

Abstract
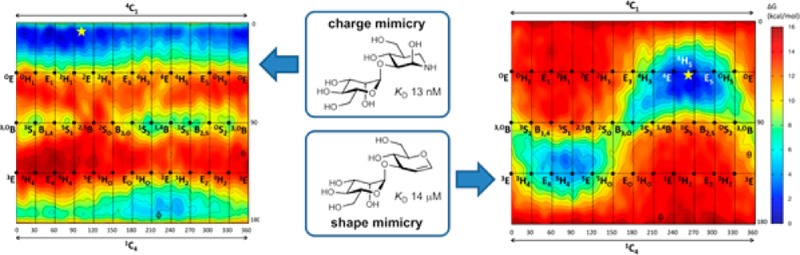
Inhibitor design incorporating features of the reaction coordinate and transition-state structure has emerged as a powerful approach for the development of enzyme inhibitors. Such inhibitors find use as mechanistic probes, chemical biology tools, and therapeutics. Endo-α-1,2-mannosidases and endo-α-1,2-mannanases, members of glycoside hydrolase family 99 (GH99), are interesting targets for inhibitor development as they play key roles in N-glycan maturation and microbiotal yeast mannan degradation, respectively. These enzymes are proposed to act via a 1,2-anhydrosugar “epoxide” mechanism that proceeds through an unusual conformational itinerary. Here, we explore how shape and charge contribute to binding of diverse inhibitors of these enzymes. We report the synthesis of neutral dideoxy, glucal and cyclohexenyl disaccharide inhibitors, their binding to GH99 endo-α-1,2-mannanases, and their structural analysis by X-ray crystallography. Quantum mechanical calculations of the free energy landscapes reveal how the neutral inhibitors provide shape but not charge mimicry of the proposed intermediate and transition state structures. Building upon the knowledge of shape and charge contributions to inhibition of family GH99 enzymes, we design and synthesize α-Man-1,3-noeuromycin, which is revealed to be the most potent inhibitor (KD 13 nM for Bacteroides xylanisolvens GH99 enzyme) of these enzymes yet reported. This work reveals how shape and charge mimicry of transition state features can enable the rational design of potent inhibitors.
Introduction
Over 500 000 gene sequences have been discovered encoding glycoside hydrolases that are grouped into more than 130 families according to the Carbohydrate Active enZyme classification (CAZy: www.cazy.org).1 Glycoside hydrolases of family 99 possess two distinct activities: endo-α-1,2-mannosidase and endo-α-1,2-mannanase. endo-α-1,2-Mannosidases are eukaryotic proteins involved in N-linked glycan maturation, folding, and quality control2−5 and are of clinical significance as they provide a means for viruses and cancer to evade the effect of exo-glucosidase inhibitors.5,6endo-α-1,2-Mannanases are produced by Bacteroides spp., bacterial residents of the gut microbiota.7 They facilitate the degradation of dietary yeast mannan consumed in bread and fermented foods, facilitating the breakdown of these complex carbohydrates, with beneficial effects on the gastrointestinal tract and, possibly, mitigating the symptoms of Crohn’s disease.8 Given the importance of family GH99 enzymes in N-linked glycan maturation and carbohydrate processing by the microbiota, the development of inhibitors has been of particular importance to allow assessment and manipulation of their roles in these complex processes. In this work, we investigate several mechanism-inspired inhibitor design concepts for family GH99 endo-α-1,2-mannanases from the gut microbiota constituents Bacteroides thetaiotaomicron and Bacteroides xylanisolvens; BtGH99 and BxGH99, respectively. Our results cast light on the importance of structural mimicry of shape and charge of species along the reaction coordinate for achieving potent inhibition of this enzyme family.
Glycosidases that cleave their substrates with retention of anomeric configuration typically operate through a two-step mechanism that proceeds via a covalent glycosyl-enzyme intermediate. Such enzymes utilize enzymatic amino acid residues that in the first step act as general acid and nucleophile to assist in departure of the anomeric substituent while simultaneously substituting the anomeric group; in the second step the first carboxylate acts as a general base to deprotonate a nucleophilic water molecule that hydrolyzes the covalent glycosyl enzyme intermediate (Figure 1A).9,10 Important exceptions include a range of β-hexosaminidases that perform catalysis through mechanisms involving neighboring group participation by the 2-acetamido group of the substrate (Figure 1B).11 These enzymes also operate through a two-step mechanism: in the first step an amino acid residue acts as a general acid to assist in departure of the leaving group while the 2-acetamido group performs a nucleophilic attack on the anomeric center, forming a bicyclic oxazoline/oxazolinium ion intermediate. In the second step the same amino acid residue acts as a general base, assisting nucleophilic attack by a water molecule that opens the oxazolinium ion ring, reforming the 2-acetamido group and completing the hydrolysis reaction.
Figure 1.

(A) Mechanism for a canonical retaining α-glycosidase that proceeds through a covalent glycosyl-enzyme intermediate. (B) Mechanism for a retaining β-hexosaminidase involving neighboring group participation by the 2-acetamido group, via an oxazolinium ion intermediate. (C) Proposed mechanism for family GH99 α-mannosidases involving neighboring group participation by the 2-OH group, via a 1,2-anhydro sugar (epoxide). Numbering is for BxGH99.
