

Abstract
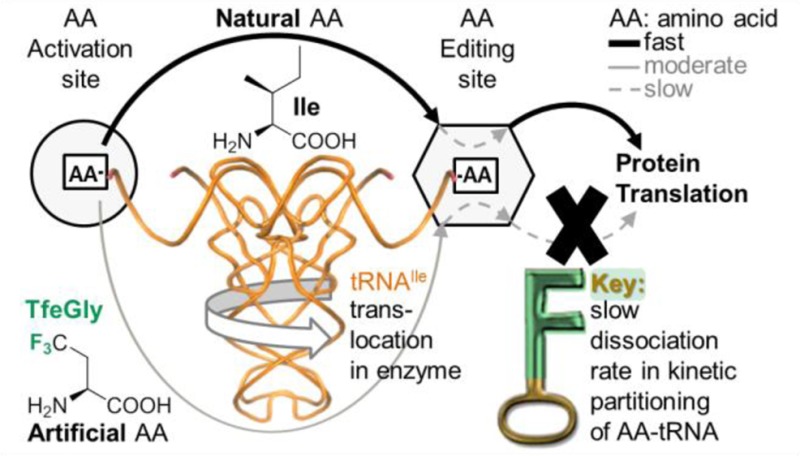
Fluorine being not substantially present in the chemistry of living beings is an attractive element in tailoring novel chemical, biophysical, and pharmacokinetic properties of peptides and proteins. The hallmark of ribosome-mediated artificial amino acid incorporation into peptides and proteins is a broad substrate tolerance, which is assumed to rely on the absence of evolutionary pressure for efficient editing of artificial amino acids. We used the well-characterized editing proficient isoleucyl-tRNA synthetase (IleRS) from Escherichia coli to investigate the crosstalk of aminoacylation and editing activities against fluorinated amino acids. We show that translation of trifluoroethylglycine (TfeGly) into proteins is prevented by hydrolysis of TfeGly-tRNAIle in the IleRS post-transfer editing domain. The remarkable observation is that dissociation of TfeGly-tRNAIle from IleRS is significantly slowed down. This finding is in sharp contrast to natural editing reactions by tRNA synthetases wherein fast editing rates for the noncognate substrates are essential to outcompete fast aa-tRNA dissociation rates. Using a post-transfer editing deficient mutant of IleRS (IleRSAla10), we were able to achieve ribosomal incorporation of TfeGly in vivo. Our work expands the knowledge of ribosome-mediated artificial amino acid translation with detailed analysis of natural editing function against an artificial amino acid providing an impulse for further systematic investigations and engineering of the translation and editing of unusual amino acids.
Short abstract
In this work, we show that the distinct chemical features of fluorinated amino acids may influence synthetic or editing pathways of aminoacyl-tRNA synthetases and contribute to protein quality control.
Numerous reports describing the influence of artificial amino acid side chains containing one, few, or several fluorine atoms on various peptide/protein properties have provided insights into the unpredictable ways in which this unique element engages in intermolecular interactions within protein environments.1 We recently reported that P1 position mutants of the protease inhibitor BPTI containing either (2S)-4,4-difluoroethylglycine (DfeGly) or (2S)-4,4,4-trifluoroethylglycine (TfeGly) exhibit wild-type level inhibitor activity against β-trypsin, whereas the hydrocarbon parent BPTI containing (2S)-ethylglycine ((2S)-2-aminobutanoic acid (Abu)) is markedly less effective at inhibiting this enzyme.2 In the protein environment of an enzyme active site, crystal structures supported a role for the fluorinated side chains in which multipolar interactions with protein amide fragments and a potential weak hydrogen bond to a structural water dominate. This is of course a very different kind of behavior than that observed, for example, by Marsh and co-workers in the environment of the hydrophobic core of a helical bundle; here, enhanced thermostability mediated by fluorine was based on increased buried hydrophobic surface area and volume.3 In another recent study using DfeGly and TfeGly, we showed that side chain fluorination has differential effects on the protease stability of a model peptide substrate depending upon the number of fluorine atoms, the position of the unnatural amino acid, and the particular enzyme studied.4
The studies described above were made possible by chemical synthesis of the fluorinated building blocks DfeGly and TfeGly, followed by automated solid-phase peptide synthesis (SPPS). However, this method has limitations with respect to the length of the polypeptide that can be synthesized. Thus, ribosomal synthesis would be a valuable tool for the synthesis of large proteins containing such artificial residues.
Ribosomal polypeptide synthesis is mediated by aminoacyl-tRNA synthetases (AARSs). These central enzymes in protein synthesis have been shown to be often unable to discriminate between sterically and biophysically similar canonical (cAAs, Figure 1b, left) and noncanonical (ncAAs, Figure 1b, right) amino acids.5 Thus, DfeGly and TfeGly have the potential to be accepted by the natural translation apparatus due to their similar calculated van der Waals volume and hydrophobicity compared to the natural substrates Ile, Leu, Val, and Met.6 AARSs display a conserved mechanism of catalysis (Figure 1a, Aminoacylation).7 First, they use the energy stored in ATP to activate the amino acid (AA), forming an aminoacyl-adenylate intermediate (AARS–AA-AMP) (Figure 1a, AA activation). In the second step, the AA is transferred to its corresponding tRNA (Figure 1a, AA transfer to tRNA). At the end of this process the charged tRNA dissociates to solution (Figure 1a, AA-tRNA dissociation from AARS) and can take part in translation at the ribosome. Fidelity of ribosomal peptide synthesis highly depends on the ability of AARSs to discriminate efficiently against noncognate AAs (Figure 1b). Some AARS families are unable to distinguish cognate (Figure 1b, left) from noncognate AAs in the aminoacylation reaction (Figure 1a, aminoacylation) alone. These enzymes possess additional editing activities in order to hydrolyze erroneously activated amino acids and misacylated tRNAs (Figure 1a, hydrolytic editing).8
Figure 1.
