

Abstract
Pembrolizumab, a potent antibody against programmed death 1 (PD‐1) receptor, has shown robust antitumor activity and manageable safety in patients with advanced solid tumors. Its pharmacokinetic (PK) properties were analyzed with population PK modeling using pooled data from the KEYNOTE‐001, −002, and −006 studies of patients with advanced melanoma, non‐small cell lung cancer (NSCLC), and other solid tumor types. Pembrolizumab clearance was low and the volume of distribution small, as is typical for therapeutic antibodies. Identified effects of sex, baseline Eastern Cooperative Oncology Group performance status, measures of renal and hepatic function, tumor type and burden, and prior ipilimumab treatment on pembrolizumab exposure were modest and lacked clinical significance. Furthermore, simulations demonstrated the model has robust power to detect clinically relevant covariate effects on clearance. These results support the use of the approved pembrolizumab dose of 2 mg/kg every 3 weeks without dose adjustment in a variety of patient subpopulations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Pembrolizumab is a potent antibody against the cellular immune “switch” PD‐1 receptor with high activity in the treatment of certain types of advanced cancer. However, an analysis of the population PKs of pembrolizumab in patients with advanced cancer had not yet been performed.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The characterization of pembrolizumab PKs and quantification of the effect of its intrinsic and extrinsic factors on exposure.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ PK profile is typical for a therapeutic monoclonal antibody with low clearance, limited volume of distribution, and low variability. Intrinsic (e.g., body weight, age, sex, tumor type and burden, and renal and hepatic impairment) and extrinsic (e.g., concomitant medications) factors have no clinically relevant impact on pembrolizumab exposure.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ A model‐based approach could be used to systematically justify the absence of dose adjustments of pembrolizumab in subpopulations relative to the proposed clinical dosing regimen.
The programmed death 1 (PD‐1) receptor pathway represents a major immune control switch, which may be engaged by tumor cells to overcome active T‐cell immune surveillance. Many tumors, including melanoma, co‐opt the PD‐1 pathway by expressing the PD ligand‐1 on the cell surface, leading to suppression of tumor‐infiltrating cytotoxic T‐cell activity.1, 2 Pembrolizumab (MK‐3475) is a potent and highly selective anti–PD‐1 humanized monoclonal antibody currently approved for the treatment of advanced melanoma and, in the United States, advanced non‐small cell lung cancer (NSCLC) previously treated with platinum‐doublet chemotherapy and that expresses PD ligand‐1. It is in clinical development for treatment of various other advanced cancers. Until recently, the standard‐of‐care treatment for patients with advanced melanoma was ipilimumab, a fully human, monoclonal antibody that blocks cytotoxic T‐lymphocyte antigen‐4, another immune checkpoint protein.3, 4 In early preclinical models, the combined blockade of PD‐1 and cytotoxic T‐lymphocyte antigen‐4 achieved more pronounced antitumor activity than blockade of either pathway alone.5, 6
In traditional drug development programs, clinical pharmacology is typically characterized through phase I studies in healthy volunteers with intensive pharmacokinetic (PK) sampling. As in most oncology programs, this approach was not possible in the pembrolizumab program because of a limited ability to administer the drug to healthy subjects. Therefore, population PK analysis of sparse data obtained in patients across multiple trials was the most efficient approach to answer traditional clinical pharmacology development questions, such as the impact of intrinsic and extrinsic factors on PK and the need for dose adjustment.
In this study, model‐based analysis of pooled data from the KEYNOTE‐001, ‐002, and ‐006 studies were used to characterize the PK of pembrolizumab in patients with advanced melanoma, NSCLC, and other cancer types. Specific objectives were to characterize serum concentration profiles over time across indications, investigate the effects of potential covariates on pertinent PK parameters, evaluate the impact of selected covariates on exposure to support dose recommendations in subpopulations, and assess the adequacy of a weight‐based dosing regimen. Results from this analysis provide an integrated evaluation of the impact of intrinsic and extrinsic factors on the systemic exposure to pembrolizumab, and present evidence for the lacking need for dose adjustment for most subpopulations of patients with advanced solid tumors.
METHODS
Analysis dataset
This analysis was based on pooled data derived from patients from KEYNOTE‐001 (ClinicalTrials.gov identifier NCT01295827), KEYNOTE‐002 (ClinicalTrials.gov identifier NCT01704287), and KEYNOTE‐006 (ClinicalTrials.gov identifier NCT01866319) treated with pembrolizumab in a dose range of 1–10 mg/kg administered as intravenous infusion.
The majority of patients included in this analysis were treated for advanced melanoma or NSCLC, but several other tumor types were represented in the dose‐escalation phase of KEYNOTE‐001. For the assessment of pembrolizumab PK, serum samples were collected at regular prespecified time points in each of the studies. These included both peak (within 30 minutes after end of infusion) and trough (within 24 hours before the next dose) samples. Pembrolizumab serum concentrations in the samples were quantified using an electrochemical luminescence‐based immunoassay method with a calibration curve range of 10–800 ng/mL after a minimum required 1:10 dilution. The interassay accuracy was 98.9–110.2% of the nominal concentration and the interassay precision was <9.0%.