Family GH99 enzymes cleave glycosides with an overall retention of anomeric configuration.12 X-ray structures are available for GH99 enzymes in complex with a variety of ligands based on sugar-shaped heterocycles.7,8,12 However, in X-ray structures of B. xylanisolvens BxGH99 with various substrate-mimicking ligands, it was not possible to identify an appropriately positioned enzymatic nucleophile within the typical <3 Å distance from the reactive anomeric center, leading to the proposal of a nonclassical mechanism.12 In particular, in structures of GlcIFG (2) and ManIFG (3) with BxGH99, there were no close contacts with a likely candidate enzymatic nucleophile,7,12 at odds with the usual observation of a carboxylate situated typically 2.6–2.7 Å away in classical retaining glycosidases. Moreover, in complexes of GlcDMJ (1) with BxGH99 a conserved carboxyl residue (E333; numbering refers to BxGH99) was located 2.7 Å from the 2-OH group;12 similar observations extend to the binding of a substrate (α-Man-1,3-α-ManMU) to the carboxamide mutant BxGH99 E333Q.7 Collectively these data supported the proposal of a two-step reaction involving in the first step the formation of a bicyclic 1,2-anhydro sugar intermediate, through E333 acting as a general base residue to deprotonate the 2-OH and facilitating a nucleophilic substitution at C1 coincident with departure of the leaving group, assisted by E336 acting as general acid (Figure 1C).12 In the second step of this proposed mechanism, E333 acts as a general acid, assisting ring opening of the epoxide, while E336 acts as a general base, promoting nucleophilic attack by a water molecule. While such a mechanism lacks precedent in enzymes, there is strong evidence that the base-catalyzed solvolysis of 4-nitrophenyl α-d-mannoside and α-mannosyl fluoride proceed through similar neighboring group participation mechanisms.13−16 The most stable conformation of 1,2-anhydro-β-d-mannose is a 4H5 half-chair; applying the principle of least nuclear motion, a 4C1 → 4E‡ → 4H5 conformational itinerary has been proposed for the first step of the family GH99 reaction coordinate.17
Intensive efforts have been invested in the rational development of glycosidase inhibitors, and many fundamental principles have been articulated inspired by our deep mechanistic understanding of this class of enzyme. Based on pioneering insights from Pauling18 and Wolfenden,19 it is recognized that a common principle underpinning catalysis is the selective affinity of an enzyme for a reaction transition state, relative to the ground state. Accordingly, inhibitor design by transition state mimicry, which can take advantage of the high transition state affinity of a glycosidase, has proven a useful guiding strategy.20 While it is widely appreciated that perfect transition state mimics are chemically unstable and thus unattainable, a general design principle is to develop analogues incorporating features that mimic the shape and charge of the transition state.20 Three features have been highlighted for consideration in the development of effective glycosidase inhibitors: configuration, conformation and charge.21 Configuration is the simplest to address and not surprisingly it is usually found that glycosidases are normally best targeted by inhibitors with stereochemistry matching that of the substrate. In the case of BxGH99, an enzyme that has the ability to cleave both α-Glc-1,3-α-Man-OR and α-Man-1,3-α-Man-OR configured substrates (with a preference for the latter),7 optimal inhibition is achieved by inhibitors matching the preferred substrate configuration. Glycosidases typically operate through transition states with substantial oxocarbenium ion character, and partial double bond development between O5 and C1, leading to a flattened conformation at the transition state. Consequently, mimicry of the flattened conformation expected at the transition state has proven a second effective strategy, with inhibitors bearing sp2-hybridized atoms at the anomeric or endocyclic oxygen positions, such as glyconolactones and -lactams, identified as fairly broad spectrum glycosidase inhibitors. Finally, partial charge development at C1 and the endocyclic oxygen at the transition state can be mimicked by the protonated forms of nitrogen-containing heterocycles, exemplified by deoxymannojirimycin (DMJ) with a nitrogen in place of the endocyclic oxygen, and isofagomine with a nitrogen in place of C1.
Motivated by the unusual mechanism proposed
for family GH99 enzymes
and the complexity of their biochemical roles, our understanding of
which should benefit from the development of potent inhibitors, we
undertook a study of several inhibitor designs inspired by the concepts
of charge and shape mimicry. GlcDMJ (1), an inhibitor
reported by Spiro and co-workers,22 was
the first effective inhibitor of mammalian endo-α-1,2-mannosidase,6 and was subsequently shown to bind to and inhibit BxGH99 and BtGH99 (Table 1).12 More potent
inhibition was achieved by GlcIFG (2),12 which also proved to be a more effective inhibitor than
GlcDMJ of mammalian endo-α-1,2-mannosidase
in cell-based studies,23 demonstrating
that varying the position of charge can provide improvements in potency.
Further, upon identification of the Bacteroides spp.
enzymes as preferential endo-α-1,2-mannanases,7,8 we were able to configurationally match the substrate and develop
the inhibitor ManIFG (3), the most potent inhibitor yet
reported for any GH99 enzyme.7 However,
ManIFG and GlcIFG lack the 2-OH group of the substrate and thus cannot
benefit from specific interactions with the putative acid/base E333.
Separately, Spiro and co-workers reported that two other neutral compounds
were almost as effective as GlcDMJ in the inhibition of mammalian endo-α-1,2-mannosidase, namely, GlcddMan (4) and GlcGlucal (6).6,24 We were intrigued
by these observations and sought to investigate whether the equivalent
configurationally matched species, ManddMan (5) and ManGlucal
(7), and the related cyclohexene derivative (8) were inhibitors of bacterial GH99 enzymes and to understand how
they bind to the enzyme.
Table 1. Dissociation Constants for GH99 endo-Mannanase Inhibitors.
|
KD values (μM) |
|||
|---|---|---|---|
| compd | BtGH99 | BxGH99 | method |
| 1 (GlcDMJ) | 24 | NDa | ITC12 |
| 2 (GlcIFG) | 0.63 | NDa | ITC12 |
| 3 (ManIFG) | 0.14 | 0.27 | ITC6 |
| 5 (ManddMan) | 53 ± 5 | 221 ± 11 | NMR |
| 7 (ManGlucal) | 15 ± 1.9 | 111 ± 11 | NMR |
| 8 (GlcChex) | no binding | no binding | NMR/ITC |
| 9 (ManNOE) | 0.03 ± 0.01 | 0.013 ± 0.002 | ITC |
ND, not determined.