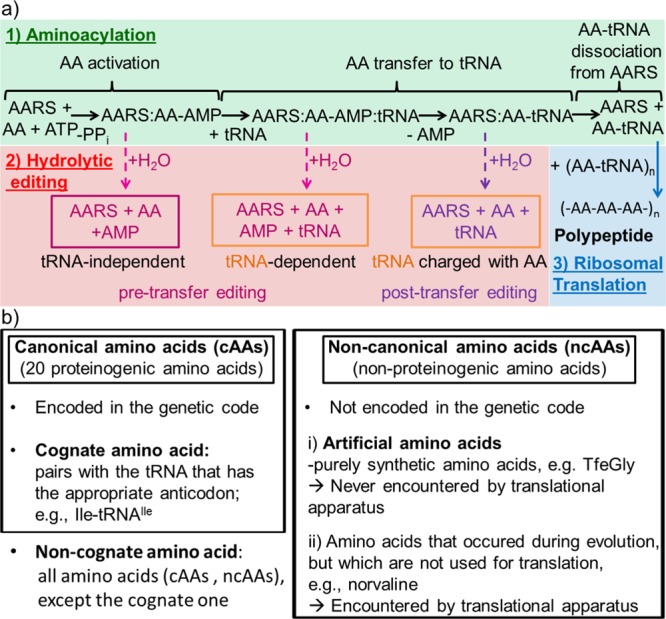
(a) Overall kinetic scheme for hydrolytic editing by AARSs. Additionally, nonenzymatic editing is possible by release of the reaction intermediates and their hydrolysis free in solution. (b) Nomenclature used in this work for referring to amino acids.
In order to enhance translation of noncanonical amino acids several approaches were taken. One is to alter the substrate specificity by reengineering of the AARSs binding pockets. Successful examples comprise incorporations of 3-iodophenylalanine, 3-chlorophenylalanine and 3-bromophenylalanie and 3-iodotyrosine by engineered PheRS and TyRS, respectively.9−12 Oki et al. further increased the substrate specificity of the engineered TyrRS toward 3-iodotyrosine by transplantation of the PheRS editing domain, which prevents translation of Tyr.13 Finally, the use of screening systems in combination with rational or computational design of binding and editing domain libraries of the AARSs has highly increased the number and selectivity of ncAA translations.14
No evolutionary pressure existed for the discrimination of AARSs against artificial AAs (Figure 1b, right (i)), to which DfeGly and TfeGly belong, because they were not encountered by the enzymes during evolution. Because of the lack of comprehensive studies, little is known about the possible proofreading activity of AARSs against such building blocks.
In all class I AARSs, the Rossmann fold that comprises the catalytic domain is split by an inserted α/β domain (connective peptide 1, CP1). CP1 is greatly enlarged in IleRS, ValRS, and LeuRS, generating a distinct catalytic site for the hydrolysis of misacylated tRNAs (Figure 1a, post-transfer editing).8,15 It has been proposed that this dedicated editing site acts as a second sieve that rejects noncognate AAs on the basis of steric issues and/or different hydrophobicity.16−20 Although the emerging evidence indicates that the mechanism is more complicated,21,22 post-transfer editing undoubtedly acts as a powerful second check that prevents stable aminoacylation of tRNAs with noncognate AAs. In ILVRSs (IleRS, LeuRS, ValRS) the synthetic and editing sites are approximately 30–40 Å away from each other.8 The AA covalently bound to tRNA is transferred for proofreading from the synthetic to the editing site by translocation of the 3′-end of AA-tRNA.8 Besides post-transfer editing, AARSs may also employ pretransfer editing (Figure 1a) within the synthetic site.8 In tRNA-independent pretransfer editing, noncognate aminoacyl-adenylate intermediates are hydrolyzed independently of the presence of tRNA.23 In some cases, like in IleRS, tRNA may induce a conformational change that organizes the synthetic site for enhanced editing (tRNA-dependent pretransfer editing (Figure 1a)).8,24 The contribution of each pathway to overall editing differs among various enzymes; for ILVRSs post-transfer editing represents the major pathway.8
Disabling of ILVRS post-transfer editing has been shown to allow the translation of several ncAAs that are efficiently discriminated against by the wild-type enzymes. Different working groups have used rational site-directed mutagenesis of the editing domain or random mutagenesis in combination with a selection system to identify LeuRS and ValRS variants, respectively, that enabled the incorporation of numerous aliphatic ncAAs.25−28 Furthermore, Pezo et al. demonstrated that disruption of the editing site in IleRS by substituting 10 residues in the post-transfer editing domain with alanine leads to a mutant (IleRSAla10) that facilitates the ribosomal translation of norvaline, O-methylserine, norleucine, S-methylcysteine, and O-methylthreonine.29
Interestingly, in none of these reports was the observation made that the crosstalk between synthetic and editing pathways of ncAAs differs from that of the 20 cAAs, and this may be true for side chains containing only elements from the cAA pool that were encountered by the AARSs during evolution. However, fluorine is absent from this pool and has unique stereoelectronic properties that arise from a combination of small size, very low polarizability, and the strongest inductive effect found among all the chemical elements30 and consequentially might affect interactions and chemical steps mediated by AARSs in ways that have not yet been observed. Therefore, we sought to probe the compatibility of several aliphatic fluorinated amino acids with the natural ribosomal translation machinery by investigating the aminoacylation and editing activities of the AARSs against them, as well as their translation into polypeptides.
Results and Discussion
Synthetic Active Site of ValRS and in Particular IleRS Shows Relaxed Substrate Tolerance toward the Selected Fluorinated AAs
Investigating the first step of aminoacylation (Figure 1a, AA activation) delivers
important information about the likelihood of ribosomal translation
of noncognate AAs. Activation of a specific substrate by an AARS is
based on optimal binding at the ground and transition state, which
can be investigated by Michaelis–Menten kinetic experiments.
Both states are strongly influenced by the steric size and physicochemical
properties of the AA substrate.18,31 The parameter 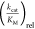 , which is the
, which is the 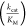 of the noncognate substrate divided by
of the noncognate substrate divided by 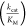 of the cognate substrate is often used
to show how efficiently the noncognate substrate is utilized compared
to the natural one; thus, a
of the cognate substrate is often used
to show how efficiently the noncognate substrate is utilized compared
to the natural one; thus, a 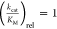 represents a substrate that is
as good
as the cognate one. In the absence of editing, a strong correlation
between AA activation and ribosomal translation can be assumed.
represents a substrate that is
as good
as the cognate one. In the absence of editing, a strong correlation
between AA activation and ribosomal translation can be assumed.
However, this correlation might, besides editing, be further complicated by other steps of aminoacylation (Figure 1a), or arise from weaker or stronger binding to elongation factor (EF-Tu in prokaryotes like Escherichiacoli (E. coli)) or perturbed interactions at the ribosome.
