.; Ma Y. Q.; Yang J.; Hirbawi J.; Plow E. F.; Qin J. Structure 2011, 19, 1664–1671. 10.1016/j.str.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S. J.; Hellmich U. A.; Ullrich S.; Glaubitz C. Nat. Chem. Biol. 2011, 7, 263–270. 10.1038/nchembio.543. [DOI] [PubMed] [Google Scholar]
- Goni F. M.; Montes L. R.; Alonso A. Prog. Lipid Res. 2012, 51, 238–266. 10.1016/j.plipres.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Zhdanov V. P.; Hook F. Biophys. Chem. 2012, 170, 17–24. 10.1016/j.bpc.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Hanson M. A.; Roth C. B.; Jo E.; Griffith M. T.; Scott F. L.; Rein hart G.; Desale H.; Clemons B.; Cahalan S. M.; Schuerer S. C.; Sanna M. G.; Han G. W.; Kuhn P.; Rosen H.; Stevens R. C. Science 2012, 335, 851–855. 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C. K.; Olefsky J. M. Cell Metab. 2012, 15, 635–645. 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R.; van der Ploeg K.; Ron D. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4628–4633. 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan C. D.; Marty M. T.; Sligar S. G.; Bailey R. C. Anal. Chem. 2013, 85, 2970–2976. 10.1021/ac3037359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. C.; Tian Y.; Yuan H.; Park H. W.; Chang Y. Y.; Kim J.; Kim H.; Neufeld T. P.; Dillin A.; Guan K. L. Nat. Cell Biol. 2013, 15, 741–750. 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaishy B.; Zhang Q.; Chung H. S.; Riehle C.; Soto J.; Jenkins S.; Abel P.; Cowart L. A.; Van Eyk J. E.; Abel E. D. J. Lipid Res. 2015, 56, 546–561. 10.1194/jlr.M055152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S.; Pande A. H.; Nemec K. N.; Tatulian S. A. J. Mol. Biol. 2004, 344, 71–89. 10.1016/j.jmb.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Burke J. E.; Dennis E. A. J. Lipid Res. 2009, 50, S237–S242. 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou Y. L.; Lin S. R.; Chang L. S. J. Pept. Sci. 2008, 14, 890–897. 10.1002/psc.1020. [DOI] [PubMed] [Google Scholar]
- Chiou Y. L.; Cheng Y. C.; Kao P. H.; Wang J. J.; Chang L. S. Toxicon 2008, 51, 270–279. 10.1016/j.toxicon.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Giger L.; Caner S.; Obexer R.; Kast P.; Baker D.; Ban N.; Hilvert D. Nat. Chem. Biol. 2013, 9, 494–498. 10.1038/nchembio.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Kiss G.; Rothlisberger D.; Dym O.; Albeck S.; Houk K. N.; Baker D.; Tawfik D. S. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 10358–10363. 10.1073/pnas.1121063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiswerk N.; Beck T.; Schulz J. D.; Milovnik P.; Mayer C.; Siegel J. B.; Baker D.; Hilvert D. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 8013–8018. 10.1073/pnas.1401073111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R. D.; Goldsmith M.; Ashani Y.; Simo Y.; Mullokandov G.; Bar H.; Ben-David M.; Leader H.; Margalit R.; Silman I.; Sussman J. L.; Tawfik D. S. Nat. Chem. Biol. 2011, 7, 120–125. 10.1038/nchembio.510. [DOI] [PubMed] [Google Scholar]
- Le Q. A. T.; Chang R.; Kim Y. H. Chem. Commun. 2015, 51, 14536–14539. 10.1039/C5CC05857K. [DOI] [PubMed] [Google Scholar]
- Mohamed M. F.; Hollfelder F. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 417–424. 10.1016/j.bbapap.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Pabis A.; Kamerlin S. C. L. Curr. Opin. Struct. Biol. 2016, 37, 14–21. 10.1016/j.sbi.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Ling M. M.; Robinson B. H. Anal. Biochem. 1997, 254, 157–178. 10.1006/abio.1997.2428. [DOI] [PubMed] [Google Scholar]
