

Abstract
Waardenburg syndrome type 4 (WS4) or Waardenburg-Shah syndrome is a rare genetic disorder with a prevalence of <1/1,000,000 and characterized by the association of congenital sensorineural hearing loss, pigmentary abnormalities, and intestinal aganglionosis. There are three types of WS4 (WS4A–C) caused by mutations in endothelin receptor type B, endothelin 3, and SRY-box 10 (SOX10), respectively. This study investigated a genetic mutation in a Chinese family with one WS4 patient in order to improve genetic counselling. Genomic DNA was extracted, and mutation analysis of the three WS4 related genes was performed using Sanger sequencing. We detected a de novo heterozygous deletion mutation [c.1333delT (p.Ser445Glnfs*57)] in SOX10 in the patient; however, this mutation was absent in the unaffected parents and 40 ethnicity matched healthy controls. Subsequent phylogenetic analysis and three-dimensional modelling of the SOX10 protein confirmed that the c.1333delT heterozygous mutation was pathogenic, indicating that this mutation might constitute a candidate disease-causing mutation.
Waardenburg syndrome (WS), also known as auditory pigmentary syndrome, is characterized by congenital sensorineural deafness, dystopia canthorum, and pigmentary abnormalities affecting the hair, skin, and eyes and occurring with a frequency of 1/40,0001,2. WS is clinically and genetically heterogeneous and is classified into four types (WS1–4) caused by mutations of paired box 3 (PAX3), melanogenesis-associated transcription factor (MITF), endothelin 3 (EDN3), endothelin receptor type B (EDNRB), snail-family transcriptional repressor 2 (SNAI2), and SRY-box 10 (SOX10)3,4. WS1 and WS2 are the most frequent types, whereas WS4 constitutes a rare disorder5,6.
WS4 is also known as Waardenburg-Shah syndrome (OMIM 277580) and is characterized by hearing loss, depigmentation, and aganglionic megacolon (Hirschsprung disease). WS4 includes three subtypes [WS4A–C (OMIM 277580, 613265, and 613266)] caused by mutations in EDNRB, EDN3, and SOX10, respectively7,8,9. Mutations in EDNRB and EDN3 are inherited in the autosomal recessive (AR) or autosomal dominant (AD) form, whereas the SOX10 mutation is inherited as AD10,11,12,13 and found in ~50% of WS4 patients1,6, with >30 WS4-related mutations reported in the Human Gene Mutation Database. SOX10 is a critical transcription factor, targeting MITF, tyrosinase, myelin protein zero, gap junction protein beta 1, ret proto-oncogene, and EDNRB during neural-crest-derived cell migration and differentiation. Additionally, SOX10 modulates the expression of its target genes and the migration of pluripotent neural crest cells from the neural tube during embryogenesis14,15.
In this study, we conducted detailed clinical and genetic analysis of a Chinese family with a WS4-afflicted child. A de novo heterozygous deletion mutation [c.1333delT (p.Ser445Glnfs*57)] in SOX10 was detected in the patient, although this mutation was absent in the unaffected parents and 40 ethnicity matched healthy controls. Our findings indicated that this mutation might be a candidate disease-causing mutation.
Methods
Subjects and clinical evaluation
The patient, his unaffected parents, and 40 unrelated healthy controls were included in this study, and ophthalmic and audiologic examinations were performed. Written informed consent was obtained from all participants, and this study was formally approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. All procedures were performed in accordance with the approved guidelines.