When creating the analysis datasets, imputation rules were defined to address issues of missing or conflicting data. These rules allowed for reasonable imputation of missing concentrations or times in data records in which there is sufficient information to reasonably impute values, and additionally provided a precise definition of circumstances in which there are insufficient or conflicting data, warranting exclusion from the analysis. For example, in cases of nonreportable serum concentrations, insufficient samples, missing bioanalysis results, or duplicate measurements, or if samples could not be related to associated clinical data, the observation records were flagged and excluded from the analysis.
Modeling approach
The population PK analysis was performed using a nonlinear mixed effects modeling approach. One‐ and two‐compartment models were evaluated, followed by testing different residual error structures. The model was then refined by testing interindividual variability for each PK parameter followed by inspection of the correlations among the random effect (η) values to guide the development of a parsimonious omega structure. Interoccasion variability of selected model parameters was explored. In addition, incorporation of allometric scaling by body weight was evaluated on all PK parameters. Model selection was based on the log‐likelihood criterion, goodness‐of‐fit plots, and scientific plausibility. The NONMEM, version VII (ICON Development Solutions, Ellicott City, MD),7, 8 Xpose version 4.4.0 (xpose.sourceforge.net),9 PsN (psn.sourceforge.net),10, 11 and R (R‐project, www.r-project.org)12 software packages were used for the exploratory analysis, model development, and post processing of NONMEM output.
Missing covariate information was imputed by the median value for continuous covariates and by the most prevalent category for categorical covariates. Overall, the level of missing continuous covariate information was limited and did not exceed 11.2%. However, for the categorical covariate describing ipilimumab pretreatment, the covariate was missing in 26.4% of subjects, and “missing” was included as a separate category.
Categorical covariates were tested if at least 5% of patients belonged to that category. Pembrolizumab serum concentrations determined to be below the limit of quantification were excluded from the analysis, because the number of samples below that limit was low (0.42% of total observations).
Covariate identification
Covariates tested for inclusion in the model included demographic factors (age, sex, and race), baseline estimated glomerular filtration rate (eGFR) as a measure of renal function, measures of hepatic function (baseline total bilirubin, aspartate aminotransferase (AST), and alkaline phosphatase), baseline albumin as general indicator of catabolism capacity, concomitant use of systemic corticosteroids (glucocorticoids), prior treatment with ipilimumab, and measures of disease severity (cancer type, baseline Eastern Cooperative Oncology Group‐Performance Status (ECOG‐PS), and baseline tumor burden (sum of longest dimensions of target lesions)). A power model was used to describe continuous covariate relationships, whereas categorical covariates were described as a proportional change.
Covariates were identified using a stepwise covariate method7 involving testing of covariate relationships in a forward inclusion (reduction in the objective function value (ΔOFV) of 6.63; P < 0.01 for 1 degree of freedom) and backward exclusion (ΔOFV of 10.8; P < 0.001 for 1 degree of freedom) procedure. In the case of categorical covariates, ΔOFV at the respective P values would be different depending on the degrees of freedom.
Model qualification
Robustness of the model parameter estimates was assessed by means of a nonparametric bootstrap evaluation. The PK parameters were estimated repeatedly by fitting the final model to 1,000 bootstrap datasets, sampled from the original dataset with replacement. The median values and 95% confidence intervals (CIs) of the population PK parameter estimates from these 1,000 bootstrap datasets were compared with the point estimates from the final model.
The predictive performance of the final model was assessed using a posterior predictive check approach comparing the observed concentrations with model predictions. A total of 1,000 simulated datasets were generated using the final model. For the different dosing regimens, the observed concentration‐time data were graphically overlaid with the median values and the 5th and 95th percentiles of the simulated concentration‐time profiles. The performance of the model was deemed adequate if the observed concentration data were appropriately distributed within the 5th and 95th percentiles of the simulated data.
Assessing clinical relevance of covariates
Parameter estimates from the bootstrap analysis were used to derive the distribution of geometric mean ratio (GMR) area under the curves (AUCs) for each covariate obtained from the covariate analysis, in addition to body weight, which was incorporated as part of the structural model. The steady‐state AUC was calculated over 6 weeks (AUCss‐6wk) to enable a consistent application of the same exposure parameter to data from different pembrolizumab dosing schedules (every 3 weeks (Q3W) and every 2 weeks (Q2W)) regimens. AUCss‐6wk was defined as:
where *actual dose (mg) = nominal dose (mg/kg) × body weight (kg).