Results and Discussion
Synthesis of ManGlucal and ManddMan
ManddMan (5) and ManGlucal (7) were prepared from α-1,3-mannobiose (10) (Scheme 1). Acetylation, followed by bromination afforded mannobiosyl bromide (12), which was converted to the protected glucal (13) using Zn in MeOH. Zemplén transesterification afforded ManGlucal (7). Alternatively, reduction of 13 using H2/Pd–C, followed by Zemplén transesterification afforded ManddMan (5). The preparation of GlcChex (8) will be described elsewhere.
Scheme 1. Synthesis of ManddMan (5) and ManGlucal (7).

Binding and 3-D Structural Analyses of Putative “Shape Mimics”
Dissociation constants for the binding of compounds 5 and 7 to BtGH99 and BxGH99 were determined by NMR spectroscopy (Figure S1). The 2D NMR SOFAST-HMQC spectra of 15N-labeled enzymes determined in the presence or absence of a saturating amount of the ligands revealed several H–N peaks that displayed significant chemical shift perturbations. For instance, new signals for an arginine residue (assigned as R295 in BxGH99 and R291 in BtGH99 on the basis of analysis of inter-residue nOes from the 3D-HSQC-NOESY spectra; see annotation to Figure 4A) appeared during the titration experiments, which were in slow exchange with the initial ones in the chemical shift time scale (Figure 2). Therefore, since the relative intensities of these signals are proportional to the populations of the bound and unbound forms (see Experimental Section and Supporting Information (SI)), the dissociation constants (KD) were readily calculated. The binding constants are shown in Table 1. No evidence for binding of GlcChex could be obtained by either NMR or ITC.
Figure 4.

(A) X-ray structure of ternary complex of BxGH99 with ManNOE (above) and α-1,2-mannobiose (below). The residue used for NMR titrations, R295, is shown in yellow. 2F0 – Fc map contoured at 1.0 σ (0.42 eÅ–3). The +1 subsite mannose residue electron density is best modeled by two mannose conformations with 0.6/0.4 occupancy, rotated by about 30° with respect to +2 mannose. (B) Stereoview of ManNOE in the active site. 2F0 – Fc map contoured at 1.5 σ (0.58 eÅ–3). Assembled using CCP4mg.29
Figure 2.
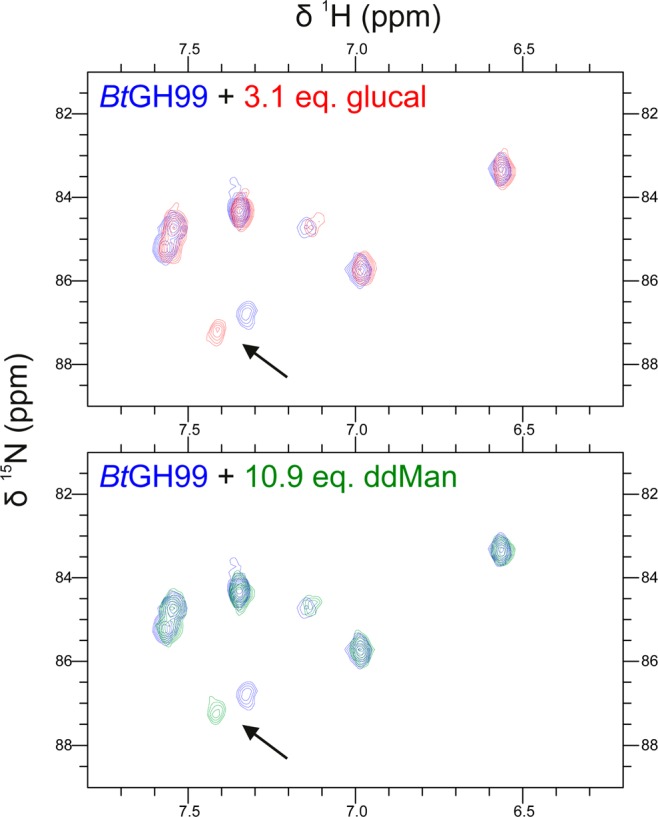
Excerpt of the side chain HN ε-arginine region in the 2D SOFAST-HMQC NMR spectra of 15N-labeled BtGH99 in the absence (blue) or presence of an excess (top) of ManGlucal (7; red) or (bottom) ManddMan (5; green). The arrow highlights the chemical shift perturbation observed for the HN-ε signal corresponding to R295.
In order to analyze the mode of binding of the conformationally restricted compounds, 3-D structures of complexes of BxGH99 with 7 and 8 were determined by X-ray crystallography at near atomic (approximately 1.0 Å) resolutions (Table 2, Figure 3A,B). ManGlucal 7 (KD 111 μM) binds to BxGH99 in the −2 and −1 subsites, with the −1 glucal ring intact in a 4H5 conformation. For reasons most likely related to its poor affinity for the enzyme, we were unable to obtain a complex of GlcChex with wildtype enzyme, but were serendipitously successful in obtaining a complex with the catalytically inactive BxGH99 E333Q mutant. In this complex GlcChex 8 also bound in the −2/–1 subsite with the cyclohexene ring in a 4H5 conformation (Figure 3B). Relative to ManGlucal, GlcChex suffers both by replacement of the endocyclic oxygen with methylene and by the presence of a nonreducing-end glucosyl moiety, the latter of which is known to reduce binding to the Bacteroides spp. enzymes by 4–10-fold.7 Owing to the unmeasurable binding of GlcChex, the synthesis of the mannose analogue was not pursued. The lack of oxygen atoms within the GlcChex ring means it cannot form hydrogen bonds with active site residues Y252, E333 or E336. MS experiments indicated that the compound is not affected by the enzyme or its variants so the reason for binding to only the inactive variant is unclear.
