

Abstract
Evaluation of pharmacokinetic/pharmacodynamic (PK/PD) properties played an important role in the early clinical development of pembrolizumab. Because analysis of data from a traditional 3 + 3 dose‐escalation design revealed several critical uncertainties, a model‐based approach was implemented to better understand these properties. Based on anticipated scenarios for potency and PK nonlinearity, a follow‐up study was designed and thoroughly evaluated. Execution of 14,000 virtual trials led to the selection and implementation of a robust design that extended the low‐dose range by 200‐fold. Modeling of the resulting data demonstrated that pembrolizumab PKs are nonlinear at <0.3 mg/kg every 3 weeks, but linear in the clinical dose range. Saturation of ex vivo target engagement in blood began at ≥1 mg/kg every 3 weeks, and a steady‐state dose of 2 mg/kg every 3 weeks was needed to reach 95% target engagement, supporting examination of 2 mg/kg every 3 weeks in ongoing trials in melanoma and other advanced cancers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Although the development of pembrolizumab has been supported by a robust program of PK/PD assessments, the data were limited. The most appropriate dose for study of clinical efficacy in large patient cohorts had not yet been determined.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ How additional cohorts could be designed to improve precision and robustness in the determination of PK/PD properties, and to help inform dose selection.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The likelihood of achieving target engagement saturation is considerably lower at pembrolizumab doses below 1 mg/kg compared with 2 mg/kg every 3 weeks. A “learn and confirm” cycle using modeling and simulation successfully supported the determination of the dose that should be tested for clinical efficacy: 2 mg/kg every 3 weeks.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study demonstrates how existing methods can be practically combined and applied to transform early PK/PD results into a robust design and assessment of a drug's clinical pharmacology properties.
Some tumors are able to escape immune detection by altering their microenvironment during development.1, 2 One immune checkpoint pathway used by tumors to suppress antitumor activity is the programmed death 1 (PD‐1) receptor pathway.2 This receptor is expressed on the surface of activated T cells3, 4 and is involved in immune tolerance and the prevention of chronic inflammation–associated tissue damage.2 Dampening of T‐cell–receptor signaling as a result of the interaction between PD‐1 and its ligands PD‐L1 and PD‐L2, results in downregulation of T‐cell activation and proliferation, and thus suppression of the T‐cell–mediated antitumor immune response.5 Understanding of this process has led to targeting immune checkpoints with a view to stimulating the anticancer immune response. The application of immune checkpoint inhibitors in advanced cancer has yielded durable responses and survival benefits.6, 7, 8, 9
Pembrolizumab (MK‐3475) is a highly selective, humanized monoclonal immunoglobulin G4‐kappa isotype antibody designed to block the interaction between PD‐1 and its ligands.10 Pembrolizumab has demonstrated robust activity in a functional ex vivo T‐cell modulation assay using human donor blood cells (data on file, Merck). Blockade of PD‐1 with pembrolizumab showed marked clinical activity in metastatic melanoma9, 11, 12, 13 as well as other tumor types, including non‐small cell lung cancer.14
The development of pembrolizumab has been supported by a robust program of pharmacokinetic/pharmacodynamic (PK/PD) assessments. The large phase I KEYNOTE‐001 study commenced with a first‐in‐human, standard 3 + 3 dose‐escalation cohort to explore the maximum tolerated dose of pembrolizumab.15 However, PK/PD data were limited, leaving uncertainties regarding the linearity of pembrolizumab's PK profile and its PDs. To enable selection of the lowest dose for study in larger patient populations, modeling and simulation were applied to guide the design of an additional cohort (A2) of KEYNOTE‐001. The resulting “learn and confirm” cycle in model‐based analysis (in line with the principles set out by Sheiner16) is described herein. The initial model development, subsequent simulation‐aided design of within‐patient dose escalation, model updating using the data obtained from the simulation‐designed studies, and simulation‐supported decision‐making are discussed.
METHODS
Study population and design
KEYNOTE‐001 is a large, international, multicohort, phase Ia study (ClinicalTrials.gov identifier NCT01295827). The study design and eligibility criteria for the initial dose‐escalation portion of the study have been described previously.15 Briefly, patients aged ≥18 years with advanced solid tumors who did not require systemic corticosteroid treatment and had not received prior treatment with a PD‐1, programmed death ligand‐1, or cytotoxic T‐lymphocyte‐associated protein 4 inhibitor were enrolled at two sites in the United States. The study protocol and all amendments were approved by the appropriate ethics bodies. Written informed consent to participate was provided by all patients before study start.
