

Abstract
Background
The prevalence of atopic dermatitis has increased over the last 10 years. Atopic dermatitis tends to run in families and commonly begins to manifest in childhood. The prevalence of atopic dermatitis is as high as 20% in children. Thus, early diagnosis and treatment of atopic dermatitis are important. Understanding its genetic basis is also needed to facilitate early detection.
Methods
To identify family-specific candidate genetic variants associated with early-onset atopic dermatitis in Koreans, we carried out whole-exome sequencing of three separate families with this condition. Additional validation was performed in 112 AD patients and 61 controls using Sanger sequencing.
Results
We focused on both common functional variants with a minor allele frequency higher than 1% and rare variants with a minor allele frequency less than 1%. The relevance of the respective variants was supported by a program that could predict whether the mutations resulted in damaged protein function. Fourteen overlapping genes were identified during exome sequencing. Three variants of the COL6A6 gene appeared in all three families and were in close proximity to atopic dermatitis-related loci on chromosome 3q21. The homozygous frequency for the rs16830494 minor allele (AA) and the rs59021909 (TT) allele and the rs200963433 heterozygous (CT) frequency were all higher in AD cases compared to controls in a population-based case-control study.
Conclusion
Identifying family-specific COL6A6 polymorphisms and genetic variants of other candidate genes associated with AD using WES is a novel approach. Our study suggests that COL6A6 variants may be risk factors for atopic dermatitis. This study provides a genetic basis for early-onset AD diagnosis in Korean patients and the development of new therapies.
Trial registration
Trial registration number: IRB NO. C2008030 (133); Name of registry: The collection research of clinical data and patient blood to identify genetic and protein biomarker of atopic dermatitis; Date of registration: 09-July-2008.
Trial registration number: IRB NO. C2015258 (1716); Name of registry: The collection study of patient blood and clinical data for the development of the prognosis prediction and early diagnosis of atopic dermatitis; Date of registration: 15-jan-2016.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-017-0368-9) contains supplementary material, which is available to authorized users.
Keywords: COL6A6, Atopic dermatitis, Sanger sequencing, Whole-exome sequencing
Background
Atopic dermatitis (AD) is a chronic, relapsing, inflammatory skin disorder characterized by eczematous lesions and dry, itchy skin. AD seems to be caused by a combination of hereditary and environmental factors. Although AD has features of a multigenic syndrome, it tends to run in families and commonly begins to manifest in childhood [1]. In a large cohort study of family history, AD was determined to be inherited in an autosomal dominant fashion [2]. There are strong genetic heritable components in many other common and complex diseases [3].
AD occurs at a rate as high as 20% in children [4]. Understanding the genetic background, early discovery, and best therapies for AD is important. Thus, identification of causal variants associated with a common complex trait like AD is needed for early detection.
Whole-exome sequencing (WES) is a technique that involves the sequencing of all protein-coding genes, known as the exome, which is comprised of about 3×107 bases. Although the exome constitutes less than 2% of the human genome, mutations in the exome can have more severe consequences than do mutations in the other 98% of the human genome [5]. The purpose of WES is to identify variations by filtering big data collected from all protein-coding regions. This data includes disease-causing mutations inherited in a Mendelian pattern or disease-predisposing single-nucleotide polymorphisms (SNPs) found in both common and complex disorders [6]. To identify familial causative variants of early-onset AD, we recruited three pedigrees from families with a history of AD and severe clinical phenotypes. We then performed WES on all involved individuals. Alleles were compared to identify causal variants and subsequently validated in a population-based case-control study using Sanger sequencing.
A considerable number of rare and common variants were found, and 14 overlapping genes were detected in the three families. The common disease-common variant (CD-CV) hypothesis can best be tested in genome-wide association studies (GWASs). However, the hypothesis that individually gathered rare variants have severe effects arose from issues of missing heritability in the CD-CV hypothesis [7]. Common variants of common diseases were recently reported to be shared among different races [8]. The importance of common and rare variants is still uncertain [9]. Thus, we aimed to identify not only rare variants, but also common variants.
Linkage analysis is used to determine the rough positions of causal genes relative to known genetic makers. An AD linkage analysis found that chromosome region 1q21 contains an epidermal differentiation complex (EDC). The EDC includes various AD-related genes (e.g., loricrin, involucrin, filaggrin, and the S100 family) [10]. We used AD-related loci to confirm the association of the 14 overlapping genes with AD [11].
Here, we report the WES results from familes with early-onset AD, and we suggest the possibility of new variants, COL6A6 polymorphisms, as novel candidate for the detection of early-onset AD.
Methods
Patients
Peripheral blood samples were obtained from three families with a history of AD. Each family consists of 2 affected and 2 unaffected individuals (Additional file 1: Figure S1). We attempted to eliminate environmental factors as much as possible by recruiting early-onset cases. This study was reviewed and approved by the Chung-Ang University Hospital Institutional Review Board. Each family member was diagnosed with AD by a dermatologist. All patients and children developed AD before 2 years of age and were selected based on high IgE level (>1000) and SCORAD score (>50). Additionally, 112 AD patients and 61 control subjects under 2 years 9 months old were enrolled to validate the association between the candidate variants and atopic dermatitis (Additional file 1: Table S1).