The effect of the covariate was judged clinically unimportant if the GMR AUCs and their computed 95% CIs remained within the established clinical bounds interval of 0.5–5, which has been defined relative to the therapeutic dose of 2 mg/kg Q3W and is based on clinical data as well as exposure response and PK‐pharmacodynamic evaluations reported elsewhere (see Chattergee et al.,13 Elassaiss‐Schaap et al.,14 and Robert et al.15), based on the range of clinical exposure experience for which there is sufficient evidence to date to demonstrate similar target engagement and efficacy, and no appreciable increase in the occurrence of adverse events. For categorical covariates, the AUC values were calculated for each of the categories. For continuous covariates, the AUC values were calculated at the 10% and 90% percentiles of the covariate distributions.
Power simulations
Simulations with the full covariate model were performed to determine the power of the analysis to detect as statistically significant (P < 0.001) effects on clearance for both categorical and continuous covariates under various hypothetical true covariate effect magnitude and sample size assumptions. These simulations were conducted to better understand the robustness of the negative covariate analysis results with respect to informing appropriate dosing guidance for intrinsic and extrinsic factors. For the evaluation of categorical covariates, a hypothetical covariate effect on clearance was included in a subset of patients. Three separate covariate frequencies (5%, 10%, and 20% of the population) and four magnitudes (10%, 20%, 50%, and 100%) of the covariate effect were evaluated. One thousand simulations of patient populations were performed with the final population PK model for both sample size and hypothetical covariate effect size using the parameter estimates, interindividual variability, and residual variability to obtain concentration data. A single parameter was added to the final model to simulate the covariate effect in a randomly selected subset of patients.
For continuous covariates, the power of the model to detect a statistically significant (P < 0.001) covariate effect on clearance was evaluated for albumin, immunoglobulin G, bilirubin, eGFR, and AST. A hypothetical covariate effect on clearance, representing a true effect size (0%, 5%, 10%, 20%, 50%, and 100%) at the level of the 10th percentile of the population, was added to the final model as a power relationship. The distribution of the continuous covariate values was based on the presence of the tested covariate in the model development dataset. The false‐positive rate (0% effect) and the smallest effect size (5%) were only evaluated for covariates included in the final model because these results were not relevant for assessment of the robustness of the negative covariate findings.
The difference in the multivariate objective values for each pair of estimations (final model with and without the covariate effect) for each replicate simulated dataset was obtained and was considered statistically significant if it was ≥10.83 (α = 0.001, degree of freedom = 1). The fraction of simulations found to be statistically significant was computed for each scenario.
RESULTS
Analysis dataset
The final analysis dataset was composed of 1,223 patients from KEYNOTE‐001, 421 from KEYNOTE‐002, and 551 from KEYNOTE‐006. The number of patients and PK observations by dose in the pooled analysis datasets are provided in Table 1. The median (range) number of PK observations per patient was 5 (range, 1–24).
Table 1.
Number of patients and PK observations by dose and dosage regimen in the analysis dataset
| Dose/dosage regimen | No. of patients a | No. of PK observations |
|---|---|---|
| 1 mg/kg Q2W | 4 | 43 |
| 1 mg/kg Q3W | 6 | 10 |
| 2 mg/kg Q3W | 433 | 2,133 |
| 3 mg/kg Q2W | 3 | 55 |
| 10 mg/kg Q2W | 665 | 4015 |
| 10 mg/kg Q3W | 1,084 | 5,915 |
PK, pharmacokinetic; Q2W, every 2 weeks; Q3W, every 3 weeks.
aSome patients in dose‐escalation cohorts received more than one dose level.
Base model development
One‐ and two‐compartment models with and without allometric scaling on the basis of body weight were selected as a starting point for base model development. Two‐compartment models were found to better describe the multiexponential disposition profile of pembrolizumab, resulting in a decrease in OFV of >4,000 points with the addition of two parameters. Inclusion of body weight scaling on clearance and volume parameters largely corrected for apparent dependencies of the η values on body weight and resulted in a decrease in OFV of 575 points with the addition of 2 parameters (Supplementary Figure S1).
Testing of interindividual variability on PK parameters led to the estimation of a shared interindividual variability (η1) on clearance and intercompartmental clearance (Q) and a shared interindividual variability (η2) on central and peripheral volumes of distribution (Vc and Vp, respectively), as well as the covariance between these two. This reduced covariance structure effectively characterized apparent correlations between the parameters with limited impact on OFV. Subsequently, exploration of various residual variability models led to the selection of the additive residual error on the log‐scale model, as this optimally characterized the distribution of residuals across the entire concentration range. The base model was stable upon perturbation of initial parameter estimates and had a low condition number (4.6). Goodness‐of‐fit plots confirmed the adequacy of the base model to describe both the total population and the individual study populations without bias (Supplementary Figure S2). ETA distributions, as shown in Supplementary Figure S3, are close to a normal distribution, reflecting the adequacy of the exponential models for IIV. A plot of conditional weighted residuals vs. population‐predicted residuals and a plot of conditional weighted residuals vs. time after first dose are shown in Supplementary Figure S4. Even when there is some fluctuation of the smooth lines of the residuals around the horizontal line representing an average of zero (no bias), there is no indication of a systematic increasing trend under prediction by the model at later points in time.