- Aharoni A.; Gaidukov L.; Yagur S.; Toker L.; Silman I.; Tawfik D. S. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 482–487. 10.1073/pnas.2536901100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalov M. V.; Dunbrack R. L. Jr. Structure 2011, 19, 844–858. 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Åqvist J.; Warshel A. Biochemistry 1989, 28, 4680–4689. 10.1021/bi00437a026. [DOI] [PubMed] [Google Scholar]
- Duarte F.; Bauer P.; Barrozo A.; Amrein B. A.; Purg M.; Åqvist J.; Kamerlin S. C. L. J. Phys. Chem. B 2014, 118, 4351–4362. 10.1021/jp501737x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra M. C.; Rath S. N.; Pradhan S. K.; Maharana J.; De S. Eur. Biophys. J. 2014, 43, 35–51. 10.1007/s00249-013-0937-6. [DOI] [PubMed] [Google Scholar]
- King G.; Warshel A. J. Chem. Phys. 1989, 91, 3647. 10.1063/1.456845. [DOI] [Google Scholar]
- Marelius J.; Kolmodin K.; Feierberg I.; Aqvist J. J. Mol. Graphics Modell. 1998, 16, 213–225. 10.1016/S1093-3263(98)80006-5. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.; Maxwell D.; Tirado-Rives J. J. Am. Chem. Soc. 1996, 118, 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Sorenson R. C.; Bisgaier C. L.; Aviram M.; Hsu C.; Billecke S.; La Du B. N. Arterioscler., Thromb., Vasc. Biol. 1999, 19, 2214–2225. 10.1161/01.ATV.19.9.2214. [DOI] [PubMed] [Google Scholar]
- Gaidukov L.; Tawfik D. S. Biochemistry 2005, 44, 11843–11854. 10.1021/bi050862i. [DOI] [PubMed] [Google Scholar]
- López-Canut V.; Roca M.; Bertrán J.; Moliner V.; Tuñón I. J. Am. Chem. Soc. 2010, 132, 6955–6963. 10.1021/ja908391v. [DOI] [PubMed] [Google Scholar]
- López-Canut V.; Roca M.; Bertrán J.; Moliner V.; Tuñón I. J. Am. Chem. Soc. 2011, 133, 12050–12062. 10.1021/ja2017575. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Weiss R. M. J. Am. Chem. Soc. 1980, 102, 6218–6226. 10.1021/ja00540a008. [DOI] [Google Scholar]
- Warshel A.Computer Modeling of Chemical Reaction in Enzymes and Solutions; Wiley: New York, 1991. [Google Scholar]
- Shurki A.; Derat E.; Barrozo A.; Kamerlin S. C. L. Chem. Soc. Rev. 2015, 44, 1037–1052. 10.1039/C4CS00241E. [DOI] [PubMed] [Google Scholar]
- Purcell J.; Hengge A. C. J. Org. Chem. 2005, 70, 8437–8442. 10.1021/jo0511997. [DOI] [PubMed] [Google Scholar]
- Caldwell S. R.; Raushel F. M.; Weiss P. M.; Cleland W. W. J. Am. Chem. Soc. 1991, 113, 730–732. 10.1021/ja00002a083. [DOI] [Google Scholar]
- Caldwell S. R.; Raushel F. M.; Weiss P. M.; Cleland W. W. Biochemistry 1991, 30, 7444–7450. 10.1021/bi00244a011. [DOI] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Calle E.; Casado J. J. Org. Chem. 2013, 78, 6868–6879. 10.1021/jo400258w. [DOI] [PubMed] [Google Scholar]
- Kaiser E. T.; Kezdy F. J. Prog. Bioorg. Chem. 1976, 4, 239–267. [Google Scholar]
- Roe D. R.; Cheatham T. E. III. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Botello-Smith D. S.; Cerutti T. E.; Cheatham T. E. III; Darden R. E.; Duke T. J.; Giese H.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Luo R.; Madej B.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Nguyen H. T.; Omelyan I.; Onufriev A.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 16; University of California: San Francisco, 2016.