Whole-exome sequencing
Genomic DNA was isolated from the peripheral blood of the members of the three families using a QIAamp DNA Mini Kit (Qiagen Inc, Valencia, CA, USA). The DNA quality and quantity were assessed with a Nanodrop spectrometer (Nanodrop Technologies, Wilmington, DE, USA) and a Qubit fluorometer (Life Technologies, Grand Island, NY, USA). WES was performed using SureSelect Human All Exon V4 + UTR 71 Mb (Agilent, CA, USA), following the manufacturer’s standard protocol. Genomic DNA was sheared using Covaris (Covaris, Woburn, MA, USA). A paired-end DNA sequencing library was prepared through shearing, end-repair, A-tailing, peak detection, PE adaptor ligation, and amplification. After the library was hybridized with bait sequences for 24 h, it was purified and amplified with an index barcode tag, and the library quality and quantity were determined. The exome library was sequenced with the 100-bp paired-end mode of the HiSeq SBS kit.
Whole-exome sequencing processing and alignment
Sequence reads in FASTQ format were mapped to the human assembly UCSC hg19 using the Burrows-Wheeler Aligner (BWA, v0.7.7) [12] with “mem” and seed value parameters “-k 45” to create SAM files with correct mate pair information. The read group tag included the sample name. Picard (v1.92) was then used to convert the SAM files to compressed BAM files and then to sort the BAM files by chromosome coordinate. The Genome Analysis Toolkit (v2.3.9Lite) [13] was used to locally realign the BAM files at intervals corresponding to potential insertion/deletion (indel) alignment errors. Insertions and deletions were identified with Mutect [14] and a GATK Somatic Indel Detector, respectively. Single-nucleotide variants and indels were annotated using snpEff (v3.6c) [15] to classify variants as synonymous, non-synonymous, missense, frameshift point mutations, or frameshift indels.
Annotation
Filter 1: SnpEff (http://snpeff.sourceforge.net/index.html) is a type of variant annotation and an effect prediction tool. It annotates and predicts the effects of variants, such as amino acid changes, on genes. Variants produce effects of different “types” (e.g., non-synonymous, stop-gained, insertion, deletion).
Filter 2: Impact prediction by SnpEff shows results of putative variants, making it easier to quickly categorize and prioritize variants (High: Splice_Site_Acceptor, Splice_Site_Donor, Start_Lost, Frame_Shift, Stop_Gained; Moderate: Non_Synonymous_coding, Codon_Change, Codon_Insertion and Deletion, etc.).
Filter 3: SIFT and Polyphen2 of dbNSFP
The SIFT score predicts whether an amino acid substitution affects protein function. SIFT prediction is based on the degree of amino acid conservation in aligned segments derived from closely related sequences, as collected through PSI-BLAST. The range was 0 to 1; substitutions with scores lower than 0.05 were predicted to be damaging (a lower score signified a greater detrimental effect), whereas substitutions with higher scores were predicted to be tolerable.
The Polyphen2 HDIV score was based on HumDiv, i.e., hdiv_prob. The score ranged from 0 to 1, and a higher score suggested a greater degree of predicted damage. A prediction of “probably damaging” corresponded to scores between 0.957 and 1, “possibly damaging” for scores ranging from 0.453 to 0.956, and “benign” for those between 0 and 0.452.
Filter 4: The PhyloP of dbNSFP detects sites under negative or positive selection while allowing for changes in evolution rate over the branches of the phylogenetic tree. A higher PhyloP score indicates a better conserved site (http://varianttools.sourceforge.net/Annotation/DbNSFP).
Filter 5: PhastCons measures the strength of purifying selection acting on a DNA sequence. A high PhastCons score (0.2) is strong evidence that a genomic region is functionally important.
Filter 6: The 1000 Genome allele frequency of dbNSFP selects variants with frequencies of less than 0.01 or those with unknown frequencies.
Filter 7: The in-house Korean database at the Theragen Etex Bio Institute selects variants with minor allele frequencies (MAFs, less than 0.02 or unknown).
Sanger sequencing
Three SNPs were selected for Sanger validation. PCR amplification of all SNPs was performed at 95 °C for 10 min, followed by 35 cycles at 95 °C for 30 s, 55–58 °C for 30 s and 72 °C for 40 s, with a final extension at 72 °C for 1 min 30 s. The PCR mixtures (total volume 50 μL) contained 25 μL of 2X EF-Taq premix (SolGent, Seoul, South Korea), 2.5 μL of oligonucleotide primer (10 pmol/μL), 18 μL of distilled water, and 2 μL of template containing 20 ng genomic DNA. The PCR products underwent purification via a PCR purification kit (Favorgen, Pingtung, Taiwan) and were sequenced on an Applied Biosystems 3500 DNA sequencer (Foster City, CA, USA) according to the manufacturer’s instructions.