Mutation screening
Peripheral blood was collected, and genomic DNA was extracted using a DNeasy blood and tissue kit from Qiagen (Hilden, Germany). Polymerase chain reaction (PCR) was performed to amplify all coding exons and intron/exon boundaries of the EDNRB, EDN3, SOX10, PAX3, MITF, and SNAI2 genes. Some of the primers used in the study were referenced from a master’s thesis (title here, Dong Siqi; Chinese PLA General Hospital, Beijing, China), and other primers were designed using Primer 5. Primers are shown in Table 1. PCR of the SOX10 exons was performed in a total volume of 50 μL containing 60 ng of genomic DNA, 400 nM each of the forward and reverse primers, 40 mM dNTPs, and 2.5 U LA Taq DNA polymerase with GC buffer I from TAKARA (Tokyo, Japan). The amplification consisted of an initial denaturation stage at 94 °C for 3 min, followed by 35 cycles consisting of denaturation at 94 °C for 30 s, annealing for 30 s at 60 °C, and extension at 72 °C for 50 s, with an extension step performed at 72 °C for 3 min. Amplification of exons for the remaining genes was performed using 2× PCR master mix under similar conditions, except for annealing at 57 °C. PCR products were purified and sequenced using an ABI 3500 Dx genetic analyser with a BigDye terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA, USA), and the sequences were analysed using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Table 1. Primers used in this study.
| Primer name | Sequence |
|---|---|
| SOX10 E1F (765 bp) | AGATGGGTTTAGCTGGAGCA |
| SOX10 E1R (765 bp) | ACCTGGTCTTCCAGCCCTAT |
| SOX10 E2F (686 bp) | GTTATTCCTTGGGCCTCACA |
| SOX10 E2R (686 bp) | CTTTGCCCAGTAGGATCAGC |
| SOX10 E3A F (686 bp) | GCTGCCAAAATGTGAAACTTA |
| SOX10 E3A R (686 bp) | GAGTGGCCATAATAGGGTCC |
| SOX10 E3BF (561 bp) | AGCCCAGGTGAAGACAGAGA |
| SOX10 E3BR (561 bp) | TCTGTCCAGCCTGTTCTCCT |
| EDN3 E1 F (407 bp) | CAGAAGCCAGAAAAGCCCGA |
| EDN3 E1 R (407 bp) | CCAGGCAAGAGTTTGCTCCC |
| EDN3 E2 F (597 bp) | TTTGCAGACATTTTGCTTGC |
| EDN3 E2 R (597 bp) | CCTGACCTGCAGAAGAGACC |
| EDN3 E3 F (480 bp) | GGTGCACAGTTCACTCCAGA |
| EDN3 E3 R (480 bp) | CCCACAGGACGACAGTAGGT |
| EDN3 E4 F (607 bp) | CGTCTGTGAAACCCAGTGTG |
| EDN3 E4 R (607 bp) | CATCACTGCCCAGAGCTACA |
| EDN3 E5 F (424 bp) | GGCTCGGAAATTGCTGAGAAG |
| EDN3 E5 R (424 bp) | TCTTTGGGTGGGTGTTCTGC |
| EDNRB E1 F(748 bp) | CTTTTGAGCGTGGATACTGG |
| EDNRB E1 R(748 bp) | AGGGAGCTAAAGGGAAGCTC |
| EDNRB E2 F (498 bp) | AACACACTTTCCTGTCCCATAC |
| EDNRB E2 R (498 bp) | TTCTACTGCTGTCCATTTTGG |