The determined 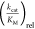 values for the activation of various fluorinated
AAs (Figure 2a) by
the different class Ia AARSs from E. coli (IleRS,
ValRS, MetRS, LeuRS) are shown in Figure 2b. The higher the bar in the figure, the
larger the
values for the activation of various fluorinated
AAs (Figure 2a) by
the different class Ia AARSs from E. coli (IleRS,
ValRS, MetRS, LeuRS) are shown in Figure 2b. The higher the bar in the figure, the
larger the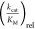 , and better the activation of the fluorinated
AAs by a certain AARS compared to its cognate cAA. Additionally, this
is represented by a color code: green, cyan, and yellow indicate a
relatively good substrate, whereas black and red indicate only marginal
or no observed activation. Table S1 shows
the detailed kinetic values. On the basis of these results, among
the class Ia AARSs, only ValRS and IleRS efficiently activate some
selected fluorinated amino acids that have a different carbon backbone
and structure than their cognate substrates (IleRS: MfeGly, DfeGly,
TfeGly, (2S,3R)-TfVal; ValRS: MfeGly,
TfeGly). We have investigated the crystal structures of the AARSs
that exist in the PDB (Supporting Information, Figure S1). From our analysis we conclude that this relaxed
substrate tolerance of ValRS and IleRS is connected to the arrangement
of the surface of the hydrophobic binding pockets of the enzymes,
which, in the cases of MetRS and LeuRS reflect the shape of their
cognate substrates, but in the cases of IleRS and ValRS the pockets
display not high shape complementarity but rather a general hydrophobic
cavity (Supporting Information, Section 1.2, Figure S1). In the case of MetRS a particularly important determinant
for its highly discriminating synthetic active site is the induced-fit
mechanism of its binding pocket, as described by Serre et al.,32 which differs from the preformed binding pockets
of IleRS, ValRS, and LeuRS.
, and better the activation of the fluorinated
AAs by a certain AARS compared to its cognate cAA. Additionally, this
is represented by a color code: green, cyan, and yellow indicate a
relatively good substrate, whereas black and red indicate only marginal
or no observed activation. Table S1 shows
the detailed kinetic values. On the basis of these results, among
the class Ia AARSs, only ValRS and IleRS efficiently activate some
selected fluorinated amino acids that have a different carbon backbone
and structure than their cognate substrates (IleRS: MfeGly, DfeGly,
TfeGly, (2S,3R)-TfVal; ValRS: MfeGly,
TfeGly). We have investigated the crystal structures of the AARSs
that exist in the PDB (Supporting Information, Figure S1). From our analysis we conclude that this relaxed
substrate tolerance of ValRS and IleRS is connected to the arrangement
of the surface of the hydrophobic binding pockets of the enzymes,
which, in the cases of MetRS and LeuRS reflect the shape of their
cognate substrates, but in the cases of IleRS and ValRS the pockets
display not high shape complementarity but rather a general hydrophobic
cavity (Supporting Information, Section 1.2, Figure S1). In the case of MetRS a particularly important determinant
for its highly discriminating synthetic active site is the induced-fit
mechanism of its binding pocket, as described by Serre et al.,32 which differs from the preformed binding pockets
of IleRS, ValRS, and LeuRS.
Figure 2.

(a) Chemical structure of natural substrates of class Ia AARSs and putative fluorinated analogues. 1: (2S)-4,4,4-trifluoroethylglycine (TfeGly), 2: (2S)-4,4-difluoroethylglycine (DfeGly), 3: (2S)-4-monofluoroethylglycine (MfeGly), 4: (2S)-4,4-difluoropropylglycine (DfpGly), 5: (2S,4S,4R)-5,5,5-trifluoroleucine (TfLeu), 6: (2S)-5,5,5,5′,5′,5′-hexafluoroleucine (HfLeu), 7: (2S,2R)-3,3,3-trifluoroalanine (TfAla), 8: (2S,3R)-4,4,4-trifluorovaline ((2S,3R)-TfVal), 9: (2S,3S)-4,4,4-trifluorovaline ((2S,3S)-TfVal), 10: (2S,2R)-4,4,4,4′,4′,4′-hexafluorovaline (HfVal), 11: (2S,2R)-6,6,6-trifluoronorleucine (TfNLeu), (b) Kinetic data that were determined in this study for the activation of these fluorinated AAs by the indicated class Ia AARSs.
Altogether, the activation rates of the fluorinated AAs by IleRS, LeuRS, and ValRS range from several hundred- to several thousand-fold lower than the values of the respective natural substrates (Figure 2, Table S1). These values clearly show that the fluorinated AAs are markedly less suitable substrates for the AARSs than their natural substrates. These activation data are in good agreement with previous reports33−38 of detailed kinetic studies for the activation of (2S,3R)-TfVal by ValRS and IleRS, and TfLeu and HfLeu by LeuRS as well as their effective ribosomal translation. The values are in a similar range compared to new amino acid activations (MfeGly, DfeGly, TfeGly) that we discovered in this study (Figure 2, Table S1). This suggests that they could be efficiently translated using the natural translation machinery in the absence of the cognate AARS substrate, i.e., in an appropriate auxotrophic genetic background; if such an attempt proved inefficient, supplementation of a vector that encodes the appropriate AARS gene, and would thus increase the amount of this biocatalyst in the cell, could improve the outcome.34
Discovery and Investigation of Editing Function of IleRS against TfeGly
We chose TfeGly as a model artificial AA in order to probe ribosomal incorporation via IleRS. However, despite detected activation with IleRS (Figure 2, Table S1) no incorporation of TfeGly is observed in an Ile auxotrophic strain with TfeGly supplementation in the presence of increased cytosolic concentrations of IleRS (see Supporting Information, Figure S3). A possible reason for this observation is that TfeGly is edited by IleRS (Figure 1a, hydrolytic editing).
To address this issue, we investigated in vitro the proofreading and two-step aminoacylation of TfeGly by E. coli IleRS. A substantial increase in TfeGly-dependent AMP accumulation clearly demonstrates that this artificial AA is edited by the wild-type enzyme (Figure 3a,b). The overall editing of TfeGly by IleRS is 100-fold reduced relative to the cAA valine (0.015 s–1 vs 1.56 s–1 for Val).39 Nevertheless, this activity prevents stable formation of TfeGly-tRNAIle, as confirmed in an independent assay that used [32P]-labeled tRNA (Figure 3d). Our data (Figure 3; Supporting Information, Section 1.7) further indicate that editing against TfeGly, the fluorinated synthetic Abu derivative, is significantly lower than editing of Abu, which is a natural ncAA40,41 that is edited with an efficiency similar to canonical noncognate substrates.27,42−46
Figure 3.