Results
Whole-exome sequencing in three families with atopic dermatitis
WES was performed on three families with AD. To limit our study to genetic factors, we recruited three pedigrees from families with severe clinical AD phenotypes and attempted to minimize environmental factors by selecting early-onset cases. The WES analysis showed that all affected individuals were heterozygous for the identified variants, while unaffected individuals were homozygous wild-type (Table 1).
Table 1.
Genotypes of overlapping common and rare variants in three families
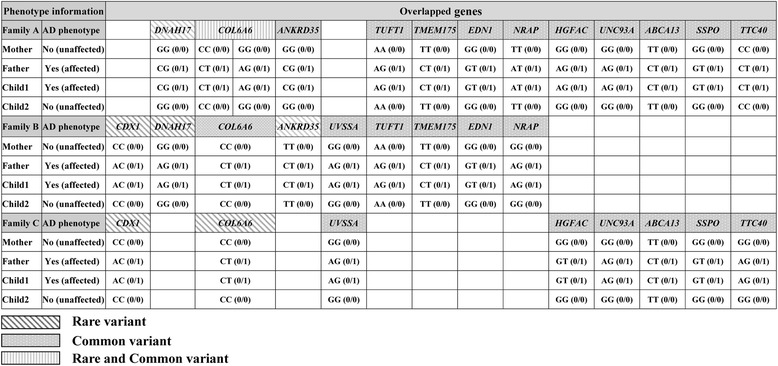
A large amount of WES data from each individual exome were filtered in a stepwise fashion to isolate candidate variants related to AD (Additional file 1: Figure S2). The variants collected from the three families were counted after each filtering process. We obtained an average of 176 common variants per family after filter 4 and an average of 48 rare variants following the 1000 Genome filter 5. An average of 44 rare variants were detected after the Korean filter (Table 2).
Table 2.
Number of variants by filter in three families using a dominant genetic model
| Filter system | Dominant model | |||
|---|---|---|---|---|
| Filter step | Filter process | Family A | Family B | Family C |
| Raw | – | 7523 | 3564 | 3227 |
| Filter 1 | EFFECT | 2799 | 2489 | 2198 |
| Filter 2 | IMPACT | 502 | 516 | 443 |
| Filter 3 | SIFT, PolyPhen2 | 278 | 258 | 231 |
| Filter 4 | phyloP | 234 | 225 | 206 |
| Filter 5 | Phastcon | 200 | 173 | 156 |
| Filter 6 | 1000 genome | 33 | 60 | 53 |
| Filter 7 | Korean | 31 | 54 | 49 |
Amino acid changes and variant types (non-synonymous SNP, stop-gained, insertion, deletion) were determined using the variant annotation from the step 1 filter. A non-synonymous SNP is a single-nucleotide change that results in a codon for a different amino acid. Considerable numbers of non-synonymous SNPs were observed in the three families (data not shown).
The variants identified by filter 5 not only indicated common variants (MAF greater than 1%), but also possible functional variants predicted to impair protein function as well as sequences that are highly conserved among 100 vertebrate species.
To find more critical variants among the many filtered genes, we confirmed the overlapping genes of common variants from filter 5 in the three families (Table 1 and Additional file 1: Figure S3). There were 14 overlapping genes in filter 5, four of which reached filter 7 and could be called “rare variants.” Risk alleles were identified in AD patients by comparison with healthy controls (Table 1). The number of overlapping genes is also depicted as a Venn diagram (Additional file 1: Figure S3). Variants of COL6A6 appeared in all three families (Tables 3, 4 and 5), and two COL6A6 SNPs were detected in Family A (Table 3).
Table 3.