| EDNRB E3 F (555 bp) | CTGTGGGAATCACTGTGCTG |
| EDNRB E3 R (555 bp) | AGCTTGAGTCATTGATCACCA |
| EDNRB E4 F (432 bp) | TGTTCAGTAAGTGTGGCCTGA |
| EDNRB E4 R (432 bp) | CAAGAAAAAGGAAATATGCTCTGG |
| EDNRB E5 F (466 bp) | CACTTCGGTTCCACTTCACA |
| EDNRB E5 R (466 bp) | CTTCCCTGTCCCTCTCAACA |
| EDNRB E6 F (493 bp) | GAGGGGGACACAGACAGAGA |
| EDNRB E6 R (493 bp) | GCAGTAGGGAGTGGCTGACT |
| EDNRB E7 F (466 bp) | AAGAGGGAAAATAAAAGAGCACTG |
| EDNRB E7 R (466 bp) | TTCTTTCCATGCCGTAAACA |
| PAX3 E1F (620 bp) | GAACATTTGCCCAGACTCGT |
| PAX3 E1R (620 bp) | TCCAAAACAACAGGGACAAGT |
| PAX3-2F (503 bp) | CCGATGTCGAGCAGTTTCAG |
| PAX3-2R (503 bp) | CGCACCTTCACAAACCTCAG |
| PAX3-3F (420 bp) | TGGGATGTGTTCTGGTCTG |
| PAX3-3R (420 bp) | TCCCAATAGCTGAGATCGA |
| PAX3-4F (383 bp) | CTGGAGAAGGATGAGGATGT |
| PAX3-4R (383 bp) | CGTCAGATCACCAATGTCAG |
| PAX3-5F (508 bp) | TACGGATTGGTTAGACTTGT |
| PAX3-5R (508 bp) | AACAATATGCATCCCTAGTAA |
| PAX3-6F (445 bp) | CAACACAGAAGGCAGAGA |
| PAX3-6R (445 bp) | ATAGGTACGTTCAGGACAA |
| PAX3-7F (586 bp) | TGTGCAGAGATAGGTGTGAC |
| PAX3-7R (586 bp) | TTTGATGAAGCCAGTAGGA |
| PAX3 E8F (543 bp) | GTTATTCTTTCAGCTGTAGGC |
| PAX3 E8R (543 bp) | GTCTCAACAATTAATAACCGC |
| MITF E1F (630 bp) | GGAGTTGCACTAGCGGTGTC |
| MITF E1R (630 bp) | GCTCCATCCGAGCTTCCTA |
| MITF E2F (628 bp) | GCCTGATAAAAATGCCTTGA |
| MITF E2R (628 bp) | AGCCACGTAAGAATTAAGGGA |
| MITF E3F (564 bp) | GCACAGTGCCTGGTACATAAC |
| MITF E3R (564 bp) | TGCTCTACACCCAATAACCC |
| MITF E4 F (310 bp) | TCATCTTTTGGTCAGATTCCAC |
| MITF E4 R (310 bp) | TGCTTAAGTTTTCAGGAAGGTG |
| MITF-5F (343 bp) | GACCATTATTGCTTTGGGTAAAA |
| MITF-5R (343 bp) | TGTGATCCTGAGATAATTCTCCATT |
| MITF-6F (425 bp) | TGAGGAGATCCTGTACCTCTCTT |
| MITF-6R (425 bp) | AAAAGTTACGTCCATGAGTTGG |
| MITF-7F (350 bp) | GCTTTTGAAAACATGCAAGC |
| MITF-7R (350 bp) | GCTGTAGGAATCAACTCTCCTCT |
| MITF E8 F (527 bp) | AAGGGCTTTGGAAATGGTAA |
| MITF E8 R (527 bp) | AGAAAGCCACCTCCTCACAA |
| MITF-9F (425 bp) | CTTATCCATGTAACCAAGCA |
| MITF-9R (425 bp) | CACACACACAGAATCCACAAA |
| MITF-10F (646 bp) | CTAATGACGCGCATCTACCA |
| MITF-10R (646 bp) | TCCTGGGCTATTGATAAAGCA |
| SNAI2 E1 F (388 bp) | CGGGCTCAGTTCGTAAAGGA |
| SNAI2 E1 R (388 bp) | GCTCCCTTTCAGGACACTGTTA |
| SNAI2 E2 AF (534 bp) | GCCCTCCTAAATGGGTCTATC |
| SNAI2 E2 AR (534 bp) | TTTTCTAGACTGGGCATCGC |
| SNAI2 E2 BF (565 bp) | GCCCCATTAGTGATGAAGAG |
| SNAI2 E2 BR (565 bp) | GATCTTTGAGACCAAACCTTC |
| SNAI2 E3 F (556 bp) | GGTTTTGCTGCTTCTCATTAT |
| SNAI2 E3 R (556 bp) | TCTCTCAATCTAGCCATCAGC |
| D22S283 F (217 bp) | FAM-ACAAACACTTCTACAGTCCTGG |