Editing and aminoacylation of TfeGly by wild-type IleRS (wt) and IleRSAla10 (10Ala): (a) AMP formation by 2 μM enzyme in the presence of 20 mM dl-TfeGly and in the absence of tRNAIle; (b) AMP formation by 2 μM enzyme in the presence of 20 mM dl-TfeGly and 10 μM tRNAIle; (c) single-turnover deacylation of preformed TfeGly-tRNAIle using 10 μM enzyme or no enzyme (nonenzymatic); (d) aminoacylation of tRNAIle with dl-TfeGly in the presence of 1 μM enzyme.
Kinetic analysis demonstrates that IleRS edits TfeGly via a tRNA-independent route (Figure 3a) in a manner similar to valine.39 Post-transfer editing can be measured independently of overall editing as enzyme deacylation activity against the preformed misacylated tRNA. Using this assay under single-turnover conditions (excess enzyme), we were able to show that IleRS also deacylates TfeGly-tRNAIle (Figure 3c), yet with a rate that is 1300-fold slower than that reported24 for Val-tRNAIle (0.048 s–1 for TfeGly-tRNAIle and 64 s–1 for Val-tRNAIle).
To measure the contribution of post-transfer editing to TfeGly proofreading, IleRSAla10 (Supporting Information, Section 1.5), previously reported29 as a post-transfer editing inactive mutant, was produced and kinetically characterized (Supporting Information, Section 1.7). We confirmed a complete loss of TfeGly-tRNAIle deacylation activity in this variant (Figure 3c), justifying its further use in this analysis. As expected tRNA-independent editing, located in the synthetic site, is not significantly disturbed (Figure 3a). Importantly, IleRSAla10 charges tRNAIle with TfeGly (Figure 3d), confirming that post-transfer editing prevents TfeGly-tRNAIle accumulation by the wild-type enzyme. Independently measured transfer of TfeGly to tRNAIle yields a value of 0.5 s–1 or higher, which is about 10-fold lower than that of the cognate AA Ile (Supporting Information, Figure S8). Kinetic analysis thus provides evidence that TfeGly is not well discriminated at the level of kcat (Supporting Information, Table S1) and ktrans in the IleRS synthetic pathway.
It has been shown that the highly conserved aspartate (Asp 342 in E. coli IleRS) forms a salt bridge with the α-NH3+ group of the AA attached to tRNA, acting as the main determinant of the IleRS post-transfer editing site.47 Its substitution with alanine abolishes hydrolysis of Val-tRNAIle.24,48 To address the determinants of TfeGly editing, we measured deacylation of TfeGly-tRNAIle by IleRS D342A under single-turnover conditions. Interestingly, the mutant exhibits the same activity as the wild-type enzyme, showing that D342 does not significantly participate in the post-transfer editing of TfeGly (Supporting Information, Table S2). Steric constraints imposed by the structurally distinct side chain may promote distinct positioning of TfeGly-tRNAIle in the editing site. This may be further facilitated by abrogation of the “anchoring” salt bridge due to the electron withdrawing effect of the CF3 group, which decreases the pKa value of the α-NH3+ group of TfeGly.49 Our finding thus supports the notion that AARS proofreading of artificial AAs may rely on different active site determinants compared to cAAs.
Wild-type IleRS does not accumulate TfeGly-tRNAIle (Figure 3d), despite the fast synthetic steps and slow deacylation of TfeGly-tRNAIle. This was intriguing because the measured rate of deacylation was actually at the level of post-transfer editing of the cognate Ile-tRNAIle (0.058 s–1),24 which evades editing and readily accumulates in solution. We have previously50 shown that the fate of aminoacylated tRNA is determined by its kinetic partitioning between dissociation from the enzyme and deacylation at the editing domain (Figure 4). In the case of TfeGly-tRNAIle, slow deacylation is still kinetically favored, presumably due to reduced efficiency of the dissociation step.
Figure 4.
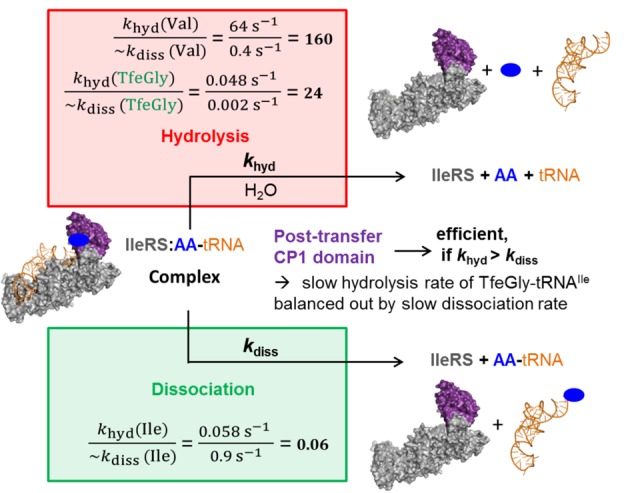
Kinetic partitioning of IleRS post-transfer proofreading. The values for both processes (dissociation and hydrolysis) obtained for TfeGly-tRNAIle in this study are compared with values of our previous studies with IleRS and Ile-tRNAIle and Val-tRNAIle.24,51
In contrast to TfeGly, the cognate product readily accumulates in solution, because kinetic partitioning of Ile-tRNAIle is poised to favor product dissociation. The presumed slow dissociation of TfeGly-tRNAIle from the enzyme is further supported with the observed slow TfeGly-tRNAIle accumulation by IleRSAla10. This variant activates TfeGly (0.3 s–1) and transfers it to tRNAIle (0.5 s–1) with rates that are significantly faster than the steady-state rate of its misaminoacylation (0.002 s–1; Figure 3d). This suggests that product (TfeGly-tRNA) dissociation is the rate-limiting step, and its rate may be approximated by the misaminoacylation rate.