Covariate analysis
Before performing the covariate search, the η‐shrinkage was assessed for individual PK parameter estimates. The η‐shrinkage for the final base model was 13.6% for clearance or Q and 28.6% for central or peripheral volume.
The clear effect of body weight already was incorporated as part of the structural model. For the covariates of sex, albumin, baseline tumor burden, cancer type, and baseline ECOG‐PS, potential minor effects on clearance are present. The more formal assessment of covariate effects was conducted through an stepwise covariate method, of which the results are summarized in Table 2. Statistically significant effects per the prespecified criteria were identified for sex, eGFR, albumin, tumor burden, ipilimumab prior therapy status, and ECOG‐PS on clearance and for sex, albumin, and ipilimumab prior therapy status on central volume of distribution.
Table 2.
Summary of covariates included in stepwise covariate analysis
| Covariate | Range or N (%) a | Median b | % Missing | Stepwise covariate model | |||
|---|---|---|---|---|---|---|---|
| Clearance | Central volume | ||||||
| Inclusion | P value c | Inclusion | P value | ||||
| Age, y | 15–94 | 62 | 0 | No | 0.246 | No | 0.95 |
| Sex | 0 | Yes | <0.0001 | Yes | <0.0001 | ||
| Male | 1,293 (59.1) | NE | |||||
| Female | 895 (40.9) | ||||||
| Cancer type | 0 | Yes | <0.0001 | No | 0.896 | ||
| Melanoma | 1,612 (73.7) | ||||||
| NSCLC | 554 (25.3) | NE | |||||
| Other cancer type | 22 (1.01) | ||||||
| ECOG‐PS | 0.2 | Yes | <0.0001 | No | 0.366 | ||
| 0 (asymptomatic) | 1,256 (57.4) | ||||||
| 1 (symptomatic) | 927 (42.4) | NE | |||||
| Tumor burden, mm | 10–895 | 86 | 11.2 | Yes | <0.0001 | No | 0.0557 |
| IPI status | 26.4 | Yes | <0.0001 | Yes | <0.0001 | ||
| IPI‐naive | 856 (39.1) | ||||||
| IPI‐treated | 755 (34.5) | NE | |||||
| eGFR, mL/min/1.73 m2 | 25.4–403.0 | 88.7 | 1.2 | Yes | <0.0001 | No | 0.173 |
| Bilirubin, μmL/L | 1–87.2 | 8.55 | 1.6 | No | 0.0028 | No | 0.113 |
| AST, IUL | 5–197 | 21 | 1.6 | No | 0.0405 | No | 0.176 |
| ALB, g/L | 15–59 | 40 | 1.8 | Yes | <0.0001 | Yes | 0.0004 |
| Coadministered GCs | 0 | No | 0.358 | No | 0.037 | ||
| Yes | 326 (14.9) | ||||||
| No | 1,862 (85.1) | NE | |||||
ALB, albumin; AST, aspartate aminotransferase; ECOG‐PS, Eastern Cooperative Oncology Group‐Performance Status; eGFR, estimated glomerular filtration rate; GCs, glucocorticoids; IPI, ipilimumab; NE, not evaluated; NSCLC, non‐small cell lung cancer.
aRange (min‐max) is presented for continuous covariates and number of subjects and percentage of total for each category for categorical covariates. bMedian are included only for continuous covariates. cLast P value from stepwise covariate method is reported. All P values are from forward inclusion expected for bilirubin, which is from backward elimination. Bilirubin is the only covariate that was removed during the backward elimination.
Additional refinements to the model were explored, including reassessment of the covariance structure for random effects, but did not produce a further improvement of model fit. Therefore, the final covariate model was considered the final model. The condition number (calculated as the ratio of the largest eigenvalue to the smallest eigenvalue) for the final covariate model was 40.9. Figure 1 illustrates the typical pembrolizumab concentration time profile during multiple dosing at different dose regimens. As is usual for a monoclonal antibody, the relatively long half‐life results in a gradual approach to steady state (time to steady state was ∼129 days) and a modest accumulation (∼2.2‐fold based on AUC) with Q3W dosing.
Figure 1.
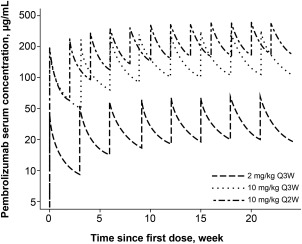
Predicted pembrolizumab concentration‐time profiles during multiple dosing with the regimens included in the clinical studies for melanoma and non‐small cell lung cancer. Q2W, every 2 weeks; Q3W, every 3 weeks.