| D22S283 R (217 bp) | TGAGCCACGGAGATCTTTC |
| D22S1177 F (186 bp) | FAM-GCCACTCTGGCACCAT |
| D22S1177 R (186 bp) | AGCTGTNAGCAAGCAGG |
| D22S1045 F (153 bp) | FAM-GCTAGATTTTCCCCGATGAT |
| D22S1045 R (153 bp) | ATGTAAAGTGCTCTCAAGAGTGC |
| D22S272 F (132 bp) | FAM-GAGTTTTGTTTGCCTGGCAC |
| D22S272 R (132 bp) | AATGCACGACCCACCTAAAG |
| D22S423 F (123 bp) | FAM-CACACTGGTACACACATACACA |
| D22S423 R (123 bp) | AAACCAACTGACTCGTTTAGG |
| rs139885 F (625 bp) | CACCCATGCCTACTGTCTTC |
| rs139885 R (625 bp) | GAGACCCTGGACCACATACA |
| rs3952 F (263 bp) | CTTGCTGTAGCCTTGGGAATA |
| rs3952 R (263 bp) | GTAGAGGGAGGTGGCGAGA |
| rs5756908 F (306 bp) | AGTTTCCCAAAGATACTGTCCC |
| rs5756908 R (306 bp) | CCAGTTAGTCCCTCCTCCAA |
| rs4821733 F (434 bp) | GCAGGCATTGGCATCACC |
| rs4821733 R (434 bp) | AAATTGCTTGAATGCGGGAG |
| rs139873 F (374 bp) | AAAAAGACTCCTGGCTTCCA |
| rs139873 R (374 bp) | CCCACAGTGCTCGGATTC |
Paternity testing and haplotype analysis
Five short tandem-repeat markers (STRs; D22S283, D22S1177, D22S1045, D22S272, and D22S423) ranging from chr22:36750705 to chr22:40382524 and five single nucleotide polymorphisms (SNPs; rs139873, rs139885, rs4821733, rs3952, and rs5756908) were selected from the UCSC Genome Browser (http://genome.ucsc.edu/), and linkage-disequilibrium analysis was performed based on LD TAG SNP selection (TagSNP; http://snpinfo.niehs.nih.gov/snpinfo/snptag.php). STR and SNP primers are shown in Table 1.
Protein structure prediction
Both the wild-type and mutated SOX10 protein sequences were used to perform protein structure prediction using I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) as previously reported16,17,18,19. In I-TASSER, the B-factor, which indicates the extent of the inherent thermal mobility of residues/atoms in proteins, is calculated from threading template proteins from the Protein Data Bank along with sequence profiles derived from sequence databases. The normalized B-factor of the target protein was defined by B = (B′ − u)/s, where B′ represents the raw B-factor value, and u and s represent the mean and standard deviation of the raw B-factors along the sequence, respectively.
Results
Clinical findings
A 1-year-old male patient was referred to our hospital with the chief complaint of Hirschsprung disease accompanied by heterochromia iridis and congenital hearing loss. Based on these clinical features, he was first suspected to be a WS4 patient. Neither parent of the patient exhibited similar symptoms (Fig. 1).
Figure 1. Clinical features of the patient.