To further address this issue, we introduced another substitution, T243R, in the background of D342A IleRS. The rationale was to reverse a kinetic partitioning of TfeGly-tRNAIle by a mutation that may promote the dissociation step. We assumed that slow post-transfer editing of TfeGly-tRNA would not be able to prevent its accumulation in this case, as it could be outcompeted by more rapid TfeGly-tRNAIle dissociation step. We have previously shown that the T243R substitution lowers affinity of the enzyme for Val-tRNAIle.39 Therefore, we produced the T243R/D342A variant and showed that it significantly accumulates TfeGly-tRNAIle (Figure 5a,b) as anticipated. Separately tested post-transfer editing of this mutant showed a deacylation rate of TfeGly-tRNA (kdeacy = 0.047 s–1; Supporting Information, Table S2) equal to that of the wild-type enzyme, which does not accumulate TfeGly-tRNAIle. This confirms that TfeGly-tRNAIle evades active T243R/D342A editing, because of its enhanced dissociation. The misacylated tRNAs that have escaped editing in cis (operating via translocation of the 3′-end of the AA-tRNA from the synthetic to the editing site) may however rebind the enzyme from solution for editing in trans.52 To analyze distribution of TfeGly-tRNAIle between the enzyme-free and enzyme-bound forms, activated EF-Tu was added to the misaminoacylation mixture of T243R/D342A. EF-Tu–TfeGly-tRNAIle complex formation shifted the equilibrium toward the enzyme-free form and promoted misacylation (Figure 5a,b). In another experiment, we followed misaminoacylation using a higher concentration of T243R/D342A (5 μM) to promote association of TfeGly-tRNAIle with the enzyme and editing (Figure 5d). No misaminoacylated tRNA was detected under these conditions (Figure 5d). This confirms that T243R/D342A opposite behavior relative to the wild-type enzyme is a consequence of reversed kinetic partitioning that favors TfeGly-tRNAIle dissociation. Similarly, EF-Tu does not show any significant effect on the rate or plateau of TfeGly-tRNA formation by wild-type IleRS or any of the single mutants (Figure 5c). T243R IleRS performs all reactions with TfeGly equally to the wild-type enzyme (Supporting Information, Table S2).
Figure 5.

(a and b) Misaminoacylation of tRNAIle with TfeGly by 1 μM T243R/D342A IleRS in the presence or absence of activated EF-Tu on different time scales. (c) Misaminoacylation of tRNAIle with TfeGly by 2 μM wild-type IleRS in the presence or absence of activated EF-Tu. (d) Misaminoacylation and AMP formation by 5 μM T243R/D342A IleRS in the presence of TfeGly.
The presented data support a model wherein kinetic partitioning of aminoacylated tRNA (Figure 4) dictates the contribution of synthetic and editing pathways. In general, rapid editing of noncognate AAs outcompetes dissociation of the respective misacylated tRNAs, while the opposite is true for the cognate systems; i.e., editing here is significantly slower than AA-tRNA dissociation. Perturbation of the partitioning of aminoacylated tRNAs, introduced by a protein mutation (Cvetesic et al.50), or by a novel chemical feature of the artificial AA substrate (this work), may significantly influence misaminoacylation activity of the enzyme. Here we show that the unique stereoelectronic properties of fluorine govern distinct TfeGly-tRNAIle partitioning within the editing site, permitting the weak AA-tRNA deacylation activity to functionally contribute to protein quality control.
In Vivo Translation of TfeGly with Post-Transfer Inactive IleRS
In the previous section we discussed that wild-type IleRS hydrolyzes TfeGly-tRNAIle, which prevents TfeGly from being translated in the place of Ile in vivo. Our experiments show that the mutant IleRSAla10 is totally defective in post-transfer editing and thus accumulates TfeGly-tRNAIle in vitro. Therefore, we further tested both the wild-type and this mutant for their in vivo translation of TfeGly into our model peptide VW18Ile53 (Supporting Information, Section 1.3) that was expressed in the absence of Ile and in the presence of 1 mM TfeGly.
Using this approach, we detected ribosomal translation of TfeGly in place of Ile only in the presence of the post-transfer inactive IleRS mutant (Figure 6). To the best of our knowledge, this is the first report of successful ribosomal translation of TfeGly. We also tested several other fluorinated amino acids. We detected an increase in the rate of aminoacylation and in vivo misincorporation of TfNVal using IleRSAla10 relative to the wild-type enzyme (Supporting Information, Figures S6 and S7). This indicates that TfNVal influences the partitioning of AA-tRNA in a manner similar to TfeGly, making inactivation of post-transfer editing important for its efficient misacylation. We did not observe this effect with TfIle and TfVal (Supporting Information, Figures S6 and S7) that have been commonly employed for ribosomal translation relying on IleRS.
Figure 6.
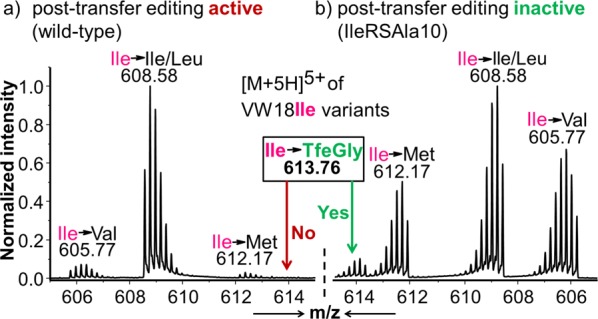
Effect of in vivo post-transfer editing of TfeGly. LC-ESI-MS experiment of VW18Ile variants expressed in the absence of Ile with supplementation of 1 mM TfeGly. For translation either the wild-type IleRS (a) or the post-transfer editing deficient mutant IleRSAla10 (b) was coexpressed. The monoisotopic masses of the 5-fold charged variants of the model peptide VW18Ile, that has just one Ile position, are displayed. Calculated monoisotopic masses for Ile substitution: Val = 605.78, Leu/Ile = 608.57, TfeGly = 613.75, Met = 612.19.