Table 2. X-ray Data and Structure Summary.
| 5 (ManddMan) | 7 (ManGlucal) | 8 (GlcChex) | 10 (ManNOE) | 10 + M2 (ManNOE + 1,2-α-mannobiose) | |
|---|---|---|---|---|---|
| resolution (outer shell) (Å) | 76.77–1.03 (1.05–1.03) | 76.85–1.07 (1.09–1.07) | 39.54–1.2 (1.22–1.2) | 76.73–1.14 (1.16–1.14) | 57.21–1.05 (1.07–1.05) |
| Rmerge (outer) | 0.052 (0.989) | 0.052 (1.748) | 0.059 (0.955) | 0.051 (1.158) | 0.054 (1.314) |
| Rcryst/Rfree | 0.117/0.130 | 0.124/0.141 | 0.119/0.137 | 0.124/0.143 | 0.115/0.133 |
| rmsd bonds (Å) | 0.010 | 0.011 | 0.012 | 0.011 | 0.013 |
| rmsd angles (deg) | 1.53 | 1.54 | 1.59 | 1.50 | 1.67 |
| PDB code | 5M17 | 5M5D | 5MEL | 5LYR | 5M03 |
Figure 3.

Stereoview of active site of B. xylanisolvens family GH99 enzyme complexes. (A) BxGH99 with ManGlucal (7), (B) BxGH99-E333Q with GlcChex (8), and (C) BxGH99 with ManddMan (5). Depicted electron density maps are REFMAC28 maximum-likelihood/σA weighted 2F0 – Fc syntheses contoured at 1.5σ (0.57, 0.59, and 0.62 eÅ–3, respectively). All panels were assembled using CCP4mg.29
Glycals are often effective inhibitors of classical retaining glycosidases. Inhibition is typically found to be time-dependent, owing to a chemical reaction in which the conjugate acid of the nucleophile protonates the enol ether of the glycal, and forms a 2-deoxy-glycosyl enzyme.25,26 This mode of reactivity has been exploited to allow the identification of the catalytic nucleophile by peptide sequencing,25 and the 2-deoxyglycosyl-enzymes are sufficiently stable to be studied by X-ray crystallography.27 To date, only two classes of retaining glycosidases have been identified upon which glycals bind as competitive inhibitors without exhibiting this mode of reactivity. These are N-acetylhexosaminidases that use neighboring group participation and retaining sialidases, and in both cases these enzymes lack a typical carboxylate nucleophile. Retaining N-acetylhexosaminidases use a 2-acetamido group, which is able to catalyze the slow hydration of the enol ether.30 Retaining sialidases lack a carboxylate nucleophile capable of protonating the glycal, and instead use a less acidic tyrosine residue as the catalytic nucleophile.31 In the case of sialidases, the corresponding glycal 2,3-didehydro-2-deoxy-N-acetylneuraminic acid has been elaborated into the extraordinarily potent inhibitor zanamivir,32 a clinically used antiinfluenza drug. Despite its potency, quantitative examination of transition state mimicry by zanamivir reveals it to be a poor transition state mimic.33 The observation that ManGlucal binds to BxGH99 without a chemical reaction despite the retaining mechanism of the enzyme provides further evidence in favor of the unique neighboring group participation mechanism proposed for this family.
Binding and 3-D Structural Analyses of ManddMan: Probing the Role of O2 Interactions
In order to harness the tighter binding of the bacterial enzymes with a mannoside in the −2 subsite7 ManddMan (5) was synthesized (Scheme 1). NMR titration revealed ManddMan to bind to BxGH99 and BtGH99 with KD values of 221 and 53 μM, respectively (Table 1). By comparison, GlcddMan (4) is an inhibitor for rat endomannosidase6 with an IC50 value of 3.8 μM for inhibition of cleavage of 14C-labeled GlcMan9GlcNAc, only slightly worse than that of GlcDMJ (1) (IC50 = 1.7 μM). The structure of BxGH99 in complex with ManddMan was determined at 1.03 Å resolution. Compound 5 binds in the −2/–1 subsites of the enzyme in an undistorted 4C1 conformation. This conformation matches that of the ground state of the substrate, and while this is consistent with the modest dissociation constant, it is noteworthy that the tight-binding inhibitor ManIFG 3 also binds in a 4C1 chair.
Design, Synthesis, and Characterization of ManNOE as a GH99 Inhibitor
As ManIFG (3), ManddMan (5), and ManGlucal (7) all lack an O2 group, in considering the contribution of shape and charge to inhibition, it would appear that the cationic nature of ManIFG contributes most significantly to inhibition, in spite of its conformational resemblance to the ground state. We therefore decided that it would be appropriate to investigate a charged inhibitor based on ManIFG that was able to make the correct O2 interactions, in particular with E333. Inspired by the work of Bols and co-workers on the development of noeuromycin, a 2-hydroxy analogue of isofagomine that binds 2–4000 times more tightly than isofagomine to various glycosidases,34 we therefore synthesized the noeuromycin derivative, ManNOE. This inhibitor was synthesized by the regioselective mannosylation of the nitrile diol 16(35) by trichloroacetimidate 15Scheme 2.36 The sole acetate group of the glycoside 17 was cleaved by treatment with HCl/MeOH to afford alcohol 18, and the nitrile group was reduced using BH3·Me2S, followed by protection as the Boc derivative 19. Hydrogenolysis of the benzyl ethers of 19 using H2/Pd–C, and then cleavage of the Boc group with HCl, afforded ManNOE.HCl (9) [as a mixture of α-hydroxypiperidine and pyranose isomers (not drawn); see the SI].
Scheme 2. Synthesis of ManNOE (9).

Isothermal titration calorimetry revealed ManNOE (9) to bind to BxGH99 with KD = 13 nM, and to BtGH99 with a KD = 30 nM, 17- and 5-fold more tightly than ManIFG (3) to the respective enzymes, commensurate with improvements seen for binding of IFG versus NOE for other enzymes,34,37 and demonstrating that better matching of the substrate by reinstatement of the 2-OH group absent in the latter compound provides more effective inhibition (Table 1, Figure S4). 3-D structures were solved of a binary complex of BxGH99–ManNOE, and a ternary complex of BxGH99–ManNOE−α-1,2-mannobiose at resolutions of 1.14 and 1.05 Å, respectively (Figure 4). The poses of ManNOE in both complexes were essentially identical and the more informative ternary complex, with ManNOE in the −2/–1 subsites, and α-1,2-mannobiose in the +1/+2 subsites will therefore be discussed. The NOE heterocycle binds in a 4C1 conformation, similar to that seen for ManIFG with the same enzyme. A close contact with E333 Oδ···O2 of 2.58 Å is evident, similar to that seen in the complex of BxGH99 with GlcDMJ (2.54 Å, PDB 4AD3).