Dose escalation (part A) was conducted using a traditional 3 + 3 design, with 3‐patient cohorts sequentially given pembrolizumab 1, 3, or 10 mg/kg administered intravenously over 30 minutes on day 1, day 28, and every 2 weeks thereafter. Seven additional patients were enrolled in a first expansion cohort (part A1) and treated with 10 mg/kg every 2 weeks. Part A2, an additional cohort of KEYNOTE‐001, was implemented to better estimate the PK/PD properties of pembrolizumab. A model‐based approach was utilized in the design of this study part, as described below.
Assessments
Venous blood samples for PK analysis were collected predose, postdose (<30 minutes after infusion), and 6 (± 2), 24 (± 2), and 48 (± 2) hours after the start of the first infusion; on days 8, 15, 22, and 29 of treatment cycle 1; predose and postdose in cycle 2, and every other cycle thereafter for the first 12 months; and 30 days after the last pembrolizumab dose. Serum pembrolizumab concentrations were quantified using a validated electrochemiluminescent assay (lower limit of quantitation, 10 ng/mL).
Target engagement pharmacodynamics (PD) were assayed using the interleukin‐2 (IL‐2) stimulation ratio in venous blood samples drawn before dosing, 24 (± 2) hours and 7 days after the start of the first infusion, predose at treatment cycles 2 and 3, and predose every fourth cycle thereafter. The IL‐2 stimulation ratio assay resembles the inhibitory nature of PD‐1–binding interaction and was run as follows: after a 1:10 dilution, 2 aliquots were removed from each sample and incubated with staphylococcal enterotoxin B either with or without the addition of 25 µg/mL pembrolizumab for 4 days at 37°C (a description of this standard immune‐assay can be found in patent WO2012018538 A2).17 The IL‐2 concentration was measured in both aliquots (lower limit of quantitation, 4 pg/mL). The stimulation ratio was calculated by dividing the IL‐2 concentration in the pembrolizumab‐supplemented aliquot by that in the aliquot treated with staphylococcal enterotoxin B alone. This reflects the staphylococcal enterotoxin B‐stimulated response in an aliquot of whole blood to which an excess of pembrolizumab has been added to ensure maximal response, relative to an aliquot of the same whole blood sample containing only the pembrolizumab circulating at the time of sampling. For example, with samples containing a very low pembrolizumab concentration, stimulation ratios centered around a ratio value of ∼2 would reflect the ability of the added pembrolizumab to elicit a doubling of response. By comparison, data from samples with high concentrations of pembrolizumab are centered around a ratio value of ∼1, indicating that circulating pembrolizumab is already achieving the maximal functional blockade.
Model development
Pharmacokinetic and PK/PD models were developed in NONMEM, version 7.2.0 (ICON Development Solutions, Ellicott City, MD)18 with the Intel FORTRAN Compiler, version 11.1 (Intel, Santa Clara, CA). The estimation method was first order conditional estimation with interaction. Xpose, version 4.4.0 (xpose.sourceforge.net),19 PsN 3.5.3 (psn.sourceforge.net),20, 21 and R version 2.14.1 (R‐project, www.r-project.org)22 were used for postprocessing of the NONMEM output (e.g., to generate goodness‐of‐fit plots). The robustness of the estimates was evaluated using the bootstrap implemented in PsN with 1,000 replicates. Between‐subject variability (BSV) components were modeled as having a log‐normal distribution; fixed effects were either normally or log‐normally specified (discussed below). The coefficient of variation of the log‐normal BSV components was calculated as:
where Ω is the NONMEM estimate of BSV variance.
Model‐based design optimization
Part A2 was included in the study to understand any nonlinearity in PKs as well as the PK/PD relationship for target engagement, as reflected by the IL‐2 stimulation ratio. Adherence to the following criteria were required regarding the design of this study extension: (1) the design should be robust under an array of assumptions on pembrolizumab properties that were not determined by previous analyses; (2) patient exposure to concentrations unlikely to be effective should be minimized; (3) the number of patients should be minimized; and (4) the design should not be sensitive to dropout within a severely ill patient population.
Design optimization was conducted in two phases. The first phase was the assessment of PK properties and is described in Supplementary Table S1. The second phase of design evaluation focused on how well a potential design would refine the quantitative PK/PD relationship derived from part A1 data. Simulation scenarios were selected (based on quantitative uncertainties as well as biological plausibility) to assess robustness of different potential designs for part A2. These scenarios covered a range of different potency (the concentration of pembrolizumab required to cause 50% inhibition of the IL‐2 stimulation ratio [IC50]) values, steepness values (Hill coefficient), values for within‐subject variability (WSV) and BSV, and the type of PKs (linear or nonlinear). Scenarios were compared using the median and largest (“extreme”) root mean‐squared error (RMSE% [http://psn.sourceforge.net/pdfdocs/sse_userguide.pdf]) of each parameter, relative to input values (“truth” as defined by the simulation). Stochastic simulation and estimation (SSE) was performed 100 times for each scenario to calculate these criteria.