Conclusions
It can be assumed that no evolutionary pressure existed for the discrimination of AARSs against artificial AAs, because they were not encountered by these enzymes during evolution, suggesting that these AAs may escape translational quality control mechanisms. The lack of comprehensive studies prompted us to investigate the details of AARS misaminoacylation and editing mechanisms against artificial AAs, using TfeGly and IleRS as a model system. Fluorinated AAs such as TfeGly can increase the pharmacokinetics of peptide pharmaceuticals and display potential in the engineering of enzyme-based catalysts that are resistant to organic solvents, so-called “Teflon-proteins”.54−56 Surprisingly, we observed efficient proofreading of TfeGly by IleRS, which hinders its incorporation into polypeptides. However, we demonstrated that post-transfer editing of IleRS against TfeGly-tRNAIle is also strongly defective. At the same time, rapid transfer of TfeGly to tRNA prevents significant pretransfer editing contributions to overall proofreading. Our finding that IleRS deacylation activity against TfeGly-tRNAIle is reduced to the level displayed by IleRS against its cognate product Ile-tRNAIle, and at the same time prevents its misaminoacylation, was puzzling. This finding supports the assumption that the proofreading mechanism of natural AARSs is generally not very effective for artificial AAs, as a consequence of the lack of evolutionary pressure to edit these out.57 Using various kinetic approaches we have provided clear evidence that the effectiveness of TfeGly-tRNAIle synthesis and subsequent TfeGly incorporation into peptides in vivo are not determined solely by the deacylation step (Figure 3c), which is typically rapid in the case of editing substrates, but slow in the case of TfeGly. Instead, the essential factor is the ratio between the rates of TfeGly-tRNAIle dissociation and hydrolysis that determine the kinetic partitioning of the aminoacylated tRNA within the IleRS editing domain (Figure 4). In this context, both features differ remarkably for TfeGly in comparison to those known thus far for the proofreading of editing substrates such as valine. We hypothesize that the dramatically reduced dissociation rate of TfeGly-tRNAIle results from specific interactions between TfeGly and residues of the isoleucine-AARS binding pocket induced by the fluorine substituents. This effect has not been reported for tRNAs with AAs containing only elements from the cAA pool. Finally, using the IleRS variant with abolished post-transfer editing we have observed, for the first time, the ribosomal translation of TfeGly. This work expands our understanding of unnatural protein translation with the discovery and investigation of a natural editing function against artificial amino acids and demonstrates that in such cases the routinely used approach of AARS overexpression for the incorporation of artificial AAs is not always sufficient. This work provides an impulse for further systematic investigations and engineering of the translation and editing of unusual amino acids.
Acknowledgments
J.V. is a member of the graduate school “Fluorine as key element” (GRK 1582) of Deutsche Forschungsgemeinschaft (DFG) and the Cluster of Excellence “UniCat” and thankful for financial support. T.B. acknowledges funding by the EU (SYNPEPTIDE). I.G.-S. acknowledges support from the Croatian Science Foundation (Grant 09.01/293). Stella Vukelic is acknowledged for providing TfNVal, Holger Erdbrink for providing TfVal, and Patrick J. Loll and Anne Diehl for providing the vector pETHSUL58(SI). The authors thank Dr. Allison Ann Berger for proofreading of the manuscript.
Glossary
Abbreviations
- ILVRSs
IleRS, LeuRS, ValRS
- cAA
canonical amino acid
- ncAA
noncanonical amino acid
- AA
amino acid
- LC-ESI-MS
liquid chromatography electrospray ionization mass spectrometry
- CP1
connective polypeptide 1 (IleRS post-transfer editing domain)
- AARS
aminoacyl-tRNA synthetase
- AA-tRNA
aminoacyl-tRNA
- Abu
α-aminobutyric acid
- ATP
adenosine-5′-triphosphate
- MfeGly
4-monofluoroethylglycine
- DfeGly
4,4-difluoroethylglycine
- TfeGly
4,4,4-trifluoroethylglycine
- TfIle
5,5,5-trifluoroisoleucine
- TfNVal
5,5,5-trifluoronorvaline
- TfVal
4,4,4-trifluorovaline
- E. coli
Escherichia coli
- kcat
turnover number
- KM
Michaelis–Menten constant
- kobs
observed rate constant
- rel
relative
- tRNA
transfer RNA
- mRNA
messenger RNA
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00339.
Detailed materials and methods, nucleotide sequences, amino acid sequences, vector and strain information, analysis of AARS binding pockets, details about the model peptide VW18Ile, incorporation attempts of TfeGly in the presence of increased cytosolic concentrations of IleRS or ValRS, respectively, representation of the location of the mutations in the post-transfer editing mutant IleRSAla10, in vitro aminoacylation and in vivo translation of TfNVal, TfIle, TfVal, aminoacylation and proofreading of TfeGly with the various CP1 IleRS variants (PDF)
Author Contributions
§ These authors (J.V and M.D.) contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Salwiczek M.; Nyakatura E. K.; Gerling U. I. M.; Ye S.; Koksch B. Fluorinated amino acids: compatibility with native protein structures and effects on protein-protein interactions. Chem. Soc. Rev. 2012, 41, 2135–2171. 10.1039/C1CS15241F. [DOI] [PubMed] [Google Scholar]
- Ye S.; Loll B.; Berger A. A.; Mülow U.; Alings C.; Wahl M. C.; Koksch B. Fluorine teams up with water to restore inhibitor activity to mutant BPTI. Chem. Sci. 2015, 6, 5246–5254. 10.1039/C4SC03227F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buer B. C.; Levin B. J.; Marsh E. N. G. Influence of fluorination on the thermodynamics of protein folding. J. Am. Chem. Soc. 2012, 134, 13027–13034. 10.1021/ja303521h. [DOI] [PubMed] [Google Scholar]
- Asante V.; Mortier J.; Wolber G.; Koksch B. Impact of fluorination on proteolytic stability of peptides: a case study with alpha-chymotrypsin and pepsin. Amino Acids 2014, 46, 2733–2744. 10.1007/s00726-014-1819-7. [DOI] [PubMed] [Google Scholar]
- Budisa N.Engineering the Genetic Code: Expanding the Amino Acid Repertoire for the Design of Novel Proteins; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- Samsonov S. A.; Salwiczek M.; Anders G.; Koksch B.; Pisabarro M. T. Fluorine in protein environments: a QM and MD study. J. Phys. Chem. B 2009, 113, 16400–16408. 10.1021/jp906402b. [DOI] [PubMed] [Google Scholar]