Family-specific common and rare variants in Family A
| Common variant | Functional prediction program | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | RS# | aChr | bPOS | cAmino acid change | Type | dSIFT | ePolyphen2 | fPhyloP | gPhastCons | hGlobal | East Asian | iKorean |
| COL6A6 | 16830494 | chr3 | 130,361,856 | R1739Q | cSNP | 0.04 | 0.272 | 1.703 | 0.995 | 0.12 | 0.19 | 0.188 |
| ANKRD35 | 11579366 | chr1 | 145,562,293 | E661Q | cSNP | 0.38 | 0.971 | 5.285 | 1 | 0.39 | 0.26 | 0.233 |
| TUFT1 | 3828054 | chr1 | 151,512,895 | Q18R | cSNP | 0.77 | 0.688 | 1.688 | 0.288 | 0.1 | 0.03 | 0.047 |
| TMEM175 | 34311866 | chr4 | 951,947 | M393T | cSNP | 0.01 | 0 | 1.299 | 0.999 | 0.12 | 0.12 | 0.155 |
| EDN1 | 5370 | chr6 | 12,296,255 | K198N | cSNP | 0.08 | 0.454 | 0.049 | 0.001 | 0.21 | 0.28 | 0.258 |
| NRAP | 2270182 | chr10 | 115,392,919 | N519I | cSNP | 0.19 | 0.958 | 2.477 | 1 | 0.26 | 0.20 | 0.205 |
| HGFAC | 16844401 | chr4 | 3,449,652 | R509H | cSNP | 0.22 | 0.943 | 2.662 | 0.593 | 0.07 | 0.10 | 0.115 |
| UNC93A | 2235197 | chr6 | 167,709,702 | W151 | STOP GAINED | . | . | 4.525 | 1 | 0.1 | 0.15 | 0.163 |
| ABCA13 | 1771229 | chr7 | 48,313,881 | F1540L | cSNP | . | 0.997 | 2.325 | 0.614 | 0.11 | 0.17 | 0.255 |
| SSPO | 1005603 | chr7 | 149,516,881 | S4028I | cSNP | . | . | . | . | 0.2 | 0.19 | 0.181 |
| TTC40 | 12781609 | chr10 | 134,748,331 | S264N | cSNP | . | . | . | . | 0.37 | 0.36 | 0.412 |
| Rare variant | ||||||||||||
| DNAH17 | . | chr17 | 76,567,792 | I204M | cSNP | 0.11 | . | 1.926 | 0.992 | . | . | |
| COL6A6 | 200963433 | chr3 | 130,289,976 | R906C | cSNP | 0 | 1 | 4.596 | 1 | 0.0014 | 0.01 | 0.017 |
aChr = Chromosome
bPos = Position
cAmino acid changes = single-letter codes for amino acids are presented, cSNP = single-nucleotide polymorphisms in coding regions
dSIFT, prediction scores for amino acid substitutions that affect protein function (damaging < 0.05, tolerance > 0.05; scores range from 0 to 1)
ePolyphen2, prediction of the possible impact of amino acid substitutions (0.957 < probably damaging < 1, 0.453 < possibly damaging < 0.956, 0 < benign < 0.452; scores range from 0 to 1)
fPhyloP, prediction of conserved sites across species; a higher score indicates a more conserved site (values > 0)
gPhastCons, predicts the possibility that a nucleotide belongs to a conserved element in the phylogenetic tree (Values > 0.2)
hGlobal frequency, variants with MAFs as low as 1% or with an unknown frequency (value < 0.01)
iKorean frequency, variants with MAFs as low as 2% or in unknown genes (values > 0.02)
Table 4.
Family-specific common and rare variants in Family B
| Common variant | Functional prediction program | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | RS# | Chr | POS | Amino acid change | Type | SIFT | Polyphen2 | PhyloP | PhastCons | Global | East Asian | Korean |
| COL6A6 | 59021909 | chr3 | 130,285,929 | P556S | cSNP | 0.11 | 0.999 | 2.136 | 0.997 | 0.09 | 0.08 | 0.119 |
| UVSSA | 2276904 | chr4 | 1,349,029 | R391H | cSNP | . | . | . | . | 0.23 | 0.42 | 0.374 |
| TUFT1 | 3828054 | chr1 | 151,512,895 | Q18R | cSNP | 0.77 | 0.688 | 1.688 | 0.763 | 0.1 | 0.03 | 0.047 |
| TMEM175 | 34311866 | chr4 | 951,947 | M393T | cSNP | 0.01 | 0 | 1.299 | 0.563 | 0.12 | 0.12 | 0.155 |
| EDN1 | 5370 | chr6 | 12,296,255 | K198N | cSNP | 0.08 | 0.454 | 0.049 | 0.895 | 0.21 | 0.28 | 0.258 |
| NRAP | 868738 | chr10 | 115,381,747 | R884C | cSNP | 0.01 | 0.986 | 4.833 | 1 | 0.24 | 0.17 | 0.198 |
| Rare variant | ||||||||||||
| CDX1 | 370852694 | chr5 | 149,546,819 | A127E | cSNP | 0.87 | 0.458 | 1.235 | 0.996 | . | . | 0.027 |
| DNAH17 | 78098467 | chr17 | 76,510,974 | A1332V | cSNP | 0.6 | . | 4.052 | 1 | 0.01 | 0.04 | 0.037 |
| ANKRD35 | 146839643 | chr1 | 145,560,094 | C194R | cSNP | 0.01 | 1 | 3.419 | 1 | 0.0037 | 0.01 | 0.016 |
Table 5.