RESULTS
Parts A and A1
Detailed outcomes of cohorts A and A1 are presented elsewhere.15 The pembrolizumab half‐life was in the range of 14–22 days, but could not be determined accurately because of the short sampling window after the first dose (28 days). The area under the concentration vs. time curve at the 1‐mg/kg dose seemed to be disproportionally lower than that obtained at either 3 or 10 mg/kg. This lack of dose proportionality was also evident in a concentration vs. time plot on a semilogarithmic scale (Figure 1 a). However, the number of patients contributing to this finding was very low (n = 3 per dose) and the half‐lives at 1 and 10 mg/kg were similar. This suggested that normal variability could potentially explain these data.
Figure 1.
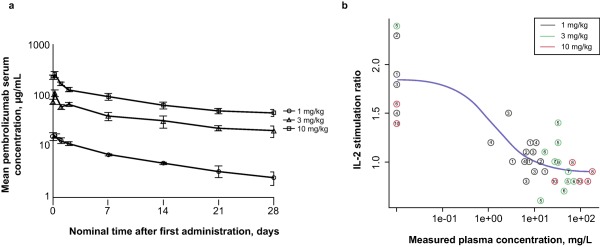
Initial pharmacokinetic (PK) and pharmacodynamic (PD) results from part A1 (reprinted with permission from Patnaik A. et al.15). (a) Arithmetic mean ± SE of the concentration‐time profiles of pembrolizumab following intravenous administration at 1, 3, or 10 mg/kg to patients with solid tumors in cycle 1 of parts A and A1 (linear‐log scale). (b) The interleukin 2 (IL‐2) stimulation ratio as a function of plasma concentration of pembrolizumab. The numbers in the circles are subject numbers.
The IL‐2 stimulation ratios determined in samples from parts A and A1 indicated saturation of blood target engagement at all three dose levels for at least two consecutive time points (time‐dependence data not shown). At later time points, some concentration dependency was observed (Figure 1 b) because of the lower concentrations just before the second dose, which was administered 4 weeks after the first. Figure 1 b also demonstrates the limited data available for characterization of pembrolizumab PD at or below the concentration of pembrolizumab required to cause 50% inhibition of the IL‐2 stimulation ratio (IC50).
Pharmacokinetic and PK/PD models were developed using data from parts A and A1 (Figure 2 a; see also Supplementary Methods and Results). This revealed a direct relationship between pembrolizumab serum concentration and the IL‐2 stimulation ratio (as this is ex vivo stimulation). The pembrolizumab IC50 for the IL‐2 stimulation ratio was estimated at 1.3 mg/L, with an SE of 24%. Because most of the observed concentration‐effect data were at the plateau of the response, this potency estimate should be interpreted with care. Furthermore, a high level of uncertainty was found for several other parameters (body weight–clearance relationship and the BSVs on clearance and relative bioavailability), indicating that the model was potentially overparameterized (but within reasonable limits for the intended simulations); the model structure was retained to ensure more realistic variability in the subsequent simulation step.
Figure 2.
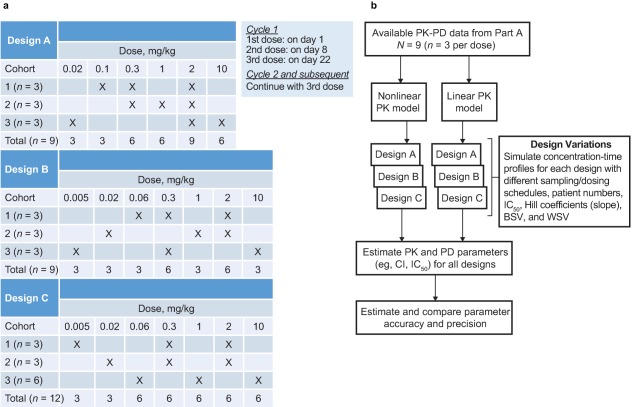
Overview of trial design variations and method for evaluating them. (a) Overview of the three study designs. Each design started with a low dose (0.005 or 0.02 mg/kg) and escalated to either 2 or 10 mg/kg every 3 weeks. Two designs (A and B) differed with respect to the starting dose (0.02 or 0.005 mg/kg, respectively) and the subsequent dose (0.1 mg/kg or 0.06 mg/kg, respectively). In the third design (C), 3 additional patients were included in one group in order to achieve a balanced number per dose level, or 12 patients in total (4 per group), whereas the other designs included 9 patients. For design A, an additional 6 patients per group, and for design B, an additional 4, 6, and 8 patients per group were evaluated to establish the influence of sample size. Thus, the total number of design variations evaluated was seven. (b) Flow chart of design evaluation. With 20 scenarios per design variation, a total number of 140 sets were simulated and reestimated, 100 times each (in total, 14,000 trial simulations). BSV, between‐subject variability; CI, confidence interval; IC50, the concentration of pembrolizumab required to cause 50% inhibition of the IL‐2 stimulation ratio; PK‐PD, pharmacokinetic‐pharmacodynamic, WSV, within‐subject variability.