Conformational Analyses of Glucal, Chex, ddMan, and NOE
In order to understand the intrinsic conformational preferences of the D-glucal, 1,2-dideoxymannose (ddMan), Chex, and noeuromycin (NOE) inhibitor warheads, so as to help rationalize the conformations observed on-enzyme, we calculated their conformational free energy landscapes (FELs). FELs were computed by ab initio metadynamics (see Experimental Section), and the Cremer–Pople puckering coordinates θ and ϕ were used as collective variables, yielding a Mercator representation38 for each inhibitor FEL (Figure 5). The same procedure has been previously used to analyze the conformational preferences of related GH inhibitors (mannoimidazole, glucoimidazole, and IFG).39,40
Figure 5.

Conformational free-energy landscapes (FELs, Mercator projection) of isolated D-glucal (A), Chex (B), ddMan (C), and NOE (D), contoured at 1 kcal mol–1. FELs have been annotated (yellow star) with the inhibitor conformations of ManGlucal 7 (for A), GlcChex 8 (for B), ManddMan 5 (for C), and ManNOE 9 (for D) that have been observed on-enzyme in this work.
The FELs of both D-glucal and Chex (Figure 5A,B) exhibit a main energy minimum centered at 4H5. The free energy well is quite large and extends through 4E–4H5–E5, with an “arm” toward B3,O, indicating substantial conformational freedom around 4H5. There is also a local minimum in the southern hemisphere, centered at 5H4. However, this is 3 kcal mol–1 higher in energy and thus less populated at room temperature. Interconversion between the two minima is hindered by an 8 kcal mol–1 energy barrier. Therefore, both D-glucal and Chex display only one main accessible conformation at room temperature, 4H5, but can readily adopt the nearby 4E conformation predicted for the transition states leading to and from the proposed 1,2-anhydrosugar intermediate. These data suggest that D-glucal and Chex provide good shape mimicry of the transition state or intermediate for the GH99 catalyzed reaction.
That both D-glucal and Chex adopt a 4H5 conformation when complexed to BxGH99 (Table 2 and yellow star in Figure 5A,B) indicates that the conformational preference of the isolated molecules are not significantly perturbed on-enzyme. This is also the case for ddMan, for which the FEL (Figure 5C) is strongly biased toward the 4C1 chair (the local minima on the equator are ≈8 kcal/mol higher in energy), as observed in the X-ray structure on-enzyme. However, the FEL of ddMan does not exhibit any stable minimum around 4H5, thus it cannot be considered a GH99 transition state shape mimic, and should instead be considered a mimic of the substrate conformation in the Michaelis complex.
NOE differs from the other inhibitors considered here as it is a basic molecule and was therefore considered as both the neutral (Figure 5D) and protonated species (Figure S5). While the topographies of the FELs for the two protonation states are broadly similar, they differ greatly in relative energies, most importantly for the global and local minima, such that the most stable state in NOE, a 4C1 chair, becomes the “ring-flipped” 1C4 conformer in protonated NOE. In that case, the two most stable species, the 1C4 and 1S5 states, are characterized by the presence of transannular hydrogen bonds between NH2+ and O6 or O3, respectively. What then is the most appropriate FEL to consider in relation to the enzyme-bound state, which is expected to be protonated, but which in a 4C1 conformation lacks a transannular hydrogen bond? It is known that in the absence of solvation (in the gas phase) flexible molecules in low charge states tend to compensate charge effects by forming stabilizing intramolecular interactions that do not take place in other environments (e.g., in solution). For example, low-charge state proteins in the gas phase tend to adopt more compact structures owing to increased intramolecular interactions.41 On balance, we consider the FEL of neutral NOE to be a more relevant representation of the enzyme-bound conformations, because interactions of the inhibitor with active site residues prevent the formation of intramolecular hydrogen bonds that dominate the conformations of charged species. Accordingly, the FEL displays a wide main minimum situated close to 4C1 (Figure 5D), and is thus similar to that of ddMan. The NOE FEL is also reminiscent of the one previously computed for the closely related neutral IFG inhibitor39 (which differs from NOE by the absence of the 2-hydroxyl group). Interestingly, the transition state region between the north pole and the equator in NOE (4H3/4E) is shifted by 60° in ϕ in ddMan. This is likely due to a vicinal intramolecular hydrogen bond formed between the 2-OH and the 3-OH, which stabilizes the 4H3/4E conformations in NOE; this interaction is not present in ddMan or IFG as they both lack a 2-OH. Overall, NOE most closely resembles the conformation of the substrate in the Michaelis complex and in its protonated state on-enzyme provides mimicry of an oxocarbenium-ion-like transition state; it is therefore best considered a “charge”-mimicking inhibitor with no significant shape mimicry of the transition state.
Conclusions
The proposed GH family 99 neighboring group participation mechanism, via a 1,2-anhydro sugar, allows prediction of a 4C1 → 4E‡ → 4H5 conformational itinerary for the first step of the reaction coordinate. The FELs for Glucal and Chex suggest that when these inhibitor warheads are extended to ManGlucal and GlcChex, their flattened conformations provide mimicry of the 4E transition state and 4H5 intermediate conformations. Their nonbasic nature provides shape but not charge mimicry of the transition state. X-ray structures of these compounds in complex with BxGH99 revealed them to bind in a 4H5 conformation, most closely matching the proposed intermediate conformation. The modest dissociation constant of ManGlucal, and lack of detectable binding for GlcChex, suggests that shape mimicry of the intermediate or transition state provides only weak affinity for the enzyme. On the other hand the FEL for the neutral sugar ddMan reveals a preference for a 4C1 conformation, and the corresponding complex of ManddMan with BxGH99 revealed this compound to bind in the same conformation, albeit with an affinity relative to ManGlucal 8- or 18-fold worse for Bx or BtGH99 enzymes, respectively. As ManGlucal, GlcChex and ManddMan all lack a 2-OH group, these ratios provide an estimate for the contribution of shape-mimicry of the TS or intermediate to enzyme binding.