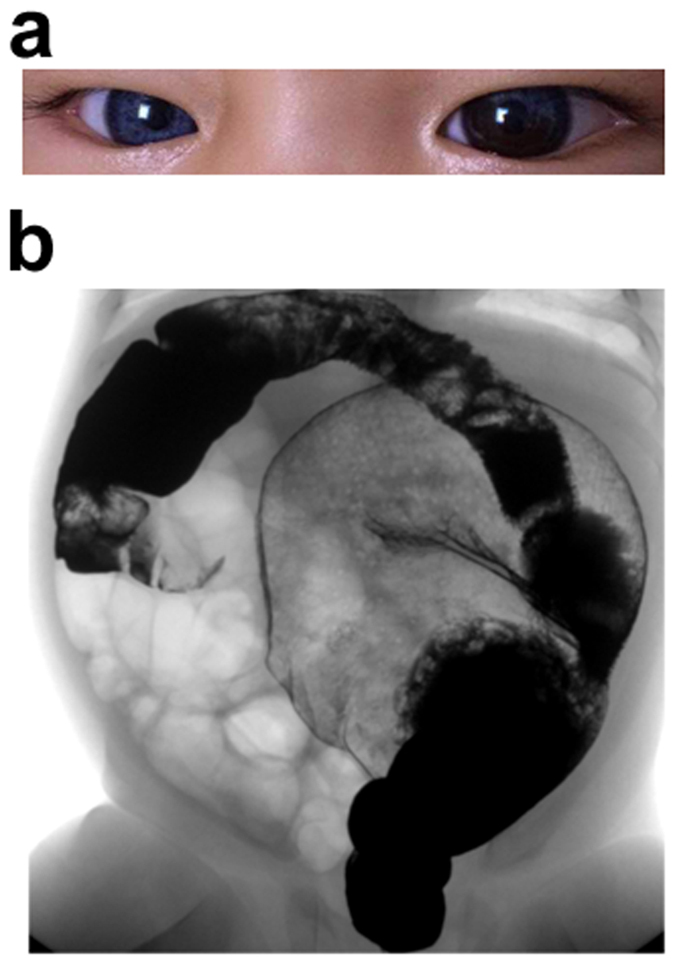
(a) Photograph of the patient presented with blue iride of the right eye and two different colours of the left eye. (b) The barium enema examination of the colon of the patient showed megacolon congenitum.
Identification of a novel SOX10 heterozygous deletion mutation
A heterozygous deletion mutation (c.1333delT) in SOX10 was identified in the patient, resulting in replacement of the 445th Ser with Gln and a shift in the reading frame to produce a longer protein consisting of 501 amino acids (p.Ser445Glnfs*57) as compared with the wild-type SOX10 protein (467 amino acids; Fig. 2, Table 2). We subsequently verified that this mutation did not exist in any of the widely used genomic databases, confirming that c.1333delT constitutes a novel deletion mutation. Moreover, this mutation was not found in the unaffected parents or in 40 unrelated healthy control subjects. However, a heterozygous missense mutation (c.1363C > A) in MITF was found in both the patient and his father, but not in his mother (Fig. 2). This mutation was found in the dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) databases (rs78962087) and is reportedly benign. Furthermore, no mutation was found in the EDN3, EDNRB, PAX3, or SANI2 genes. These results suggested that the heterozygous deletion mutation (c.1333delT) in SOX10 might be associated with the WS4 phenotype of the patient.
Figure 2. Identification of a novel SOX10 heterozygous deletion mutation.

Sequence chromatographs of the SOX10 and MITF genes of the Chinese family. (a) The heterozygous mutation in SOX10 [c.1333delT (p.Ser445Glnfs*57)] was only found in the patient, but not his father or mother. (b) The heterozygosis mutation in MITF [c.1363C > A (p.Leu455Ile)] found in the patient and his father, but not his mother.
Table 2. Genetic variants found in this family with WS4.
| Gene | Variant | Protein level | Type | Father | Mother | Report |
|---|---|---|---|---|---|---|
| Sox10 | c.1333delT | p.Ser445Glnfs*57 | heterozygous | Normal | Normal | No |
| MITF | c.1363C > A | p.Leu455Ile | heterozygous | heterozygous | Normal | Yes |
Paternity testing and haplotype analysis
SOX10c.1333delT is located in chr22:38369570. To confirm the paternity of the father, five STRs (D22S283, D22S1177, D22S1045, D22S272, and D22S423) ranging from chr22:36750705 to chr:40382524 and five SNPs (rs139873, rs139885, rs4821733, rs3952, and rs5756908) ranging from chr22:38359666 to chr:38476579 were selected from the UCSC Genome Browser (http://genome.ucsc.edu/) based on their proximity to the mutation site. Paternity testing by haplotype analysis confirmed that these were the biological parents of the patient with WS4 (Figs 3 and 4).
Figure 3. Paternity testing and haplotype analysis.