Part A2
Stochastic simulations and estimations were used to optimize the design of the subsequent part A2. A first phase of design optimization focused on ensuring robust characterization of the PKs (see Supplementary Methods and Results). In this first phase, a design with three patients per group, full sampling, and two dose steps per patient seemed to be robust across scenarios in terms of determining dose proportionality at or below 1 mg/kg. These results were then used as the starting point for designs tested for their ability to estimate PD properties. Furthermore, the initially observed IL‐2 assay potency of pembrolizumab supported evaluation of a lower dose level, 2 mg/kg every 3 weeks, as it suggested that IL‐2 assay saturation would be maintained at steady state (simulations not shown). These findings led to evaluation of seven design variations (Figure 2 a).
The low dynamic range and high variability in the IL‐2 stimulation assay could have resulted in inefficient designs due to potential biases in estimated PD properties. Therefore, simulation scenarios were applied to account for possible alternative properties and (to be conservative) to cover even unlikely cases. Four different potency (IC50) values (0.02, 1, 20, and 100 mg/L), two slope values (Hill coefficient = 1 and 5), and two types of PK (linear and nonlinear) were considered—16 in total. WSV and BSV were also unknown; WSV values of 24% (measured during assay validation) and 45% and BSV values of 44% (measured during assay validation) and 70% were selected. The highest values of WSV and BSV were selected to represent high but credible numbers. These four additional scenarios were implemented at IC50 = 1, Hill coefficient = 1, and linear PK. This gave 20 scenarios (see Supplementary Table S3), with a resulting 14,000 trial simulations (20 scenarios, 7 design variations, n = 100 each). An overview of the evaluations is shown in Figure 2 b.
In order to determine the optimal design for PD property estimation in terms of precision and robustness, simulation and subsequent reestimation results were evaluated for effects on median and extreme RMSE values. Five of the 14,000 runs did not yield an applicable result, and therefore were ignored during downstream analysis. The median RMSEs of the seven design variations were quite similar, but the means of extreme RMSEs (Table 1) were more dependent on design. The extreme RMSEs tended to decrease with larger sample size, but all were within a manageable range (25–43%). Similar tabulation of the mean IC50 RMSE across designs revealed differences of additional relevance, decreasing from 17.4% for scenarios with three subjects to 3.5% for scenarios with eight (Table 1). Design B performed better than design A for this metric. Even at three, design B performed better with an 8.4% mean across sizes than design C with a 13.4% mean across sizes, even though design C contained design B plus three additional subjects. The apparent difference between design B with three or four and design C was therefore attributed to random variation. The highest median RMSE among all parameters was found for the Hill coefficient with an average of 65.7%, but only under the most extreme (and unlikely) scenario for potency (IC50 = 100 mg/mL).
Table 1.
Extreme RMSE and mean RMSE of IC50 per design
| No. of patients per group | Mean across sizes | ||||||
|---|---|---|---|---|---|---|---|
| 3 | 4a | 6 | 8a | ||||
| Extreme RMSE% | |||||||
| Design A | 42.8 | — | 27.5 | — | 35.2 | ||
| Design B | 36.4 | 31.0 | 33.1 | 25.7 | 31.6 | ||
| Design C | — | 38.3 | — | — | 38.3 | ||
| Mean across designs | 39.6 | 34.7 | 30.3 | 25.7 | 33.5 | ||
| Mean RMSE% | |||||||
| Design A | 26.3 | — | 9.5 | — | 17.9 | ||
| Design B | 8.4 | 6.3 | 10.6 | 3.5 | 8.4 | ||
| Design C | — | 13.4 | — | — | 13.4 | ||
| Mean across designs | 17.4 | 9.8 | 10.0 | 3.5 | 11.1 | ||
Only simulations with a within‐subject variability of 24 and a between‐subject variability of 44% were tabulated. IC50, concentration of pembrolizumab required to cause 50% inhibition in vitro; RMSE, root mean squared error.
Four or 8 is the average number of patients per cohort in design C (2 cohorts of n = 3 and 1 of n = 6 in the base design, or twice those numbers for the n = 8 scenario).