- Hiblot J.; Gotthard G.; Elias M.; Chabriere E. PLoS One 2013, 8, e75272. 10.1371/journal.pone.0075272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; An J.; Yang G.-Y.; Bai A.; Zheng B.; Lou Z.; Wu G.; Ye W.; Chen H.-F.; Feng Y.; Manco G. PLoS One 2015, 10, e0115130. 10.1371/journal.pone.0115130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby S. D.; Norris J. R.; Richard Smith J.; Bahnson B. J.; Cerasoli D. M. Chem.-Biol. Interact. 2013, 203, 181–185. 10.1016/j.cbi.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Hu X.; Jiang X.; Lenz D. E.; Cerasoli D. M.; Wallqvist A. Proteins: Struct., Funct., Bioinf. 2009, 75, 486–498. 10.1002/prot.22264. [DOI] [PubMed] [Google Scholar]
- Josse D.; Xie W.; Renault F.; Rochu D.; Schopfer L. M.; Masson P.; Lockridge O. Biochemistry 1999, 38, 2816–2825. 10.1021/bi982281h. [DOI] [PubMed] [Google Scholar]
- Bar-Even A.; Milo R.; Noor E.; Tawfik D. S. Biochemistry 2015, 54, 4969–4977. 10.1021/acs.biochem.5b00621. [DOI] [PubMed] [Google Scholar]
- Aharoni A.; Gaidukov L.; Khersonsky O.; Gould S. M.; Roodveldt C.; Tawfik D. S. Nat. Genet. 2005, 37, 73–76. 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- Bloom J. D.; Romero P. A.; Lu Z.; Arnold F. H. Biol. Direct 2007, 2, 17. 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobeli I.; Favia A. D.; Thornton J. M. Nat. Biotechnol. 2009, 27, 157–167. 10.1038/nbt1519. [DOI] [PubMed] [Google Scholar]
- Kaltenbach M.; Emond S.; Hollfelder F.; Tokuriki N. PLoS Genet. 2016, 12, e1006305. 10.1371/journal.pgen.1006305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Sharma P. K.; Prasad R. B.; Warshel A. Q. Rev. Biophys. 2013, 46, 1–132. 10.1017/S0033583512000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürgi H. B.; Dunitz J. D.; Lehn J. M.; Wipff G. Tetrahedron 1974, 30, 1563–1572. 10.1016/S0040-4020(01)90678-7. [DOI] [Google Scholar]
- Chen S. L.; Fang W. H.; Himo F. J. Phys. Chem. B 2007, 111, 1253–1255. 10.1021/jp068500n. [DOI] [PubMed] [Google Scholar]
- Kim J.; Tsai P. C.; Chen S. L.; Himo F.; Almo S. C.; Raushel F. M. Biochemistry 2008, 47, 9497–9504. 10.1021/bi800971v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. Y.; Gao J. Biochemistry 2007, 46, 13352–13369. 10.1021/bi700460c. [DOI] [PubMed] [Google Scholar]
- López-Canut V.; Ruiz-Pernía J. J.; Castillo R.; Moliner V.; Tuñón I. Chem. - Eur. J. 2012, 18, 9612–9621. 10.1002/chem.201103615. [DOI] [PubMed] [Google Scholar]
- Zheng F.; Zhan C.-G.; Ornstein R. L. J. Chem. Soc., Perkin Trans. 2 2001, 2355–2363. 10.1039/b101747k. [DOI] [Google Scholar]