- Berg J. M.; Tymoczko J. L.; Stryer L.. Stryer Biochemie, 7. Aufl; Springer-Spektrum-Lehrbuch; Springer: Berlin, 2013. [Google Scholar]
- Perona J. J.; Gruic-Sovulj I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top. Curr. Chem. 2013, 344, 1–41. 10.1007/128_2013_456. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum K.; Carrico I. S.; Tirrell D. A. Biosynthesis of proteins incorporating a versatile set of phenylalanine analogues. ChemBioChem 2002, 3, 235–237. . [DOI] [PubMed] [Google Scholar]
- Ibba M.; Hennecke H. Relaxing the substrate specificity of an aminoacyl-tRNA synthetase allows in vitro and in vivo synthesis of proteins containing unnatural amino acids. FEBS Lett. 1995, 364, 272–275. 10.1016/0014-5793(95)00408-2. [DOI] [PubMed] [Google Scholar]
- Sharma N.; Furter R.; Kast P.; Tirrell D. A. Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 2000, 467, 37–40. 10.1016/S0014-5793(00)01120-0. [DOI] [PubMed] [Google Scholar]
- Kiga D.; Sakamoto K.; Kodama K.; Kigawa T.; Matsuda T.; Yabuki T.; Shirouzu M.; Harada Y.; Nakayama H.; Takio K.; et al. An engineered Escherichia coli tyrosyl-tRNA synthetase for site-specific incorporation of an unnatural amino acid into proteins in eukaryotic translation and its application in a wheat germ cell-free system. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9715–9720. 10.1073/pnas.142220099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki K.; Sakamoto K.; Kobayashi T.; Sasaki H. M.; Yokoyama S. Transplantation of a tyrosine editing domain into a tyrosyl-tRNA synthetase variant enhances its specificity for a tyrosine analog. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13298–13303. 10.1073/pnas.0803531105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Yadavalli S. S.; Ibba M. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv. Protein Chem. Struct. Biol. 2012, 86, 1–43. 10.1016/B978-0-12-386497-0.00001-3. [DOI] [PubMed] [Google Scholar]
- Fukunaga R.; Yokoyama S. Structural basis for substrate recognition by the editing domain of isoleucyl-tRNA synthetase. J. Mol. Biol. 2006, 359, 901–912. 10.1016/j.jmb.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Fukunaga R.; Yokoyama S. Structural basis for non-cognate amino acid discrimination by the valyl-tRNA synthetase editing domain. J. Biol. Chem. 2005, 280, 29937–29945. 10.1074/jbc.M502668200. [DOI] [PubMed] [Google Scholar]
- Fersht A.Enzyme Structure and Mechanism, 2nd ed.; W.H. Freeman: New York, 1985. [Google Scholar]
- Lincecum T. L.; Tukalo M.; Yaremchuk A.; Mursinna R. S.; Williams A. M.; Sproat B. S.; van Den Eynde W.; Link A.; van Calenbergh S.; Grøtli M.; Martinis S. A.; Cusack S. Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol. Cell 2003, 11, 951–963. 10.1016/S1097-2765(03)00098-4. [DOI] [PubMed] [Google Scholar]
- Dock-Bregeon A.-C.; Rees B.; Torres-Larios A.; Bey G.; Caillet J.; Moras D. Achieving error-free translation; the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol. Cell 2004, 16, 375–386. 10.1016/j.molcel.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Hussain T.; Kamarthapu V.; Kruparani S. P.; Deshmukh M. V.; Sankaranarayanan R. Mechanistic insights into cognate substrate discrimination during proofreading in translation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22117–22121. 10.1073/pnas.1014299107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S.; Muthukumar S.; Kuncha S. K.; Routh S. B.; Yerabham A. S. K.; Hussain T.; Kamarthapu V.; Kruparani S. P.; Sankaranarayanan R. Specificity and catalysis hardwired at the RNA-protein interface in a translational proofreading enzyme. Nat. Commun. 2015, 6, 7552. 10.1038/ncomms8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruic-Sovulj I.; Rokov-Plavec J.; Weygand-Durasevic I. Hydrolysis of non-cognate aminoacyl-adenylates by a class II aminoacyl-tRNA synthetase lacking an editing domain. FEBS Lett. 2007, 581, 5110–5114. 10.1016/j.febslet.2007.09.058. [DOI] [PubMed] [Google Scholar]
- Cvetesic N.; Bilus M.; Gruic-Sovulj I. The tRNA A76 Hydroxyl Groups Control Partitioning of the tRNA-dependent Pre- and Post-transfer Editing Pathways in Class I tRNA Synthetase. J. Biol. Chem. 2015, 290, 13981–13991. 10.1074/jbc.M115.648568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mursinna R. S.; Martinis S. A. Rational design to block amino acid editing of a tRNA synthetase. J. Am. Chem. Soc. 2002, 124, 7286–7287. 10.1021/ja025879s. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Tirrell D. A. Attenuation of the editing activity of the Escherichia coli leucyl-tRNA synthetase allows incorporation of novel amino acids into proteins in vivo. Biochemistry 2002, 41, 10635–10645. 10.1021/bi026130x. [DOI] [PubMed] [Google Scholar]
- Döring V.; Mootz H. D.; Nangle L. A.; Hendrickson T. L.; de Crécy-Lagard V.; Schimmel P.; Marlière P. Enlarging the amino acid set of Escherichia coli by infiltration of the valine coding pathway. Science 2001, 292, 501–504. 10.1126/science.1057718. [DOI] [PubMed] [Google Scholar]
- Cvetesic N.; Semanjski M.; Soufi B.; Krug K.; Gruic-Sovulj I.; Macek B. Proteome-wide measurement of non-canonical bacterial mistranslation by quantitative mass spectrometry of protein modifications. Sci. Rep. 2016, 6, 28631–28643. 10.1038/srep28631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezo V.; Metzgar D.; Hendrickson T. L.; Waas W. F.; Hazebrouck S.; Döring V.; Marlière P.; Schimmel P.; de Crécy-Lagard V. Artificially ambiguous genetic code confers growth yield advantage. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8593–8597. 10.1073/pnas.0402893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buer B. C.; Marsh E. N. G. Fluorine: a new element in protein design. Protein Sci. 2012, 21, 453–462. 10.1002/pro.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona J. J.; Hadd A. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry 2012, 51, 8705–8729. 10.1021/bi301180x. [DOI] [PubMed] [Google Scholar]