Further evaluation of the median RMSE using various designs and simulated IC50 values confirmed that although the designs performed similarly, design A, in which the 0.005‐mg/kg dose was not used, was the least able to quantify low IC50 values (see Supplementary Figure S1). Therefore, it was concluded that overall, the designs with a lower dose (B and C) were preferred over that without (A), and that the benefit of a sample size of six or eight per group did not outweigh the downside in terms of resource requirements and recruitment time. Design C was selected over design B because of its appealing symmetry.
Part A2 was run using design C, and 12 patients provided samples up to and including full escalation for further bioanalysis. A type of nonlinear PK behavior different from that observed in part A1 was readily detected by exploratory analysis.15 A new PK model was therefore developed that accounted for concentration‐dependent clearance at lower dose levels. Furthermore, the BSV structures of the PK and PK/PD models (Supplementary Methods and Results) were optimized with, among others, interoccasion variability in bioavailability. Adding BSV to the maximum elimination rate was necessary for successful estimation, but did not visually contribute to the model fit as determined by graphical analyses. The IC50 was higher than the concentration at which the nonlinear clearance component was half‐maximal, enabling independent estimation of these properties. The models for PK/PD were therefore estimated separately for increased estimation stability. The final PK model parameters confirmed a low clearance of about 0.2 L/day and a limited volume of distribution of approximately 6 L (Table 2). The PK model fit the data adequately, including the nonlinearity (Figure 3 and Supplementary Figure S2). The bootstrap method showed that parameter estimation was robust, as parameters were consistent with NONMEM estimates, even with few subjects and a highly structured dataset. The nonlinear components in the PK model were challenging, and 37% of bootstrap runs did not converge successfully, compared with 5% for the PD model.
Table 2.
Final PK and PD parameters
| Parameter | Estimate | RSE, %a | BSV, CV%b | IOV, % |
|---|---|---|---|---|
| CLlin (L/d) | 0.168 | 11.1 | – | – |
| Vc (L) | 2.88 | 5.90 | – | – |
| Q (L/d) | 0.384 | 31.3 | – | – |
| Vp (L) | 2.85 | 16.5 | – | – |
| Vmax (mg/d) | 0.114 | 31.5 | 22.7 | – |
| Km (μg/mL) | 0.0784 | 49.1 | – | – |
| F | 1c | – | – | 37.7 |
| RUV PK (%) | 29.6 | 25.4 | – | |
| Base | 2.09d | 5.6 | 12.0 | – |
| Imax | 0.961d | 7.1 | – | |
| IC50 (μg/mL) | 0.535d | 75.0 | – | |
| RUV PD | 0.209 | 20.1 | – |
Base, baseline; BSV, between‐subject variability; CLlin, linear clearance; CV, coefficient of variation; F, bioavailability; IC50, concentration of pembrolizumab required to cause 50% inhibition in vitro; Imax, maximal inhibitory activity; IOV, interoccasion variability; Km, concentration at half‐maximal activity of the nonlinear clearance component; PD, pharmacodynamic; PK, pharmacokinetic; Q, intercompartmental clearance; RSE, relative standard error; RUV, residual unidentified variability; Vc, central volume of distribution; Vmax, maximal activity of the nonlinear clearance component; Vp, peripheral volume of distribution.
%RSE calculated as SE*100 for log‐transformed parameters and SE*100/estimate for parameters that were estimated on the normal domain.
CV% calculated as:
Fixed value.
Exponent of the estimated parameter.
Figure 3.
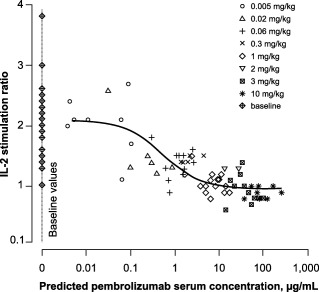
Pharmacodynamic (PD) observations (symbols by dose) and population‐predicted (solid line) programmed death 1 (PD‐1) receptor modulation as a function of pembrolizumab exposure under the extended dose range. Model‐predicted serum concentrations were used to allow inclusion of all PD observations through interpolation and extrapolation of exposure where no observed values were available. IL‐2, interleukin 2. Reprinted with permission from Patnaik A, et al.15
Simulations using fixed‐effects parameters were conducted to explore the nonlinear aspects of PK models. The contributions of linear and nonlinear components were concentration‐dependent, with the nonlinear component dominating at lower concentrations (Figure 4). Total clearance was higher in this region, consistent with the increased curvature in the PK profile at low doses. The nonlinear and linear clearances were in balance at a concentration of 0.68 mg/L, above which the linear clearance became the determinant of pembrolizumab PK (Supplementary Figure S3). Steady‐state simulations were performed, demonstrating that linear clearance dominated at doses above about 0.3 mg/kg every 3 weeks.