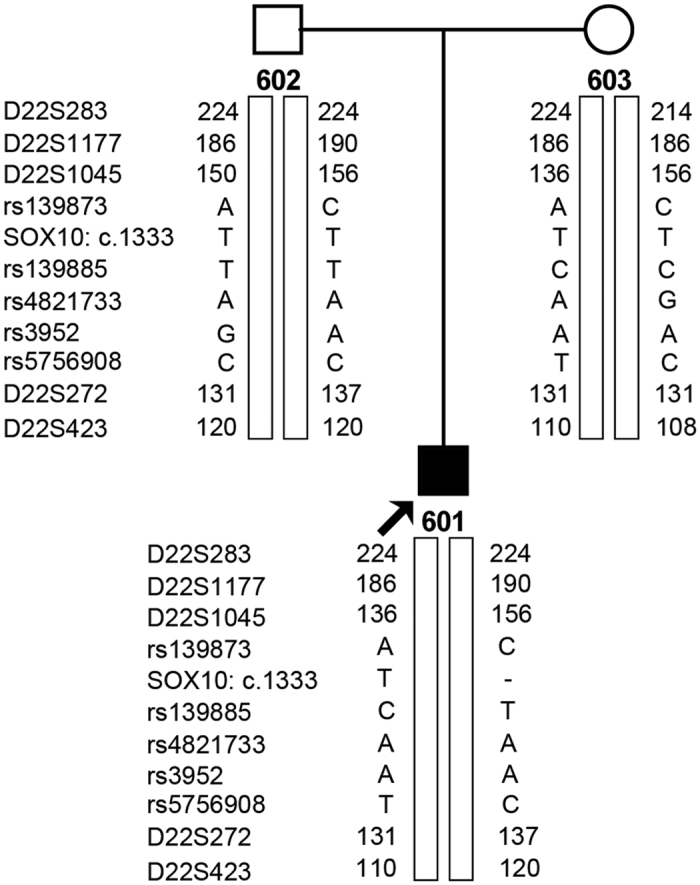
601, patient; 602, father; 603, mother.
Figure 4. SNP analysis of the Chinese family with WS4.

Five SNPs (rs139873, rs139885, rs4821733, rs3952, and rs5756908) were selected.
Protein structure prediction
The wild-type SOX10 protein consists of 467 amino acids and contains three helices, whereas the SOX10 deletion mutation (c.1333delT) results in a protein consisting of 501 amino acids with four helices (Fig. 5). The wild-type and mutant variants shared identical sequences in the first 444 amino acids, with differences occurring after this point.
Figure 5. Protein structure prediction.

(a) Wild-type SOX10 protein structure. (b) The mutated SOX10 protein structure.
Discussion
WS is classified into four primary phenotypes. WS1 is caused by mutations in PAX3 and distinguished by the presence of dystopia canthorum (lateral displacement of the inner canthi). WS2 is caused by mutations in MITF, SOX10, or SNAI2 and distinguished from type 1 by the absence of dystopia canthorum. WS3 is caused by mutations in PAX3, with patients presenting both dystopia canthorum and upper limb abnormalities. WS4 is caused by mutations in EDNRB, EDN3, or SOX10, with patients presenting with phenotypes associated with Hirschsprung disease1,20,21,22,23. Here, we described a Chinese patient with clinical features of WS4 and identified a novel heterozygous deletion mutation [c.1333delT (p.Ser445Glnfs*57)] in SOX10 that was absent in his unaffected parents and 40 ethnicity matched healthy controls. To the best of our knowledge, this constitutes the first report of this mutation, suggesting it as a candidate disease-causing mutation.
SOX10 is located on chromosome 22 and encodes an essential DNA-binding nuclear transcription factor consisting of 467 amino acids and belonging to the SOX family involved in modulating embryonic development and determining cell fate. SOX10 may act as a transcriptional activator upon forming a complex with other proteins and/or as a nucleocytoplasmic shuttle protein critical for neural crest and peripheral nervous system development24,25,26,27. Mutations in this gene are associated with WS4 and are present in ~50% of WS4 patients6,28.