Based on the above analysis, and combined with the previous discovery that the best inhibitor for GH99 endo-α-mannanases is ManIFG,7 which provides charge mimicry, but poor transition state shape mimicry, we were inspired to reinstate the 2-hydroxyl group missing in this compound. ManNOE was synthesized and shown to bind to Bx and BtGH99 with KD values of 30 and 13 nM, the most tightly binding ligand for these enzymes yet reported, and a 20-fold enhancement of affinity relative to ManIFG. The X-ray structure of ManNOE in complex with BxGH99 reveals a 4C1 ground-state conformation mimicking the Michaelis complex, and we conclude that this inhibitor acts primarily to mimic the charge of the transition state.
Experimental Section
X-ray Crystallography
Data reduction was performed using xia242 or XDS.43 Data set HKL index was matched to a previous solution using Aimless44,45 software and FreeR set was generated from BxGH99–ManddMan data, and then used for every other data set. Initial polypeptide chain model was obtained from the same previous solution, refined against BxGH99–ManddMan data and used as starting model for other structure solutions. Refmac528 with ProSmart was used for restrained refinement and Coot46 for real-space refinement. During model rebuilding the Fobs – Fcalc difference map was examined at 3σ. Validation was performed using Coot and edstats.47 Sugar and pseudopyranose conformations and density correlation were validated by Privateer.48,49
Isothermal Titration Calorimetry
ManNOE binding to Bt and BxGH99 was measured using a MicroCal AutoITC200 instrument (Malvern Instruments, formerly GE Healthcare) at 25 °C in 25 mM HEPES pH 7.0, 100 mM NaCl buffer. Protein concentration was kept at 5 μM and ligand concentration at 50 μM. As ManNOE exists as a 5:2 ratio of d-gluco and d-manno NOE isomers; the effective ManNOE concentration was adjusted accordingly.
2D NMR Titrations
Two-dimensional 1H–15N SOFAST-HMQC50 spectra were recorded at 298 K for 1 h using 15N-labeled BtGH99 and BxGH99 on a Bruker AVANCE III 800 MHz spectrometer with cryoprobe. Upon binding of ManGlucal or ManddMan, chemical shift perturbations were observed in slow exchange regime. A signal corresponding to Nε-Hε of R295 (numbering based on BxGH99), and which is close to the enzyme active site, was chosen as binding reporter. In the case of BxGH99, this signal shifts from δ 7.33, 87.5 ppm (1H,15N) in the free state to δ 7.41, 87.2 ppm (1H,15N) in the bound state. Bound and free protein populations at different protein/ligand ratios were calculated from peak intensities. NMR measurements were made in 50 mM potassium phosphate pH 7.0, 50 mM KCl with 5% D2O added. Protein concentration was determined by measuring 280 nm UV absorbance after denaturing the solution in 6 M guanidinium chloride (average protein concentration: 58 μM). Ligand concentrations were cross verified by integrating the 1H peaks against the internal standard TSP-d4 (Sigma-Aldrich). The NMR data were processed and integrated using NMRPipe.51 The dissociation constants (KD values) were estimated using in-house Matlab 2015b scripts, using the following equation:
where L is the free ligand concentration and ([PL]/PT) is the ligand-bound protein fraction. Duplicate experimental points were used for error-bar estimation. Errors were propagated using a Monte Carlo algorithm to estimate the uncertainty in the KD values. A distribution of KD values (n = 10 000) were obtained from data sets randomly varying within the error bars, and the standard deviation of was used for the KD error estimation.
Quantum Chemical Calculations
To obtain the conformational free energy landscapes of ddMan, Glucal, Chex, and NOE, quantum mechanical calculations were performed using density functional theory based molecular dynamics (MD), according to the Car–Parrinello (CP) method.52 Each molecule was enclosed in an isolated cubic box of 12.0 Å × 12.0 Å × 12.0 Å. A fictitious electron mass of 700 au was used for the CP Lagrangian and a time step of 0.12 fs was used in all CPMD simulations. The Kohn–Sham orbitals were expanded in a plane wave (PW) basis set with a kinetic energy cutoff of 70 Ry. Ab initio pseudopotentials, generated within the Troullier–Martins scheme, were employed.53 The Perdew, Burke, and Ernzerhoff generalized gradient-corrected approximation (PBE)54 was selected in view of its good performance in previous work on isolated sugars,55 glycosidases,56 and glycosyltransferases.57
The metadynamics algorithm,58 provided by the Plumed 2 plugin,59 was used to explore the conformational free energy landscape of the systems, taking as collective variables θ and φ of the puckering coordinates of Cremer and Pople,60 in the spirit of the pioneering work by Dowd, French, and Reilly.61 Initially, the height of these Gaussian terms was set at 0.6 kcal mol–1 and a new Gaussian-like potential was added every 250 MD steps. Once the whole free energy space was explored, the height of the Gaussian terms was reduced to half of its initial value (0.3 kcal·mol–1) and a new Gaussian-like potential was added every 500 MD steps. The width of the Gaussian terms was set to 0.10 Å. The simulations were stopped when energy differences among wells remain constant, which was further confirmed by a time-independent free energy estimator.62 For all molecules, the phase space was fully explored in less than 60 ps and the simulations were further extended up to 140 ps for Chex and Glucal, 160 ps for ddMan, and 240 ps for NOE. The errors in the principal minima, taken as a standard deviation (SD) from the last 60 ps, are below 0.6 kcal mol–1 (Figure S6).