Family-specific common and rare variants in Family C
| Common variant | Functional prediction program | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | #RS | Chr | POS | Amino acid change | Type | SIFT | Polyphen2 | PhyloP | PhastCons | Global | East Asian | Korean |
| UVSSA | 2276904 | chr4 | 1,349,029 | R391H | cSNP | . | . | . | . | 0.23 | 0.42 | 0.374 |
| HGFAC | 3748034 | chr4 | 3,446,091 | A218S | cSNP | 0.38 | 0.659 | 2 | 1 | 0.15 | 0.28 | 0.305 |
| UNC93A | 2235197 | chr6 | 167,709,702 | W151 | STOP GAINED | . | . | 4.525 | 1 | 0.1 | 0.15 | 0.163 |
| ABCA13 | 17712299 | chr7 | 48,313,881 | F1540L | cSNP | . | 0.997 | 2.325 | 0.85 | 0.11 | 0.17 | 0.255 |
| SSPO | 1005603 | chr7 | 149,516,881 | S4028I | cSNP | . | . | . | 1 | 0.2 | 0.19 | 0.181 |
| TTC40 | . | chr10 | 134,679,632 | T1596M | cSNP | . | . | . | . | . | . | |
| Rare variant | ||||||||||||
| COL6A6 | 200963433 | chr3 | 130,289,976 | R906C | cSNP | 0 | 1 | 4.596 | 1 | 0.0014 | 0.01 | 0.017 |
| CDX1 | 370852694 | chr5 | 149,546,819 | A127E | cSNP | 0.87 | 0.458 | 1.235 | 0.996 | . | . | 0.027 |
The genotypes of the 14 genes were further assessed in an exome analysis. Affected fathers and children were heterozygous, while unaffected mothers and children were homozygous wild type for all 14 candidate genes (Table 1).
The chromosome position, amino acid change, and functional prediction score for each of the 14 overlapping genes are presented for each family. The 14 candidate genes were deemed functionally interesting and supported by the SIFT scores (probability of being damaging/deleterious) and the results of the PhyloP analysis (highly conserved among 100 vertebrate species). The significantly low SIFT and high PhyloP and Phastcon scores of COL6A6 found in all three families signify deleterious protein function. Furthermore, when the Korean filter was applied to COL6A6, all three families showed a genetic variant with an MAF range of 1.7–18%, as measured in 800 Koreans subjects (Tables 3, 4 and 5).
To reduce errors from WES, COL6A6 was analyzed by Sanger sequencing to detect SNPs in all three families. The variants we identified were in positions consistent with those of the WES results. In Family A, the missense mutations c.5216G > A, p.Arg1739Glu (common variant), and c.2716C > T, p.Arg906Cys (rare variant) were detected in the affected family members, but not in unaffected family members (Fig. 1a and b). In Family B, the missense mutation c.1666C > T, p.Pro555Ser (common variant) was only found in the affected members (Fig. 1c). In Family C, the missense mutation c.2716C > T, p.Arg906Cys (rare variant) was also only detected in the affected members (Fig. 1d).
Fig. 1.
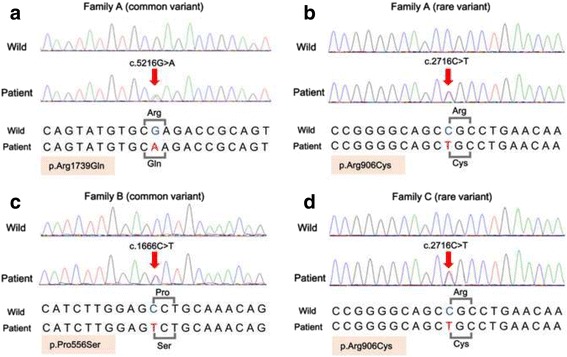
Single-nucleotide polymorphisms (SNPs) in coding regions of COL6A6 in three families. The missense mutation was detected by Sanger sequencing analysis in each family (a-d)
Additionally, to investigate the possibility of COL6A6 variants (rs16830494, rs59021909, and rs200963433) as candidate risk factors for AD, Sanger sequencing was also performed in a case-control study involving 112 patients with AD and 61 control subjects. The allele and genotype frequencies were counted (Table 6). Odds ratios (OR) and 95% confidence intervals (CI) were estimated for all risk factors (Additional file 1: Table S2). No significant associations were found in these three variants under either dominant or recessive models. However, a minor allele (A) of the common variant rs16830494 showed a tendency toward lower frequency. Homozygosity for the rs16830494 minor allele (AA) and for the rs59021909 (TT) allele were more frequent in AD cases compared to controls. The rs200963433 is a rare variant; the 3.5% heterozygous frequency (CT) of rs200963433 seen in the AD cases was double the 1.6% frequency observed in healthy controls (Table 6). The frequency of rs200963433 in Sanger sequencing was also compared with the frequency of 1000 global, 286 East Asian, and 800 Korean subjects. The 1.7% MAF of rs200963433 in the 800 Koreans surveyed nearly matched that of the 61 health controls in Sanger sequencing, and the MAF was elevated in AD cases (Tables 3 and 6).
Table 6.