Model qualification
The final model was fitted to 1,000 bootstrap replicate datasets to evaluate its stability and performance. Among the 1,000 runs, 23 (2.3%) had a failed covariance step due to a parameter estimate near the boundary. Parameter estimates of the final model, along with 95% CIs obtained from successfully converged bootstrapping runs, are shown in Table 3 and were found to be consistent with the parameter estimates of the final model.
Table 3.
Parameter estimates of the final model along with 95% CIs obtained from successfully converged bootstrapping runs
| Estimate | Mean bootstrap | 95% CI | %RSE | Shrinkage | |
|---|---|---|---|---|---|
| Parameter | |||||
| CL, L/day a | 0.22 | 0.22 | 0.211–0.229 | 2.14 | |
| Vc, L b | 3.48 | 3.48 | 3.42–3.53 | 0.891 | |
| Q, L/day | 0.795 | 0.793 | 0.73–0.858 | 4.01 | |
| Vp, L | 4.06 | 4.06 | 3.91–4.23 | 2.01 | |
| ɑ for CL and Q c | 0.595 | 0.595 | 0.506–0.686 | 7.95 | |
| ɑ for Vc or Vp c | 0.489 | 0.488 | 0.431–0.545 | 6.05 | |
| ALB on CL | −0.907 | −0.909 | −1.05 to −0.759 | 8.39 | |
| BSLD on CL | 0.0872 | 0.0870 | 0.0652–0.108 | 12.2 | |
| eGFR on CL | 0.135 | 0.135 | 0.0802–0.197 | 23.2 | |
| Sex on CL | −0.152 | −0.152 | −0.184 to −0.117 | 11.6 | |
| Cancer type on CL | 0.145 | 0.142 | 0.0922–0.193 | 17.0 | |
| Baseline ECOG‐PS on CL | −0.0739 | −0.0736 | −0.108 to −0.0374 | 22.7 | |
| IPI status d on CL | 0.140 | 0.140 | 0.0948–0.186 | 18.5 | |
| ALB on Vc | −0.208 | −0.209 | −0.304 to −0.112 | 22.7 | |
| Sex on Vc | −0.134 | −0.134 | −0.158 to −0.109 | 9.33 | |
| IPI status d on Vc | 0.0736 | 0.0736 | 0.0416–0.106 | 23.5 | |
| Random effect | |||||
| IIV on CL or Q | 0.134 (38% e ) | 0.133 | 0.12–0.147 | 5.28 | 13.6 |
| IIV on Vc or Vp | 0.0417 (21% e ) | 0.0413 | 0.0341–0.0488 | 9.14 | 28.6 |
| Residual error | 0.272 (27% d ) | 0.272 | 0.263–0.282 | 1.87 | 11.7 |
95% CI, 95% confidence interval of parameter estimate based on bootstrap results; ALB, albumin; BSLD, baseline tumor burden; CL, clearance; ECOG‐PS, Eastern Cooperative Oncology Group‐Performance Status; eGFR, estimated glomerular filtration rate; IIV, interindividual variability; IPI, ipilimumab; Q, intercompartmental flow; RSE, relative standard error; Vc, central volume; Vp, peripheral volume.
aCL = 0.202 (weight [WGT]/76.8)0.578 × (ALB/39.6)(−0.854) × (BSLD/89.6)0.0926 × (eGFR/88.47)0.139 × [(1 ‐ 0.152) if female] × [(1 + 0.145) if NSCLC] × [(1 + 0.0739) if baseline Eastern Cooperative Oncology Group numeric = 1] × [(1 + 0.140) if IPI = prior treatment]. bVc = 3.48 (WGT/76.8)0.492 × (ALB/39.6)(−0.178) × [(1 ‐ 0.134) if female] × [(1 + 0.0736) if IPI = prior treatment)]. cɑ = power value for weight‐based scaling. dIPI prior treatment status: naive or treated. ePercentage of coefficient of variation.
Visual predictive checks (VPCs) were stratified for trough and peak sample concentrations for the different dosage regimens (2 mg Q2W and Q3W, 10 mg Q2W and Q3W) and all other sampling concentrations (Figure 2). Overall, the plots indicate that the final model was able to predict the observed pembrolizumab concentrations reasonably well, both for the median and 90% CI of observations. However, some minor trends of increasing concentrations beyond 20 weeks were visible in the VPC plot at the last portion of the concentration time curves; this was clearer for some treatment groups than for others.
Figure 2.