Acknowledgments
We thank the Australian Research Council (FT130100103; DP120101396), the BBSRC (BB/G016127/1) and the ERC (ERC-2012-AdG-322942), the Spanish Ministry of Economy and Competitiveness (CTQ2014-55174, CTQ2015-64597-C2-1P, CTQ2015-68756-R) and the Generalitat de Catalunya (2014SGR-987). We thank Diamond Light Source for access to beamlines i02, i04, i04-1, and i24 (mx-9948) that contributed to the results presented here. We are grateful to Sivanandam Veeramuthu for his help with NMR. In-house crystal screening and testing was performed on X-ray equipment provided, in part, by the Wellcome Trust. The authors gratefully acknowledge the computer resources at MareNostrum and the technical support provided by BSC-CNS (RES-QCM-2016-3-00017). L.R. thanks the University of Barcelona for an APIF predoctoral fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b10075.
Experimental procedures, analytical data (1H and 13C NMR, MS) for all new compounds; detailed crystallographic experimental and data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. Nucleic Acids Res. 2014, 42, D490–5. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas W. A.; Spiro R. G. J. Biol. Chem. 1987, 262, 3775–81. [PubMed] [Google Scholar]
- Lubas W. A.; Spiro R. G. J. Biol. Chem. 1988, 263, 3990–8. [PubMed] [Google Scholar]
- Moore S. E.; Spiro R. G. J. Biol. Chem. 1990, 265, 13104–12. [PubMed] [Google Scholar]
- Moore S. E.; Spiro R. G. J. Biol. Chem. 1992, 267, 8443–51. [PubMed] [Google Scholar]
- Hiraizumi S.; Spohr U.; Spiro R. G. J. Biol. Chem. 1993, 268, 9927–35. [PubMed] [Google Scholar]
- Hakki Z.; Thompson A. J.; Bellmaine S.; Speciale G.; Davies G. J.; Williams S. J. Chem. - Eur. J. 2015, 21, 1966–77. 10.1002/chem.201405539. [DOI] [PubMed] [Google Scholar]
- Cuskin F.; Lowe E. C.; Temple M. J.; Zhu Y.; Cameron E. A.; Pudlo N. A.; Porter N. T.; Urs K.; Thompson A. J.; Cartmell A.; Rogowski A.; Hamilton B. S.; Chen R.; Tolbert T. J.; Piens K.; Bracke D.; Vervecken W.; Hakki Z.; Speciale G.; Munoz-Munoz J. L.; Day A.; Pena M. J.; McLean R.; Suits M. D.; Boraston A. B.; Atherly T.; Ziemer C. J.; Williams S. J.; Davies G. J.; Abbott D. W.; Martens E. C.; Gilbert H. J. Nature 2015, 517, 165–9. 10.1038/nature13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott M. L. Chem. Rev. 1990, 90, 1171–202. 10.1021/cr00105a006. [DOI] [Google Scholar]
- Zechel D. L.; Withers S. G. Acc. Chem. Res. 2000, 33, 11–8. [DOI] [PubMed] [Google Scholar]
- Vocadlo D. J.; Davies G. J. Curr. Opin. Chem. Biol. 2008, 12, 539–55. 10.1016/j.cbpa.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Williams R. J.; Hakki Z.; Alonzi D. S.; Wennekes T.; Gloster T. M.; Songsrirote K.; Thomas-Oates J. E.; Wrodnigg T. M.; Spreitz J.; Stutz A. E.; Butters T. D.; Williams S. J.; Davies G. J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 781–6. 10.1073/pnas.1111482109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasman R. C.; Johnson D. C. J. Org. Chem. 1966, 31, 1830–8. 10.1021/jo01344a037. [DOI] [Google Scholar]
- Micheel F.; Borrmann D. Chem. Ber. 1960, 93, 1143–7. 10.1002/cber.19600930524. [DOI] [Google Scholar]
- Kyosaka S.; Murata S.; Tanaka M. Chem. Pharm. Bull. 1983, 31, 3902–5. 10.1248/cpb.31.3902. [DOI] [Google Scholar]
- Speciale G.; Farren-Dai M.; Shidmoossavee F. S.; Williams S. J.; Bennet A. J. J. Am. Chem. Soc. 2016, 138, 14012–9. 10.1021/jacs.6b07935. [DOI] [PubMed] [Google Scholar]
- Speciale G.; Thompson A. J.; Davies G. J.; Williams S. J. Curr. Opin. Struct. Biol. 2014, 28, 1–13. 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L. Am. Sci. 1948, 36, 51–8. [PubMed] [Google Scholar]
- Wolfenden R. Acc. Chem. Res. 1972, 5, 10–8. 10.1021/ar50049a002. [DOI] [Google Scholar]
- Heightman T. D.; Vasella A. T. Angew. Chem., Int. Ed. 1999, 38, 750–70. . [DOI] [PubMed] [Google Scholar]
- Gloster T. M.; Vocadlo D. J. Nat. Chem. Biol. 2012, 8, 683–94. 10.1038/nchembio.1029. [DOI] [PubMed] [Google Scholar]
- Spohr U.; Bach M.; Spiro R. G. Can. J. Chem. 1993, 71, 1928–42. 10.1139/v93-240. [DOI] [Google Scholar]
- Alonzi D. S.; Kukushkin N. V.; Allman S. A.; Hakki Z.; Williams S. J.; Pierce L.; Dwek R. A.; Butters T. D. Cell. Mol. Life Sci. 2013, 70, 2799–814. 10.1007/s00018-013-1304-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spohr U.; Bach M.; Spiro R. G. Can. J. Chem. 1993, 71, 1919–27. 10.1139/v93-239. [DOI] [Google Scholar]
- Legler G.; Roeser K. R.; Illig H. K. Eur. J. Biochem. 1979, 101, 85–92. 10.1111/j.1432-1033.1979.tb04219.x. [DOI] [PubMed] [Google Scholar]
- Arribas J. C.; Herrero A. G.; Martin-Lomas M.; Canada F. J.; He S.; Withers S. G. Eur. J. Biochem. 2000, 267, 6996–7005. 10.1046/j.1432-1327.2000.01784.x. [DOI] [PubMed] [Google Scholar]