- Gaidukov L.; Rosenblat M.; Aviram M.; Tawfik D. S. J. Lipid Res. 2006, 47, 2492–2502. 10.1194/jlr.M600297-JLR200. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; De Oliveira C. A. F.; McCammon J. A. J. Mol. Graphics Modell. 2011, 29, 773–776. 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Blum M. M.; Chen J. C. Chem.-Biol. Interact. 2010, 187, 373–379. 10.1016/j.cbi.2010.02.043. [DOI] [PubMed] [Google Scholar]
- Blum M. M.; Löhr F.; Richardt A.; Rüterjans H.; Chen J. C. J. Am. Chem. Soc. 2006, 128, 12750–12757. 10.1021/ja061887n. [DOI] [PubMed] [Google Scholar]
- Hiblot J.; Gotthard G.; Chabriere E.; Elias M. Sci. Rep. 2012, 2, 779. 10.1038/srep00779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhooke J. L.; Benning M. W.; Raushel F. M.; Holden H. M. Biochemistry 1996, 35, 6020–6025. 10.1021/bi960325l. [DOI] [PubMed] [Google Scholar]
- Greenblatt D. M.; Dvir H.; Silman I.; Sussman J. L. J. Mol. Neurosci. 2003, 20, 369–383. 10.1385/JMN:20:3:369. [DOI] [PubMed] [Google Scholar]
- Yang H.; Carr P. D.; McLoughlin S. Y.; Liu J. W.; Horne I.; Qiu X.; Jeffries C. M. J.; Russell R. J.; Oakeshott J. G.; Ollis D. L. Protein Eng., Des. Sel. 2003, 16, 135–145. 10.1093/proeng/gzg013. [DOI] [PubMed] [Google Scholar]
- Afriat Jurnou L.; Jackson C. J.; Tawfik D. S. Biochemistry 2012, 51, 6047–6055. 10.1021/bi300694t. [DOI] [PubMed] [Google Scholar]
- Barrozo A.; Duarte F.; Bauer P.; Carvalho A. T. P.; Kamerlin S. C. L. J. Am. Chem. Soc. 2015, 137, 9061–9076. 10.1021/jacs.5b03945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo B.; Jonas S.; Babtie A. C.; Benjdia A.; Berteau O.; Hyvönen M.; Hollfelder F. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2740–2745. 10.1073/pnas.0903951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H.; Hong S.-B.; Raushel F. M. J. Biol. Chem. 1998, 273, 17445–17450. 10.1074/jbc.273.28.17445. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg G. M.; Mednick M. L.; Maddox J.; Rice R.; Cramer J. J. Med. Chem. 1975, 18, 1056–1061. 10.1021/jm00245a002. [DOI] [PubMed] [Google Scholar]
- Shin S.; Roth L. G.; Chen C.-H. Int. J. Biochem. 1991, 23, 1131–1136. 10.1016/0020-711X(91)90154-F. [DOI] [PubMed] [Google Scholar]
- Strub C.; Alies C.; Lougarre A.; Ladurantie C.; Czaplicki J.; Fournier D. BMC Biochem. 2004, 5, 9. 10.1186/1471-2091-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith M.; Ashani Y.; Simo Y.; Ben-David M.; Leader H.; Silman I.; Sussman J. L.; Tawfik D. S. Chem. Biol. 2012, 19, 456–466. 10.1016/j.chembiol.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Rezaei K.; Jenab E.; Temelli F. Crit. Rev. Biotechnol. 2007, 27, 183–195. 10.1080/07388550701775901. [DOI] [PubMed] [Google Scholar]
- Hendil-Forssell P.; Martinelle M.; Syrén P.-O. Chem. Commun. 2015, 51, 17221–17224. 10.1039/C5CC07162C. [DOI] [PubMed] [Google Scholar]
- Ladbury J. E. Chem. Biol. 1996, 3, 973–980. 10.1016/S1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.