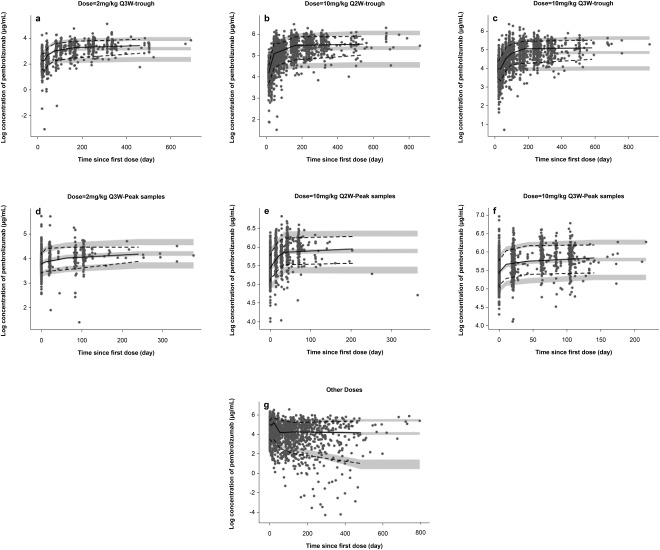
Visual predictive check for (a) 2 mg/kg every 3 weeks (Q3W) dosing group trough concentrations, (b) 10 mg/kg every 2 weeks (Q2W) dosing group trough concentrations, (c) 10 mg/kg every 2 weeks (Q2W) dosing group trough concentrations, (d) 2 mg/kg every 3 weeks (Q3W) dosing group postdose concentrations, (e) 10 mg/kg every 2 weeks (Q2W) dosing group postdose concentrations, (f) 10 mg/kg every 3 weeks (Q3W) dosing group postdose concentrations, and (e) all other samplings concentrations. Black circles represent observations; solid black lines represent median of observations; dashed lines span medians of the 90% confidence interval (CI) of observations; and the gray area represents 90% CI of the median.
Assessing clinical relevance of covariates
In order to establish the impact of the selected covariates on pembrolizumab exposure, AUC values were simulated for different values of the covariates included in the model. Intrinsic and extrinsic effects were not considered clinically relevant when the mean exposure (AUCss‐6wk) and its 90% CI varied within the (0.5–5.0) interval of the GMR relative to the typical exposure at 2 mg/kg Q3W. Any covariates causing more than a 0.5‐fold reduction or 5‐fold increase in GMR of the exposure were considered clinically relevant. The effects on AUC GMR for the covariates found to have a statistically significant effect on clearance are shown in Figure 3. When assessing the effect sizes relative to established exposure margin, it can be concluded that none of the covariates that were included in the model as a result of the stepwise covariate method have a clinically relevant impact on pembrolizumab exposure.
Figure 3.
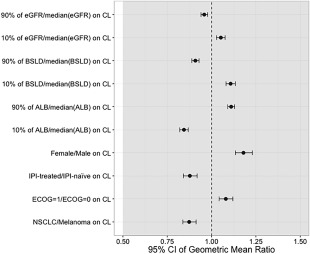
Magnitude of exposure effects on the area‐under‐the curve (AUC) geometric mean ratio (GMR) for statistically significant covariates on clearance (CL) in the final model. The dashed vertical line represents the GMR for a typical AUC value. Circles represent the GMR for the AUC given certain covariate values, with error bars denoting the 95% confidence interval (CI) of the GMR estimate. The gray region illustrates the clinical equivalence range that extends from a GMR of 0.5–5.0. All visual predictive checks (VPCs) were performed with six bins of equal width based on the independent variable.14, 15 ALB, albumin; BSLD, baseline tumor burden; CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; IPI, ipilimumab; NSCLC, non‐small cell lung cancer.
Power analysis
The power to identify a statistically significant (P < 0.001) effect of a continuous and categorical covariate on the clearance of pembrolizumab is shown in Supplementary Table S1. The power was >99.9% to identify a true continuous covariate effect of ≥10% at the 10th percentile of the population. For a clinically relevant effect size of 50% or greater, all simulations identified the effect as statistically significant, indicating a power of >99.9% to rule out a clinical meaningful effect through a negative covariate finding.
The false‐positive rate for the continuous covariates, albumin, bilirubin, baseline tumor burden, and eGFR, was found to be minimal, at 0.1% (P < 0.001). For categorical covariates, >80% of the simulations were identified as statistically significant, with an assumed effect size of ≥20% increase in clearance in ≥20% of patients. The power was >99.9% to identify a true categorical covariate effect of ≥50% present in ≥10% of the population, and the power was 99% to identify a true categorical effect of 50% in 5% of the population. These results support a very high power to rule out a clinically meaningful effect through a negative continuous covariate finding.
DISCUSSION
Pembrolizumab concentration‐time data obtained from doses of 1–10 mg/kg administered Q2W or Q3W were described adequately by a two‐compartment model with linear clearance. Nonlinearity in pembrolizumab PK has been observed at doses well below 1 mg/kg (see Elassaiss‐Schaap et al.14). The present analysis, which was restricted to data above 1 mg/kg, was consistent with those results and did not show an indication for nonlinearity in the clinical dose range. Goodness‐of‐fit plots showed no clear systematic trends or biases, confirming the adequacy of the model to describe the observed concentration‐time profiles of pembrolizumab. Nevertheless, trends of increasing concentrations beyond 20 weeks were visible in the VPC plot at the last portion of the concentration time curves; this was clearer for some treatment groups than for others. The number of patients decreased toward later sections of the profiles as a result of varying enrollment dates and as a result of withdrawal, which may have contributed to the apparent modest misspecification. No trends as a function of dose were visible in the VPC or in other diagnostic plots (Figure 2). Overall, the model adequately captures dose linearity and time dependency in the PK profile of pembrolizumab.