SOX10 contains a highly conserved high mobility group (HMG) DNA-binding domain and a C-terminal transactivation (TA) domain that is enriched in serine, proline, and acidic residues29,30. Additionally, SOX10 contains two separate TA domains, with one localized in the C-terminal region and the other in the central region of the structure. The C-terminal TA domain is frequently involved in various interactions, whereas the TA domain located in the centre of the structure is only involved in TA-related activity in certain cell types and under certain developmental conditions31. SOX10 binds to the promoters of its target genes via the HMG domain, with several studies reporting the importance of the TA domain for inducing transcriptional activation of its target genes32. Wang et al.32 identified a c.1063C > T (p.Q355*) mutation in SOX10 in a family with WS4 and reported that the mutated SOX10 variant retained nuclear localization and DNA-binding capabilities comparable to those observed in wild-type SOX10; however, the mutated SOX10 variant was unable to activate transcription of MITF via its promoter and acted as a dominant-negative repressor as compared with activity associated with wild-type SOX107,33. In this study, we detected a c.1333delT (p.Ser445Glnfs*57) mutation in SOX10 in a family with WS4, with the mutated SOX10 variant sharing sequence homology with only the N-terminal 444 amino acids of the wild-type protein. Furthermore, we identified an additional helix in the C-terminal region of the mutated SOX10 variant (Fig. 4), which may affect its normal biological function.
In conclusion, here, we described a de novo heterozygous deletion mutation [c.1333delT (p.Ser445Glnfs*57)] in SOX10 identified in a Chinese family with WS4. Our analyses indicated that this mutation might constitute a candidate disease-causing mutation associated with WS4.
Additional Information
How to cite this article: Wang, X. et al. A de novo deletion mutation in SOX10 in a Chinese family with Waardenburg syndrome type 4. Sci. Rep. 7, 41513; doi: 10.1038/srep41513 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
This work was partially supported by the National Natural Science Foundation of China (No. 81500925).
Footnotes
The authors declare no competing financial interests.
Author Contributions H.L. and Y.L. designed this work, X.W. and Y.Z. performed sequencing and analysis, N.S., C.W., and J.P. prepared figures, and X.W. wrote the manuscript.
References
- Pingault V. et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat 31, 391–406, doi: 10.1002/humu.21211 (2010). [DOI] [PubMed] [Google Scholar]
- Zaman A., Capper R. & Baddoo W. Waardenburg syndrome: more common than you think! Clin Otolaryngol 40, 44–48, doi: 10.1111/coa.12312 (2015). [DOI] [PubMed] [Google Scholar]
- Song J. et al. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet, doi: 10.1111/cge.12631 (2015). [DOI] [PubMed] [Google Scholar]
- Toriello H. V. Pigmentary anomalies and hearing loss. Adv Otorhinolaryngol 70, 50–55, doi: 10.1159/000322471 (2011). [DOI] [PubMed] [Google Scholar]
- Doubaj Y. et al. A novel mutation in the endothelin B receptor gene in a moroccan family with shah-waardenburg syndrome. Mol Syndromol 6, 44–49, doi: 10.1159/000371590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez R. M. et al. Waardenburg syndrome type 4: report of two new cases caused by SOX10 mutations in Spain. Am J Med Genet A 164A, 542–547, doi: 10.1002/ajmg.a.36302 (2014). [DOI] [PubMed] [Google Scholar]
- Wang H. H. et al. Identification and functional analysis of a novel mutation in the SOX10 gene associated with Waardenburg syndrome type IV. Gene 538, 36–41, doi: 10.1016/j.gene.2014.01.026 (2014). [DOI] [PubMed] [Google Scholar]
- Mahmoudi A. et al. Shah-Waardenburg syndrome. Pan Afr Med J 14, 60, doi: 10.11604/pamj.2013.14.60.1543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthey K. et al. SOX10 mutation with peripheral amyelination and developmental disturbance of axons. Muscle Nerve 45, 284–290, doi: 10.1002/mus.22262 (2012). [DOI] [PubMed] [Google Scholar]