Figure 4.
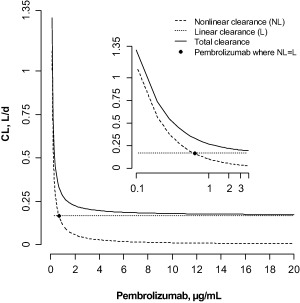
Dependence of total, linear, and nonlinear clearance on pembrolizumab concentrations. Inset: log‐linear plot. CL, clearance.
After including the new part A2 data, the PK/PD relationship between pembrolizumab and the IL‐2 stimulation ratio remained consistent with the initial data, and only limited model development was required to optimize the fixed‐effects representation (now using log‐transformation for parameters) and to estimate BSV (Table 1). Resulting model fits were adequate to describe the data (Figure 3 and Supplementary Figure S4), and bootstrap estimates were consistent with the model (Supplementary Figure S5). The final pembrolizumab IC50 estimate was 0.54 mg/L, with a 95% confidence interval (CI) of 0.12–2.3 mg/L. This is lower than the estimate based only on part A1 data, but still within the limits of the CI of the initial estimate. The new dataset allowed a better characterization of the IC50 because of the denser data sampling at lower concentrations (see Figures 3 and 4d in Patnaik et al.15). Simulations were run using the final PK/PD model to obtain IL‐2 stimulation ratio–derived target engagement properties in blood with parameter credibility intervals (Figure 5). Target engagement increased monotonically, with the 95% level reached at trough concentrations after dosing at 0.8 mg/kg every 3 weeks to steady state. A steady‐state dose of 2 mg/kg every 3 weeks was needed to reach 90% probability of 95% target engagement.
Figure 5.
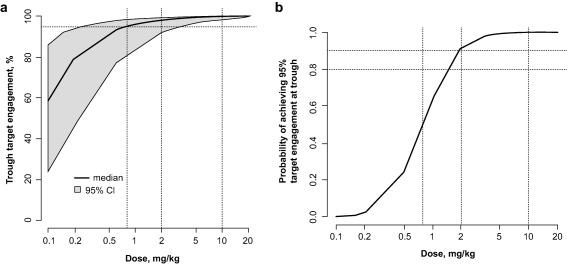
(a) Target engagement as a function of concentration at steady state. Percentage of target engagement with a band denoting the 95% confidence interval (95% CI) for an every‐3‐weeks dosing regimen, based on simulations taking into account the uncertainty in the pharmacodynamic parameter estimates. (b) Probability (percentage of subjects within a simulated population) of achieving 95% target engagement at trough for different doses given every 3 weeks.
DISCUSSION
Assessments of the dose‐escalation cohorts of KEYNOTE‐001 revealed that pembrolizumab has linear PK in the dose range 0.3–10 mg/kg given every 2 weeks or every 3 weeks, with a clearance of approximately 0.2 L/day and a volume of distribution of approximately 6 L. Nonlinear clearance becomes progressively more important at doses below 0.3 mg/kg every 3 weeks. Pembrolizumab potency, as measured with an IL‐2 stimulation ratio assay, was 0.54 mg/L (95% CI = 0.12–2.3 mg/L). The design that enabled this understanding was based on a full modeling and simulation cycle that included development of a preliminary model with nine patients and evaluations of potential designs using extensive clinical trial SSE.
The PK data from these first nine patients led to uncertainties about the dose‐proportionality of exposure after intravenous dosing. The limited deviation from proportionality in exposure precluded fitting target‐mediated drug disposition (TMDD) PK models to these data, whereas differences in half‐life are within expected variability. Empirical models were applied in order to describe the deviations from linearity in exposure, with parameterizations in (relative) bioavailability, either as an abrupt shift or as a continuous dose‐dependency. This approach allowed establishment of a design that was optimized to distinguish between these two possibilities, albeit without a direct link to physiology.23 As discussed later, the final PK model no longer required these empirical parameters.
An optimally designed trial had two objectives: (1) it had to robustly establish the extent of the linearity of pembrolizumab PK; and (2) more reliably determine target engagement PD. Within‐patient dose escalation is commonly applied in other disease areas, but is more challenging in antibody‐based drug development in oncology because of a combination of the long half‐life of the antibody and the life‐threatening nature of the disease. Intensive multidisciplinary consultation rounds led to the establishment of a paradigm of fast within‐patient dose escalation, which kept the duration of patients' exposure to potentially ineffective concentrations short enough to be unlikely to have clinical impact. A pragmatic and iterative approach was taken to design optimization using SSE, focusing on short turnaround times and easily communicable results. Although an attractive alternative approach would have been to use D‐optimality evaluations, this was hampered by the lack of clear boundaries in design and of a cost‐utility function. Furthermore, given the many uncertainties and scenarios implemented, the D‐optimality approach would have had virtually no benefit in terms of results presentation, and lacked the efficiency associated with unsupervised computation provided by SSE.