Typical for a monoclonal antibody, pembrolizumab has a low clearance (0.22 L/day), and a small volume of distribution consistent with limited distribution beyond extracellular space. Given the site of action of the drug on circulating T cells, this reflects sufficient availability of the drug to bind its target. Exchange between compartments was characterized by Q. The elimination half‐life of pembrolizumab derived from these primary PK parameters was 27.3 days. Overall, these characteristics are consistent with PK values for other therapeutic monoclonal antibodies,16 and result in a concentration‐time profile that is amenable to maintaining clinically meaningful concentrations out to 3 weeks postdose (Figure 1).
Body weight is known to influence the clearance and volume of distribution of therapeutic monoclonal antibodies.16 Therefore, body weight was included in the pembrolizumab PK model as a covariate during base model development. Both clearance and Q were found to depend on body weight according to an allometric relationship, with an exponent of 0.595, whereas the exponent of both central and peripheral volumes was found to be 0.489. These intermediate exponent values between values of zero (no relationship with body weight) and one (linear relationship with body weight) are within the range of reported exponent values for monoclonal antibodies, in which both fixed dosing and body weight‐based dosing approaches have been shown to perform similarly.17
Statistically significant effects of sex were found on clearance and central volume. Clearance was found to be lower (17%; P < 0.0001), translating into a 20% increase in AUC in female subjects (N = 900), which is small in relation to the exposure margins of (0.5–5.0) GMR and, therefore, of no clinical relevance (see Figure 3). The central volume of distribution was lower (15%; P < 0.0001) in female compared with male subjects (N = 1295) similar to findings for other monoclonal antibodies (Supplementary Figure S5).15
Therapeutic antibodies, such as pembrolizumab, are primarily eliminated by protein catabolism in a multitude of tissues, and, thus, clearance is not dependent on a single organ.18 Consequently, intrinsic factors, such as organ dysfunction or age, typically have limited impact on the exposure of therapeutic antibodies,16 and were, therefore, not anticipated to affect the exposure of pembrolizumab to a clinically meaningful extent (most effect sizes were <20%). Albumin is also being cleared by similar mechanisms as antibodies, such that the covariate effect of albumin likely reflects the underlying general catabolic rate. The overall covariate analysis results, indicating that intrinsic factors do not significantly affect exposure to pembrolizumab, confirmed this expectation.
Because therapeutic antibodies are too large to pass through the glomerular membrane of the kidney, renal insufficiency was not expected to significantly impact pembrolizumab exposure. Although the eGFR was found to have a statistically significant effect on clearance based on formal testing of the covariate relationship, the effect size was small and well within clinical bounds (0.5–5.0), indicating that renal impairment has no clinically relevant effect.
AST and total bilirubin as measures of hepatic function on pembrolizumab exposure had no clinically relevant impact on clearance. Consequently, no substantial changes in pembrolizumab exposure are anticipated in the setting of hepatic impairment.
A number of disease‐related factors were found to have statistically significant effects on pembrolizumab pharmacokinetics. Relative to ECOG‐PS 1, ECOG‐PS 0 was associated with a 7.3% increase in clearance. Similarly, cancer type (14.5% increase for patients with NSCLC) and ipilimumab status (13.9% increase in clearance for patients pretreated with ipilimumab) had a statistically significant effect on clearance. Last, a significant correlation between baseline tumor burden and clearance was observed; at the 90th percentile of baseline tumor burden distribution, clearance was increased by 8.79% (95% CI, 6.5–11%) relative to a typical subject, translating to a 9.17% reduction in AUC. Theoretically, this finding could be related to an increased extent of target PD‐1‐mediated clearance in patients with a high tumor burden. However, the total level of pembrolizumab outside the blood is low based on the limited volume of distribution, and tumor volume represents only a fraction of total body volume. Therefore, tumors have a limited potential to contribute to total body clearance of pembrolizumab. Because the resulting effect on pembrolizumab exposure for each of these factors was well within clinical bounds (AUC GMR: 0.5–5.0), none of them is considered to be clinically relevant.
Systemic corticosteroids (glucocorticoids) are anticipated to be one of the more frequent combination treatments administered with pembrolizumab, either directly or indirectly as an antiemetic during chemotherapy or as a treatment for immune‐related adverse events. Therefore, prolonged (>1 week) corticosteroid administration was assessed as a potential covariate in the population PK analysis. No relationship was observed between prolonged use of systemic corticosteroids and pembrolizumab exposure, and no significant impact of corticosteroid use on pembrolizumab exposure was identified (P = 0.4).