- Puffenberger E. G. et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell 79, 1257–1266 (1994). [DOI] [PubMed] [Google Scholar]
- Baynash A. G. et al. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 79, 1277–1285 (1994). [DOI] [PubMed] [Google Scholar]
- Pingault V. et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet 18, 171–173, doi: 10.1038/ng0298-171 (1998). [DOI] [PubMed] [Google Scholar]
- Syrris P., Carter N. D. & Patton M. A. Novel nonsense mutation of the endothelin-B receptor gene in a family with Waardenburg-Hirschsprung disease. Am J Med Genet 87, 69–71 (1999). [PubMed] [Google Scholar]
- Bondurand N. & Sham M. H. The role of SOX10 during enteric nervous system development. Dev Biol 382, 330–343, doi: 10.1016/j.ydbio.2013.04.024 (2013). [DOI] [PubMed] [Google Scholar]
- Wegner M. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res 27, 1409–1420 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods 12, 7–8, doi: 10.1038/nmeth.3213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. & Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43, W174–181, doi: 10.1093/nar/gkv342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Kucukural A. & Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738, doi: 10.1038/nprot.2010.5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40, doi: 10.1186/1471-2105-9-40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milunsky J. M. In GeneReviews (R) (eds Pagon, et al.) (1993). [Google Scholar]
- Nayak C. S. & Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann Otol Rhinol Laryngol 112, 817–820 (2003). [DOI] [PubMed] [Google Scholar]
- Newton V. E. Clinical features of the Waardenburg syndromes. Adv Otorhinolaryngol 61, 201–208 (2002). [DOI] [PubMed] [Google Scholar]
- Dourmishev A. L., Dourmishev L. A., Schwartz R. A. & Janniger C. K. Waardenburg syndrome. Int J Dermatol 38, 656–663 (1999). [DOI] [PubMed] [Google Scholar]
- Lecerf L. et al. An impairment of long distance SOX10 regulatory elements underlies isolated Hirschsprung disease. Hum Mutat 35, 303–307, doi: 10.1002/humu.22499 (2014). [DOI] [PubMed] [Google Scholar]
- Chen K. et al. De novo dominant mutation of SOX10 gene in a Chinese family with Waardenburg syndrome type II. Int J Pediatr Otorhinolaryngol 78, 926–929, doi: 10.1016/j.ijporl.2014.03.014 (2014). [DOI] [PubMed] [Google Scholar]
- Pingault V. et al. SOX10 mutations mimic isolated hearing loss. Clin Genet 88, 352–359, doi: 10.1111/cge.12506 (2015). [DOI] [PubMed] [Google Scholar]
- Chen K. et al. Genetic counseling for a three-generation Chinese family with Waardenburg syndrome type II associated with a rare SOX10 mutation. Int J Pediatr Otorhinolaryngol 79, 745–748, doi: 10.1016/j.ijporl.2015.03.006 (2015). [DOI] [PubMed] [Google Scholar]
- Amiel J. et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 45, 1–14, doi: 10.1136/jmg.2007.053959 (2008). [DOI] [PubMed] [Google Scholar]
- Wegner M. Secrets to a healthy Sox life: lessons for melanocytes. Pigment Cell Res 18, 74–85, doi: 10.1111/j.1600-0749.2005.00218.x (2005). [DOI] [PubMed] [Google Scholar]
- Pusch C. et al. The SOX10/Sox10 gene from human and mouse: sequence, expression, and transactivation by the encoded HMG domain transcription factor. Hum Genet 103, 115–123 (1998). [DOI] [PubMed] [Google Scholar]
- Schreiner S. et al. Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development 134, 3271–3281, doi: 10.1242/dev.003350 (2007). [DOI] [PubMed] [Google Scholar]
- Chaoui A. et al. Identification and functional analysis of SOX10 missense mutations in different subtypes of Waardenburg syndrome. Hum Mutat 32, 1436–1449, doi: 10.1002/humu.21583 (2011). [DOI] [PubMed] [Google Scholar]
- Bondurand N. et al. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet 9, 1907–1917 (2000). [DOI] [PubMed] [Google Scholar]