Another choice in the simulation approach was to repeat each scenario only 100 times. A larger number of replicates would have led to better resolution between scenarios, but this could have led to an overrepresentation of potential differences that would have been unlikely to materialize, given the limited size of the trial and the many uncertainties (at that point) in PK/PD properties. At the start of the design phase, a large number of scenarios were derived from available observations. The resulting design was sufficiently robust to capture an unanticipated PK profile. Although in hindsight a TMDD profile should have been considered initially, there were insufficient data to derive the concentration range at which the PK nonlinearity would occur. Nevertheless, future investigations might more fully consider TMDD when extrapolating to much lower PK profiles with antibodies.
The nonlinear components in the final PK model were observed at the lowest observed concentrations, and probably contributed to the suboptimal stability. This was evident from the bootstrap convergence rate. The observations were in line with TMDD patterns seen with many other antibody drugs and provided a plausible and adequate description of the data.24 Moreover, the nonlinearity is not relevant for the clinical dosing range, and therefore was not investigated further. The origin of the PK nonlinearity is uncertain because the assay is only sensitive to free pembrolizumab, and no further measurements were made on either receptor content or bound drug. Pembrolizumab is a potent antibody with very specific binding to a target of low abundance in blood. Therefore, TMDD provides the best explanation, consistent with observations, and the class of biologic agent.
The PK/PD model on the final dataset fit the data adequately, as is evident from the goodness‐of‐fit plots and bootstrap results. The pembrolizumab IC50 based on the IL‐2 stimulation ratio was within the range of the pilot model derived from parts A and A1, and the model structure after considering data from part A2 was unchanged, albeit improved by logarithmically transforming fixed‐effect parameters and including BSV. The BSV was limited in magnitude, but was also consistently underestimated in the SSE design step (results not shown); the true BSV might therefore be larger.
One of the predefined design goals was robustness for a wide range of potential PD properties. In contrast to the initial results, the final estimated potency (after part A2) was supported by observations both above the IC50 and at almost ineffective concentrations. The bootstrap performance, condition number (∼50), and mostly modest uncertainty in the final model parameters indicate that the design was sufficiently powered. However, the IC50 precision was lower, with a coefficient of variation of 75%, possibly related to IL‐2 stimulation ratios that are relatively constant for concentrations around the IC50 value (see Figure 3). This suboptimal parameter precision was accounted for in simulations, ensuring proper decision‐making support.
Predictions from the model have been interpreted as providing a direction for, but not necessarily an absolute quantification of, potentially efficacious pembrolizumab dose regimens. Important assumptions were that the IL‐2 stimulation ratio in blood is a surrogate for target engagement of PD‐1 by pembrolizumab, is a reflection of the target engagement in the tumor, and, thus, is a marker for potential antitumor efficacy. Simulations revealed a 90% probability of achieving at least 95% target engagement with doses of 2 mg/kg every 3 weeks and higher, and a 50–60% probability with a dose of 1 mg/kg every 3 weeks (Figure 5 b). The 95% criterion was used to represent receptor saturation. Because IL‐2 is a peripheral biomarker, it is possible that target engagement and associated efficacy may not translate directly inside the tumor; for example, due to different pembrolizumab concentrations and receptor densities. The optimal level of target engagement for PD‐1 inhibition in advanced cancers has not yet been established.
A complete “learn and confirm” cycle of modeling and simulation was successfully implemented to support early assessment of potential pembrolizumab dose regimens that should be tested for clinical efficacy. At doses below 1 mg/kg, the likelihood of achieving target engagement saturation decreases considerably, whereas a high probability is achieved at or above 2 mg/kg every 3 weeks. This dose level has been tested in larger, randomized trials in advanced melanoma10, 13 and non‐small cell lung cancer,25 resulting in successful characterization of the benefits and risk of pembrolizumab and its subsequent approval for the treatment of advanced melanoma and non‐small cell lung cancer.10
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
We are grateful to the patients and families who participated in our studies. Support of the KEYNOTE‐001 trial was provided by Kellie Celentano, Maxine Giannotti, and Jill A. Linda (all of Merck & Co., Inc., Kenilworth, NJ). Roger Dansey (Merck & Co., Inc.) provided critical review of the manuscript. Anna Kondic (Merck & Co., Inc.) provided critical review and support in preparation of the manuscript. Pharmacokinetic data analysis support was provided by Marielle van Zutphen (Merck & Co., Inc.). Medical writing and editorial support in the preparation of this manuscript was provided by Jacqueline Kolston, PhD, and Melanie Leiby, PhD (ApotheCom, Yardley, PA), and was funded by Merck & Co., Inc. (Kenilworth, NJ).