The power of the model to detect statistically significant covariates was shown to be sufficient to reliably detect even small (10–20%) effects in a subset of the population (10th percentile) and to have >99.9% power to detect covariates with clinically relevant effect sizes. This result confirms that the negative covariate results (i.e., age, AST, coadministered drugs, and race on clearance; cancer type, age, AST, ECOG‐PS, bilirubin, tumor burden, coadministered drugs, eGFR, and race on central volume) indeed indicate a lack of clinically important effects on the PK properties of pembrolizumab. Overall, covariates have relatively limited impact on individual exposure to pembrolizumab. Because of covariate inclusion, the between‐subject variability on clearance decreased from 49% to 37%, indicating that ∼32% of variation in clearance could be explained by the identified covariate relations.
Taken together, this analysis demonstrates that the pembrolizumab population PK model can be considered sufficiently robust to indicate the absence of covariate effects of clinical relevance, thereby supporting the approved pembrolizumab dosage of 2 mg/kg Q3W in a variety of patient subpopulations. This work illustrates the critical role that population PK analyses can play in characterizing the PKs of new anticancer treatments.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors wish to thank the patients and their families and caregivers for participating in this study. We thank Marielle van Zutphen and Krishan Sahu of Merck & Co., Inc. (Kenilworth, NJ) for technical support and Roger Dansey of Merck & Co., Inc. (Kenilworth, NJ) for critical review of the manuscript. Medical writing and editorial assistance was provided by Sarah Adai, MS, and Melanie Leiby, PhD (ApotheCom, Yardley, PA), and was funded by Merck & Co., Inc. (Kenilworth, NJ).
Author Contributions
M.A., J.A.S., R.d.G., and J.E.‐S. wrote the manuscript; M.A., R.d.G., J.E.‐S., D.P.d.A., A.K, and J.A.S. designed the research. M.A., J.E.‐S., and T.F. performed the research. M.A., M.P., C.L., and J.E.‐S. analyzed the data.
Conflict of Interest
All authors are employees of the stated companies. M.A. has nothing additional to disclose; T.F. has nothing additional to disclose. M.P. reports personal fees from Merck & Co., Inc., and qPharmetra outside the submitted work. C.L. has nothing additional to disclose. D.d.A. has nothing additional to disclose. R.d.G. reports personal fees from Merck & Co. Inc. during the conduct of the study. J.E.‐S. reports personal fees from Merck & Co., Inc. during the conduct of the study and, after leaving, started an independent consultancy company as indicated in the affiliations. A.G.K. has nothing additional to disclose. J.S. reports financial activities outside the submitted work.
References
- 1. Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Francisco, L.M. , Sage, P.T. & Sharpe, A.H. The PD‐1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hodi, F.S. et al Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robert, C. et al Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Curran, M.A. , Montalvo, W. , Yagita, H. & Allison, J.P. PD‐1 and CTLA‐4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 107, 4275–4280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Selby, M. et al Antitumor activity of concurrent blockade of immune checkpoint molecules CTLA‐4 and PD‐1 in preclinical models. J. Clin. Oncol. 31(suppl), abstract 3061 (2013). [Google Scholar]
- 7. Jonsson, E.N. & Karlsson, M.O. Automated covariate model building within NONMEM. Pharm. Res. 15, 1463–1468 (1998). [DOI] [PubMed] [Google Scholar]
- 8. Beal, S.L. & Sheiner, L.B. NONMEM Users Guide: Part V (University of California – San Francisco, San Francisco, CA, 1992).
- 9. Jonsson, E.N. & Karlsson, M.O. Xpose—an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1998). [DOI] [PubMed] [Google Scholar]
- 10. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit—a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 11. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)—a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 12.European Environment Agency. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. <http://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006>.
- 13. Chattergee, M.S. et al Population pharmacokinetic/pharmacodynamic modeling of tumor size dynamics in pembrolizumab‐treated advanced melanoma. CPT: Pharmacometrics Syst. Pharmacol; e‐pub ahead of print 2016. [DOI] [PMC free article] [PubMed]
- 14. Elassaiss‐Schaap, J. et al The pharmacokinetic‐pharmacodynamic relationship of pembrolizumab revealed using model‐based “learn and confirm” within the phase Ib KEYNOTE‐001 trial. CPT: Pharmacometrics Syst. Pharmacol (2016). [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 15. Robert, C. et al Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: a randomised dose‐comparison cohort of a phase 1 trial. Lancet 384, 1109–1117 (2014). [DOI] [PubMed] [Google Scholar]
- 16. Dirks, N.L. & Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 633–659 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Wang, D.D. , Zhang, S. , Zhao, H. , Men, A.Y. & Parivar, K. Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J. Clin. Pharmacol. 49, 1012–1024 (2009). [DOI] [PubMed] [Google Scholar]
- 18. Mahmood, I. Pharmacokinetic allometric scaling of antibodies: application to the first‐in‐human dose estimation. J. Pharm. Sci. 98, 3850–3861 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information