- Hill C. H.; Graham S. C.; Read R. J.; Deane J. E. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20479–84. 10.1073/pnas.1311990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–67. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas S.; Potterton E.; Wilson K. S.; Noble M. E. M. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 386–94. 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana A. G.; Vadlamani G.; Mark B. L.; Withers S. G. Chem. Commun. 2016, 52, 7943–6. 10.1039/C6CC02520J. [DOI] [PubMed] [Google Scholar]
- Watts A. G.; Damager I.; Amaya M. L.; Buschiazzo A.; Alzari P.; Frasch A. C.; Withers S. G. J. Am. Chem. Soc. 2003, 125, 7532–3. 10.1021/ja0344967. [DOI] [PubMed] [Google Scholar]
- von Itzstein M.; Wu W.-Y.; Kok G. B.; Pegg M. S.; Dyason J. C.; Jin B.; Phan T. V.; Smythe M. L.; White H. F.; Oliver S. W.; Colman P. M.; Varghese J. N.; Ryan D. M.; Woods J. M.; Bethell R. C.; Hotham V. J.; Cameron J. M.; Penn C. R. Nature 1993, 363, 418–23. 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- Shidmoossavee F. S.; Watson J. N.; Bennet A. J. J. Am. Chem. Soc. 2013, 135, 13254–7. 10.1021/ja405916q. [DOI] [PubMed] [Google Scholar]
- Liu H.; Liang X.; Søhoel H.; Bülow A.; Bols M. J. Am. Chem. Soc. 2001, 123, 5116–7. 10.1021/ja010240u. [DOI] [PubMed] [Google Scholar]
- Goddard-Borger E. D.; Stick R. V. Aust. J. Chem. 2007, 60, 211–3. 10.1071/CH07016. [DOI] [Google Scholar]
- Mayer T. G.; Schmidt R. R. Eur. J. Org. Chem. 1999, 1999, 1153–65. . [DOI] [Google Scholar]
- Meloncelli P. J.; Gloster T. M.; Money V. A.; Tarling C. A.; Davies G. J.; Withers S. G.; Stick R. V. Aust. J. Chem. 2007, 60, 549–65. 10.1071/CH07188. [DOI] [Google Scholar]
- Davies G. J.; Planas A.; Rovira C. Acc. Chem. Res. 2012, 45, 308–16. 10.1021/ar2001765. [DOI] [PubMed] [Google Scholar]
- Williams R. J.; Iglesias-Fernandez J.; Stepper J.; Jackson A.; Thompson A. J.; Lowe E. C.; White J. M.; Gilbert H. J.; Rovira C.; Davies G. J.; Williams S. J. Angew. Chem., Int. Ed. 2014, 53, 1087–91. 10.1002/anie.201308334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankrathok A.; Iglesias-Fernández J.; Williams R. J.; Pengthaisong S.; Baiya S.; Hakki Z.; Robinson R. C.; Hrmova M.; Rovira C.; Williams S. J.; Ketudat Cairns J. R. ACS Catal. 2015, 5, 6041–51. 10.1021/acscatal.5b01547. [DOI] [Google Scholar]
- Warnke S.; von Helden G.; Pagel K. J. Am. Chem. Soc. 2013, 135, 1177–80. 10.1021/ja308528d. [DOI] [PubMed] [Google Scholar]
- Winter G. J. Appl. Crystallogr. 2010, 43, 186–90. 10.1107/S0021889809045701. [DOI] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–32. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–92. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–14. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle I. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 454–67. 10.1107/S0907444911035918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agirre J.; Davies G.; Wilson K.; Cowtan K. Nat. Chem. Biol. 2015, 11, 303. 10.1038/nchembio.1798. [DOI] [PubMed] [Google Scholar]
- Agirre J.; Iglesias-Fernandez J.; Rovira C.; Davies G. J.; Wilson K. S.; Cowtan K. D. Nat. Struct. Mol. Biol. 2015, 22, 833–4. 10.1038/nsmb.3115. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Kupče E̅.; Brutscher B. J. Biomol. NMR 2005, 33, 199–211. 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277–93. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Car R.; Parrinello M. Phys. Rev. Lett. 1985, 55, 2471–4. 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- Troullier N.; Martins J. L. Phys. Rev. B: Condens. Matter Mater. Phys. 1991, 43, 1993–2006. 10.1103/PhysRevB.43.1993. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865–8. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Biarnes X.; Ardevol A.; Planas A.; Rovira C.; Laio A.; Parrinello M. J. Am. Chem. Soc. 2007, 129, 10686–93. 10.1021/ja068411o. [DOI] [PubMed] [Google Scholar]
- Raich L.; Borodkin V.; Fang W.; Castro-Lopez J.; van Aalten D. M.; Hurtado-Guerrero R.; Rovira C. J. Am. Chem. Soc. 2016, 138, 3325–32. 10.1021/jacs.5b10092. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Rovira C. J. Am. Chem. Soc. 2015, 137, 7528–47. 10.1021/jacs.5b01156. [DOI] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–6. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. Comput. Phys. Commun. 2014, 185, 604–13. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Cremer D.; Pople J. A. J. Am. Chem. Soc. 1975, 97, 1354–8. 10.1021/ja00839a011. [DOI] [Google Scholar]
- Dowd M. K.; French A. D.; Reilly P. J. Carbohydr. Res. 1994, 264, 1–19. 10.1016/0008-6215(94)00185-5. [DOI] [Google Scholar]
- Tiwary P.; Parrinello M. J. Phys. Chem. B 2015, 119, 736–42. 10.1021/jp504920s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.