Allele and genotype frequencies of COL6A6 polymorphisms in 112 Korean AD patients and 61 controls
| SNP | Controls, n (%) | AD, n (%) | ||
|---|---|---|---|---|
| rs16830494 | Allele | G | 99 (81.1) | 189 (84.4) |
| (c.5216G > A) | A | 23 (18.9) | 35 (15.6) | |
| Genotype | GG | 39 (63.9) | 79 (71.2) | |
| GA | 21 (34.4) | 29 (26.1) | ||
| AA | 1 (1.6) | 3 (2.7) | ||
| rs59021909 | Allele | C | 108 (88.5) | 199 (88.8) |
| (c.1666C > T) | T | 14 (11.5) | 25 (11.2) | |
| Genotype | CC | 47 (77.0) | 89 (79.5) | |
| CT | 14 (23.0) | 21 (18.8) | ||
| TT | 0 (0) | 2 (1.8) | ||
| rs200963433 | Allele | C | 121 (99.2) | 220 (98.2) |
| (c.2716C > T) | T | 1 (0.8) | 4 (1.8) | |
| Genotype | CC | 60 (98.4) | 108 (96.4) | |
| CT | 1 (1.6) | 4 (3.6) | ||
| . | TT | 0 (0) | 0 (0) |
We compared the genetic loci between AD candidate variants identified via WES and in AD-linked chromosomal regions. In previous studies, AD-related chromosome loci were detected using AD linkage analysis. The CDX1, ANKRD35, and TUFT1 genes were present at positions 5q31-33 and 1q21. COL6A6 was in close proximity to the 3q21 locus, which is known to be linked to AD (Table 7).
Table 7.
Five candidate genes within AD susceptibility loci identified through genetic linkage analysis
| Gene | Chr | POS | Locus of AD-linkage |
|---|---|---|---|
| COL6A6 | 3 | 130.2–4 Mba | 3q21 (chr3 122.2 Mb–129.5 Mb) [11] |
| CDX1 | 5 | 149.5 Mb | 5q31-33 (Chr5 131.2 Mb–160.5 Mb) [29] |
| ANKRD35 | 1 | 145.5 Mb | 1q21 (Chr1 143.2 Mb–155.1 Mb) [10] |
| TUFT1 | 1 | 151.5 Mb | 1q21 (Chr1 143.2 Mb–155.1 Mb) |
| FLG | 1 | 152.2 Mb | 1q21 (Chr1 143.2 Mb–155.1 Mb) |
aMb = mega base pairs = 1,000,000 bp
We also detected SNPs in filaggrin (FLG) and FLG2 in these three families. FLG polymorphisms at the 1q21 locus were observed in three families, respectively (Additional file 1: Table S3).
Discussion
Early-onset AD is a phenotype that may be associated with a higher risk of multiple allergies and asthma [16]. The identification of specific genes predictive of early-onset AD may lead to AD prevention and better management.
To identify familial candidate genes related to early-onset AD, we recruited three families with this phenotype. A family-based design has the advantages of being cost-effective and the ability to discover rare variants not detectable in a population study [17]. De novo gene mutations capable of influencing AD susceptibility can also be identified in affected individuals by comparison with unaffected individuals in a family.
Family-specific candidate AD genes were detected using WES. Considerable numbers of common and rare variants were identified in each of the three families. To identify highly critical genes for AD, we confirmed 14 overlapping genes in these families. Variants of the overlapping genes were predicted to be deleterious through functional prediction algorithms. The results of previous AD association studies and the functions of candidate genes were also examined.
Common and rare variants of COL6A6 were found in all three families. The COL6A6 gene encodes collagen type VI alpha 6, the α6-chain of an extracellular matrix protein that is widely expressed in human skin and maintains skeletal muscle and skin integrity [18]. COL6A6 is in close proximity to 3q21. In previous studies, AD-associated loci were confirmed by whole-genome linkage scans. The 3q21 locus has been identified as an AD susceptibility region [11]. Another genome-wide linkage study found highly significant evidence of a linkage to chromosome 3q21. Moreover, significant evidence has linked this locus with allergic sensitization presumably by paternal imprinting, further supporting the presence of an atopy-related gene in this region [19]. CD80 and CD86 are major candidate genes located on 3q21 that are expressed by antigen-presenting cells and are essential to T cell activation [20]. COL6A5, the other collagen alpha-chain, is also linked to AD. A lack of COL6A5 expression affects epidermal integrity and function [21]. Early onset-AD has a prevalence of 15–20% in industrialized countries [4]. The MAF (1.7–18%) of COL6A6 variants in a large sample of the Korean population was similar to the incidence rate of early-onset AD. Our findings suggest that variants of COL6A6 may be novel candidates for early-onset AD in Koreans.
No significant association with AD was identified among the three COL6A6 variants per the Odds ratios used in the case-control study. However, a high frequency of homo alt was detected in rs16830494 and rs59021909 in AD cases relative to controls. It was difficult to obtain p-values for rare variants using Odds ratio, a common limitation [22]. The similar frequencies of rs200963433 in both the 800 Korean population and the 61 healthy controls demonstrate the credibility of the data despite the small study size.
A rare variant of the caudal type homeobox1 (CDX1) gene was detected in two families. CDX1 is located in a candidate AD-linkage region, 5q31-33 [23]. The function of the CDX1 gene is to inhibit β-catenin/T-cell factor transcriptional activity. β-catenin regulates cell-cell adhesion, and Wnt/β-catenin signaling is associated with skin development [24, 25]. A common variant of the ultraviolet (UV)-stimulated scaffold protein A (UVSSA) gene was also identified. The function of the UVSSA gene is to repair DNA damaged by UV rays. The function of any other genes assumed the association of AD was not found.