Conflict of Interest
All authors are employees of the stated companies. J.E.‐S. reports receiving personal fees from Merck & Co., Inc., during the conduct of the study and, after leaving Merck, started an independent consultancy company, as indicated in the affiliation. S.R. has nothing additional to disclose. A.L. reports receiving personal fees from Merck Sharpe and Dohme during the conduct of the study. S.P.K. has been issued a patent for pembrolizumab. R.dG. reports receiving personal fees from Merck & Co., Inc., during the conduct of the study. J.S. reports stock ownership in other pharmaceutical and biotechnology entities outside the submitted work. D.D.A. has nothing additional to disclose.
Author Contributions
J.E.‐S., J.R.S., and D.D.A. wrote the manuscript. J.E.‐S., S.P.K., and J.R.S. designed the research. R.dG. performed the research. J.E.‐S., S.R., A.L., and J.R.S. analyzed the data.
References
- 1. Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McDermott, D.F. & Atkins, M.B. PD‐1 as a potential target in cancer therapy. Cancer Med. 2, 662–673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ishida, Y. , Agata, Y. , Shibahara, K. & Honjo, T. Induced expression of PD‐1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nishimura, H. , Nose, M. , Hiai, H. , Minato, N. & Honjo, T. Development of lupus‐like autoimmune diseases by disruption of the PD‐1 gene encoding an ITIM motif‐carrying immunoreceptor. Immunity 11, 141–151 (1999). [DOI] [PubMed] [Google Scholar]
- 5. Drake, C.G. , Jaffee, E. & Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 90, 51–81 (2006). [DOI] [PubMed] [Google Scholar]
- 6. Gettinger, S.N. et al Overall survival and long‐term safety of nivolumab (anti‐programmed death 1 antibody, BMS‐936558, ONO‐4538) in patients with previously treated advanced non‐small‐cell lung cancer. J. Clin. Oncol. 33, 2004–2012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hodi, F.S. et al Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robert, C. et al Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 (2011). [DOI] [PubMed] [Google Scholar]
- 9. Robert, C. et al Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Keytruda [package insert] (Whitehouse Station, NJ, Merck & Co., Inc, 2016). [Google Scholar]
- 11. Hamid, O. et al Safety and tumor responses with lambrolizumab (anti–PD‐1) in melanoma. N. Engl. J. Med. 369, 134–144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ribas, A. et al Pembrolizumab versus investigator‐choice chemotherapy for ipilimumab‐refractory melanoma (KEYNOTE‐002): a randomised, controlled, phase 2 trial. Lancet Oncol. 16, 908–918 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robert, C. et al Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: a randomised dose‐comparison cohort of a phase 1 trial. Lancet 384, 1109–1117 (2014). [DOI] [PubMed] [Google Scholar]
- 14. Garon, E.B. et al Pembrolizumab for the treatment of non‐small‐cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Patnaik, A. et al Phase I study of pembrolizumab (MK‐3475; anti‐PD‐1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 21, 4286–4293 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Sheiner, L.B. Learning versus confirming in clinical drug development. Clin. Pharmacol. Ther. 61, 275–291 (1997). [DOI] [PubMed] [Google Scholar]
- 17. Gregory, J.C. et al Bioassays for determining pd‐1 modulation. United States patent; WO2012018538 A2. <http://www.google.com/patents/WO2012018538A2?cl=en>. Published 9 February, 2012. Accessed 28 June 2016.
- 18. Beal, S.L. & Sheiner, L.B. NONMEM Users Guide, Part V (San Francisco, CA, University of California, 1992). [Google Scholar]
- 19. Jonsson, E.N. & Karlsson, M.O. Xpose–an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1989). [DOI] [PubMed] [Google Scholar]
- 20. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)–a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 22. European Environment Agency . R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. <http://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006>. Accessed 28 June 2016.
- 23. Kimbo, H.H.C. & Peck, C.C. Clinical Trial Simulations: Applications and Trends. 1st ed (New York, NY, Springer‐Verlag, 2011). [Google Scholar]
- 24. Cao, Y. & Jusko, W.J. Incorporating target‐mediated drug disposition in a minimal physiologically‐based pharmacokinetic model for monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 41, 375–387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rizvi, N.A. et al Safety and clinical activity of MK‐3475 as initial therapy in patients with advanced non‐small cell lung cancer (NSCLC). J. Clin. Oncol. 32(suppl.), Abstract 8007 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information