- Serre L.; Verdon G.; Choinowski T.; Hervouet N.; Risler J. L.; Zelwer C. How methionyl-tRNA synthetase creates its amino acid recognition pocket upon L-methionine binding. J. Mol. Biol. 2001, 306, 863–876. 10.1006/jmbi.2001.4408. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Tirrell D. A. Biosynthesis of a Highly Stable Coiled-Coil Protein Containing Hexafluoroleucine in an Engineered Bacterial Host. J. Am. Chem. Soc. 2001, 123, 11089–11090. 10.1021/ja016652k. [DOI] [PubMed] [Google Scholar]
- Wang P.; Fichera A.; Kumar K.; Tirrell D. A. Alternative translations of a single RNA message: an identity switch of (2S,3R)-4,4,4-trifluorovaline between valine and isoleucine codons. Angew. Chem., Int. Ed. 2004, 43, 3664–3666. 10.1002/anie.200454036. [DOI] [PubMed] [Google Scholar]
- Montclare J. K.; Son S.; Clark G. A.; Kumar K.; Tirrell D. A. Biosynthesis and stability of coiled-coil peptides containing (2S,4R)-5,5,5-trifluoroleucine and (2S,4S)-5,5,5-trifluoroleucine. ChemBioChem 2009, 10, 84–86. 10.1002/cbic.200800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert O. M.; Anker H. S. On the Incorporation of 5′,5′,5′-Trifluoroleucine into Proteins of E. coli. Biochemistry 1963, 2, 471–476. 10.1021/bi00903a013. [DOI] [PubMed] [Google Scholar]
- Montclare J. K.; Tirrell D. A. Evolving Proteins of Novel Composition. Angew. Chem. 2006, 118, 4630–4633. 10.1002/ange.200600088. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Ghirlanda G.; Petka W. A.; Nakajima T.; DeGrado W. F.; Tirrell D. A. Fluorinated Coiled-Coil Proteins Prepared In Vivo Display Enhanced Thermal and Chemical Stability. Angew. Chem., Int. Ed. 2001, 40, 1494–1496. . [DOI] [PubMed] [Google Scholar]
- Dulic M.; Cvetesic N.; Perona J. J.; Gruic-Sovulj I. Partitioning of tRNA-dependent editing between pre- and post-transfer pathways in class I aminoacyl-tRNA synthetases. J. Biol. Chem. 2010, 285, 23799–23809. 10.1074/jbc.M110.133553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biermann M.; Linnemann J.; Knüpfer U.; Vollstädt S.; Bardl B.; Seidel G.; Horn U. Trace element associated reduction of norleucine and norvaline accumulation during oxygen limitation in a recombinant Escherichia coli fermentation. Microb. Cell Fact. 2013, 12, 116–124. 10.1186/1475-2859-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt R. I.; Umbarger H. E. Isoleucine and valine metabolism in Escherichia coli. XI. Valine inhibition of the growth of Escherichia coli strain K-12. J. Bacteriol. 1962, 83, 624–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. R.; Dingwall C. Establishing the misacylation/deacylation of the tRNA pathway for the editing mechanism of prokaryotic and eukaryotic valyl-tRNA synthetases. Biochemistry 1979, 18, 1238–1245. 10.1021/bi00574a019. [DOI] [PubMed] [Google Scholar]
- Jakubowski H.; Goldman E. Editing of errors in selection of amino acids for protein synthesis. Microbiol. Rev. 1992, 56, 412–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomanbhoy T. K.; Schimmel P. R. Misactivated amino acids translocate at similar rates across surface of a tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (10), 5119–5122. 10.1073/pnas.090102197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski H.; Fersht A. R. Alternative pathways for editing non-cognate amino acids by aminoacyl-tRNA synthetases. Nucleic Acids Res. 1981, 9, 3105–3117. 10.1093/nar/9.13.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski H. Translational accuracy of aminoacyl-tRNA synthetases: implications for atherosclerosis. J. Nutr. 2001, 131, 2983–2987. [DOI] [PubMed] [Google Scholar]
- Bishop A. C.; Nomanbhoy T. K.; Schimmel P. Blocking site-to-site translocation of a misactivated amino acid by mutation of a class I tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 585–590. 10.1073/pnas.012611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson T. L.; Nomanbhoy T. K.; de Crécy-Lagard V.; Fukai S.; Nureki O.; Yokoyama S.; Schimmel P. Mutational separation of two pathways for editing by a class I tRNA synthetase. Mol. Cell 2002, 9, 353–362. 10.1016/S1097-2765(02)00449-5. [DOI] [PubMed] [Google Scholar]
- Walborsky H. M.; Lang J. H. Effects of the Trifluoromethyl Group. IV. 1,2 the pK’s of ι-Trifluoromethyl Amino Acids. J. Am. Chem. Soc. 1956, 78, 4314–4316. 10.1021/ja01598a035. [DOI] [Google Scholar]
- Cvetesic N.; Perona J. J.; Gruic-Sovulj I. Kinetic partitioning between synthetic and editing pathways in class I aminoacyl-tRNA synthetases occurs at both pre-transfer and post-transfer hydrolytic steps. J. Biol. Chem. 2012, 287, 25381–25394. 10.1074/jbc.M112.372151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic M.; Perona J. J.; Gruic-Sovulj I. Determinants for tRNA-dependent pretransfer editing in the synthetic site of isoleucyl-tRNA synthetase. Biochemistry 2014, 53, 6189–6198. 10.1021/bi5007699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J.; So B. R.; Yadavalli S. S.; Roy H.; Shoji S.; Fredrick K.; Musier-Forsyth K.; Ibba M. Resampling and editing of mischarged tRNA prior to translation elongation. Mol. Cell 2009, 33, 654–660. 10.1016/j.molcel.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerling U. I. M.; Salwiczek M.; Cadicamo C. D.; Erdbrink H.; Czekelius C.; Grage S. L.; Wadhwani P.; Ulrich A. S.; Behrends M.; Haufe G.; Koksch B. Fluorinated amino acids in amyloid formation: a symphony of size, hydrophobicity and α-helix propensity. Chem. Sci. 2014, 5, 819–830. 10.1039/C3SC52932K. [DOI] [Google Scholar]
- Meng H.; Kumar K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J. Am. Chem. Soc. 2007, 129, 15615–15622. 10.1021/ja075373f. [DOI] [PubMed] [Google Scholar]
- Niemz A.; Tirrell D. A. Self-association and membrane-binding behavior of melittins containing trifluoroleucine. J. Am. Chem. Soc. 2001, 123, 7407–7413. 10.1021/ja004351p. [DOI] [PubMed] [Google Scholar]
- Merkel L.; Schauer M.; Antranikian G.; Budisa N. Parallel incorporation of different fluorinated amino acids: on the way to ″Teflon″ proteins. ChemBioChem 2010, 11, 1505–1507. 10.1002/cbic.201000295. [DOI] [PubMed] [Google Scholar]
- Merkel L.; Budisa N. Organic fluorine as a polypeptide building element: in vivo expression of fluorinated peptides, proteins and proteomes. Org. Biomol. Chem. 2012, 10, 7241–7261. 10.1039/c2ob06922a. [DOI] [PubMed] [Google Scholar]
- Weeks S. D.; Drinker M.; Loll P. J. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expression Purif. 2007, 53, 40–50. 10.1016/j.pep.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.