Missense mutations of FLG and FLG2 were detected in three families, respectively. The effects of the FLG polymorphism on AD are not well characterized. However, Seon-Young et al. recently reported that the FLG (rs11584340) polymorphism is associated with a higher AD risk in the Korean population, and that it affects free fatty acids in serum of AD patients [26]. Loss‐of‐function mutations involving FLG strongly predispose the carrier to early-onset AD, but not to late-onset AD [27]. This study suggests that genetic screening is crucial for identifying risk variants of early-onset AD.
Individually gathered rare variants have severe effects and play important roles in complex human disorders [28]. Thus, our data will help to expand genetic studies of AD.
Conclusions
Identifying family-specific COL6A6 polymorphisms and genetic variants of other candidate genes associated with AD using WES is a novel approach. Our study suggests that COL6A6 variants may constitute candidate risk factors for AD development, as identified via family-based WES and a non-familial case-control study of 173 subjects. This study provides a genetic basis for early-onset AD diagnosis in Korean patients and the development of new therapies.
Acknowledgements
We thank all participants who made this study possible.
Funding
This study was supported by a grant from the Korean Healthcare Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C2687).
Availability of data and materials
All data generated during this study are included in this published article and its Additional file 1.
Authors’ contributions
WIH, KYP, and SJS conceived and designed the study. TJ, MKL, EHC, and NJM recruited subjects. WIH, TJ, and MKL performed all of the laboratory work. WIH, KYP, and MJK performed all data analysis. EHC, HSK, and JMB performed the statistical analysis. WIH drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was reviewed and approved by the Chung-Ang University Hospital Institutional Review Board (IRB). The recruitment and consent of participants were conducted on the basis of IRB below.
Trial registration number: IRB NO. C2008030 (133); Name of registry: The collection research of clinical data and patient blood to identify genetic and protein biomarker of atopic dermatitis; Date of registration: 09-July-2008.
Trial registration number: IRB NO. C2015258 (1716); Name of registry: The collection study of patient blood and clinical data for the development of the prognosis prediction and early diagnosis of atopic dermatitis; Date of registration: 15-jan-2016.
Abbreviations
- AD
Atopic dermatitis
- CD-CV
Common disease-common variant
- EDC
Epidermal differentiation complex
- GWASs
Genome-wide association studies
- MAF
Minor allele frequency
- SNPs
Single-nucleotide polymorphisms
- WES
Whole-exome sequencing
Additional file
Clinical characteristics of the subjects. Table S2. Odds ratios (ORs) and 95% confidence intervals (CIs) for atopic dermatitis associated with COL6A6 polymorphisms in Korean patients. Table S3. Fliaggrin variants detected in three families. Figure S1. Pedigree diagrams of three families with atopic dermatitis. Figure S2. Flow chart of the process for filtering variants. Figure S3. A Venn diagram showing the number of overlapping genes. (PDF 258 kb)
Contributor Information
Won Il Heo, Email: onebbang1234@naver.com.
Kui Young Park, Email: momo920@hanmail.net.
Taewon Jin, Email: j.magma@gmail.com.
Mi-Kyung Lee, Email: cpworld@cau.ac.kr.
MinJeong Kim, Email: moyalmin@nate.com.
Eung Ho Choi, Email: choieh@yonsei.ac.kr.
Hae-Suk Kim, Email: haesuk.kim@therabio.kr.
Jung Min Bae, Email: jminbae@gmail.com.
Nam Ju Moon, Email: njmoon@chol.com.
Seong Jun Seo, Phone: 82-2-6299-1538, Email: drseo@hanafos.com.
References
- 1.Hoffjan S, Stemmler S. Unravelling the complex genetic background of atopic dermatitis: from genetic association results towards novel therapeutic strategies. Arch Dermatol Res. 2015;307(8):659–70. [DOI] [PubMed]
- 2.Uehara M, Kimura C. Descendant family history of atopic dermatitis. Acta Derm Venereol. 1993;73(1):62–3. doi: 10.2340/00015555736263. [DOI] [PubMed] [Google Scholar]
- 3.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80(4):727–39. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nutten S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab. 2015;66(Suppl 1):8–16. doi: 10.1159/000370220. [DOI] [PubMed] [Google Scholar]
- 5.Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59(1):5–15. doi: 10.1038/jhg.2013.114. [DOI] [PubMed] [Google Scholar]
- 6.Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014;15(2):256–78. doi: 10.1093/bib/bbs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asimit J, Zeggini E. Testing for rare variant associations in complex diseases. Genome Med. 2009;1(11):24. doi: 10.1186/gm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marigorta UM, Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9(6):e1003566. doi: 10.1371/journal.pgen.1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2011;13(2):135–45. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma M, Mehla K, Batra J, Ghosh B. Association of a chromosome 1q21 locus in close proximity to a late cornified envelope-like proline-rich 1 (LELP1) gene with total serum IgE levels. J Hum Genet. 2007;52(4):378–83. doi: 10.1007/s10038-007-0118-5. [DOI] [PubMed] [Google Scholar]
- 11.Christensen U, Moller-Larsen S, Nyegaard M, Haagerup A, Hedemand A, Brasch-Andersen C, Kruse TA, Corydon TJ, Deleuran M, Borglum AD. Linkage of atopic dermatitis to chromosomes 4q22, 3p24 and 3q21. Hum Genet. 2009;126(4):549–57. doi: 10.1007/s00439-009-0692-z. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlsten C, Dimich-Ward H, Ferguson A, Watson W, Rousseau R, Dybuncio A, Becker A, Chan-Yeung M. Atopic dermatitis in a high-risk cohort: natural history, associated allergic outcomes, and risk factors. Ann Allergy Asthma Immunol. 2013;110(1):24–8. doi: 10.1016/j.anai.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Kazma R, Bailey JN. Population-based and family-based designs to analyze rare variants in complex diseases. Genet Epidemiol. 2011;35(Suppl 1):S41–7. doi: 10.1002/gepi.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabatelli P, Gara SK, Grumati P, Urciuolo A, Gualandi F, Curci R, Squarzoni S, Zamparelli A, Martoni E, Merlini L, et al. Expression of the collagen VI alpha5 and alpha6 chains in normal human skin and in skin of patients with collagen VI-related myopathies. J Invest Dermatol. 2011;131(1):99–107. doi: 10.1038/jid.2010.284. [DOI] [PubMed] [Google Scholar]
- 19.Lee YA, Wahn U, Kehrt R, Tarani L, Businco L, Gustafsson D, Andersson F, Oranje AP, Wolkertstorfer A, v Berg A, et al. A major susceptibility locus for atopic dermatitis maps to chromosome 3q21. Nat Genet. 2000;26(4):470–3. doi: 10.1038/82625. [DOI] [PubMed] [Google Scholar]
- 20.Becker KG, Barnes KC. Underlying disease specificity of genetic loci in atopic dermatitis. J Invest Dermatol. 2001;117(5):1325–7. doi: 10.1046/j.0022-202x.2001.01559.x. [DOI] [PubMed] [Google Scholar]
- 21.Soderhall C, Marenholz I, Kerscher T, Ruschendorf F, Esparza-Gordillo J, Worm M, Gruber C, Mayr G, Albrecht M, Rohde K, et al. Variants in a novel epidermal collagen gene (COL29A1) are associated with atopic dermatitis. PLoS Biol. 2007;5(9):e242. doi: 10.1371/journal.pbio.0050242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merner ND, Chandler MR, Bourassa C, Liang B, Khanna AR, Dion P, Rouleau GA, Kahle KT. Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front Cell Neurosci. 2015;9:386. doi: 10.3389/fncel.2015.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer LJ, Barnes KC, Burton PR, Chen H, Cookson WO, Deichmann KA, Elston RC, Holloway JW, Jacobs KB, Laitinen T, et al. Meta-analysis for linkage to asthma and atopy in the chromosome 5q31-33 candidate region. Hum Mol Genet. 2001;10(8):891–9. doi: 10.1093/hmg/10.8.891. [DOI] [PubMed] [Google Scholar]
- 24.Guo RJ, Huang E, Ezaki T, Patel N, Sinclair K, Wu J, Klein P, Suh ER, Lynch JP. Cdx1 inhibits human colon cancer cell proliferation by reducing beta-catenin/T-cell factor transcriptional activity. J Biol Chem. 2004;279(35):36865–75. doi: 10.1074/jbc.M405213200. [DOI] [PubMed] [Google Scholar]
- 25.Lim X, Nusse R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb Perspect Biol. 2013;5(2). [DOI] [PMC free article] [PubMed]
- 26.Kim SY, Yang SW, Kim HL, Kim SH, Kim SJ, Park SM, Son M, Ryu S, Pyo YS, Lee JS, et al. Association between P478S polymorphism of the filaggrin gene & atopic dermatitis. Indian J Med Res. 2013;138(6):922–7. [PMC free article] [PubMed] [Google Scholar]
- 27.Rupnik H, Rijavec M, Korošec P. Filaggrin loss‐of‐function mutations are not associated with atopic dermatitis that develops in late childhood or adulthood. Br J Dermatol. 2015;172(2):455–61. doi: 10.1111/bjd.13477. [DOI] [PubMed] [Google Scholar]
- 28.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141(2):210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 29.Holloway JW, Lonjou C, Beghe B, Peng Q, Gaunt TR, Gomes I, Hall IP, Dewar JC, Wilkinson J, Thomas NS, et al. Linkage analysis of the 5q31-33 candidate region for asthma in 240 UK families. Genes Immun. 2001;2(1):20–4. doi: 10.1038/sj.gene.6363723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated during this study are included in this published article and its Additional file 1.