

Abstract
Pembrolizumab is a potent immune‐modulating antibody active in advanced melanoma, as demonstrated in the KEYNOTE‐001, ‐002, and ‐006 studies. Longitudinal tumor size modeling was pursued to quantify exposure‐response relationships for efficacy. A mixture model was first developed based on an initial dataset from KEYNOTE‐001 to describe four patterns of tumor growth and shrinkage. For subsequent analyses, tumor size measurements were adequately described by a single consolidated model structure that captured continuous tumor size with a combination of growth and regression terms, as well as a fraction of tumor responsive to therapy. This revised model structure provided a framework to efficiently evaluate the impact of covariates and pembrolizumab exposure. Both models indicated that exposure to the drug was not a significant predictor of tumor size response, demonstrating that the dose range evaluated (2 and 10 mg/kg every 3 weeks) is likely near or at the plateau of maximal response.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ RECIST‐based classification of solid tumor responses is an important metric for efficacy assessments; however, categorization of tumor size can be insensitive to time dependencies in the data. Model‐based analyses of tumor size may capture changes in tumor dimensions, and identify sources of response variability and potential exposure‐efficacy relationships.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Quantification of the exposure‐response relationships for the efficacy of pembrolizumab in advanced melanoma.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The pembrolizumab‐induced longitudinal tumor growth and regression kinetics described by a nonlinear mixed‐effects framework indicated a durable response in many patients, but with a wide interpatient variability over the time course of tumor burden. A relationship between baseline disease severity and magnitude of tumor regression was observed; however, pembrolizumab exposure had no clinically meaningful impact on response rates.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The developed models may ultimately enable the correlation between tumor dynamics in melanoma and long‐term survival, impacting therapeutic decision‐making for individual patients.
Evaluation of tumor burden is central to understanding treatment outcomes in cancer. Since 2000, the Response Evaluation Criteria in Solid Tumors (RECIST)‐based classification of solid tumor response has become an important metric for such efficacy assessments.1, 2 However, categorization of tumor size can be insensitive to time dependencies in the data because it involves distilling a large number of longitudinal data points into a single outcome measure.3, 4, 5, 6, 7, 8 In the years since the initial release of the RECIST guidelines, modeling of tumor size data has become an increasingly accepted approach to augment traditional efficacy analyses.3, 9, 10, 11, 12, 13, 14 There is, to our knowledge, no publication of a tumor growth model in melanoma. Initial results that indicate clinical relevance of tumor size are just emerging (Joseph et al., manuscript in preparation). Therefore, we explored a model‐based analysis of tumor size for capturing tumor kinetics, identifying sources of response variability, and exploring potential exposure‐efficacy relationships. Model‐based efficacy analyses have been applied extensively throughout the clinical development and regulatory approval process for the anti‐programmed cell death receptor 1 antibody pembrolizumab (MK‐3475). In this report, two modeling approaches are described for characterizing melanoma tumor kinetics in patients treated with pembrolizumab in the KEYNOTE‐001, ‐002, and ‐006 studies (ClinicalTrials.gov identifiers: NCT01295827, NCT01704287, and NCT01866319, respectively).
To date, pembrolizumab has demonstrated efficacy in melanoma15, 16, 17, 18, 19, 20 as well as other tumor types, including non–small cell lung cancer.21 The US Food and Drug Administration granted pembrolizumab breakthrough status for melanoma on the basis of emerging data from the first‐in‐human study KEYNOTE‐001.15 Subsequently, two randomized controlled studies, KEYNOTE‐00219 and KEYNOTE‐006,20 demonstrated the safety and efficacy of pembrolizumab in patients who were either previously treated with ipilimumab (IPI) or were IPI‐naive.
The first cohorts in KEYNOTE‐001 showed diverse patterns of tumor growth,17 which were described using a mixture model that partitioned the subjects as “nonresponders,” “slow responders,” “fast responders,” or those with “biphasic” tumor patterns. This initial tumor size model was originally applied to support dose decisions and define exposure bounds for efficacy in the first melanoma regulatory submission. Subsequently, with additional data from KEYNOTE‐002 and ‐006, a simplified consolidated model structure was developed that adequately described the range of diverse tumor size‐time patterns via three estimated structural parameters: first‐order growth/shrinkage rates and fraction of total target lesion responsive to therapy. This “consolidated model” facilitated the conduct of an integrated exposure‐response analysis across three protocols, accounting for imbalances in dose assignment between patient subgroups exhibiting differing degrees of treatment sensitivity. In this article, both approaches are presented, and their application in the development of pembrolizumab for melanoma is discussed.
METHODS
Patients included in the tumor‐size model datasets
The first mixture‐modeling approach utilized advanced melanoma tumor size data obtained from the KEYNOTE‐001 trial, with a cutoff date of October 2013. This dataset included 364 patients randomized to receive pembrolizumab at doses of 10 mg/kg every 2 weeks (Q2W; n = 51), 10 mg/kg every 3 weeks (Q3W; n = 167), and 2 mg/kg Q3W (n = 146). Of the entire cohort, 168 were IPI‐naive; the remaining patients were previously treated with IPI. Only patients providing at least one measurable lesion at baseline and were evaluable for pharmacokinetic evaluation were included in the analyses.
The second consolidated exposure‐efficacy analysis evaluated a pooled dataset (N = 1,366) representing all available melanoma tumor‐size data from patients treated with pembrolizumab as of April 2015 in KEYNOTE‐001, KEYNOTE‐002, and KEYNOTE‐006 (summarized by protocol, cohort, dose, and schedule in Table 1). The clinical characteristics detailed in Table 1 highlight the imbalance in the doses represented in the IPI‐naive subpopulation relative to the IPI‐experienced subpopulation. Sixty‐four patients, or 8.6% of the total pooled population, were IPI‐naive and enrolled in the 2 mg/kg Q3W dose group, whereas 237, or 38.2% of the total pooled population, were IPI‐experienced and enrolled in the 2 mg/kg Q3W dose group.
Table 1.
Number of patients in the consolidated tumor size modeling dataset with available pharmacokinetic data, categorized by treatment (dose and schedule) and protocol/cohort (N = 1,366)
| IPI status | Treatment | No. of patients (%) | KEYNOTE‐001 a | KEYNOTE‐002 | KEYNOTE‐006 | |||
|---|---|---|---|---|---|---|---|---|
| B1 | B2 | B3 | D | |||||
| Naive | 10 mg/kg Q2W | 324 (43.43) | 36 | 0 | 56 | 0 | 0 | 232 |
| 10 mg/kg Q3W | 358 (47.99) | 17 | 0 | 57 | 47 | 0 | 237 | |
| 2 mg/kg Q3W | 64 (8.58) | 19 | 0 | 0 | 45 | 0 | 0 | |
| Experienced and refractory | 10 mg/kg Q2W | 73 (11.77) | 14 | 0 | 59 | 0 | 0 | 0 |
| 10 mg/kg Q3W | 310 (50) | 26 | 76 | 48 | 0 | 160 | 0 | |
| 2 mg/kg Q3W | 237 (38.23) | 0 | 79 | 0 | 0 | 158 | 0 | |
IPI, ipilimumab; Q2W, every 2 weeks; Q3W, every 3 weeks
aKEYNOTE‐001 cohorts: B1 = IPI‐naive and ‐treated patients with melanoma enrolled to pembrolizumab at 2 or 10 mg/kg Q2W, or 10 mg/kg Q3W (sequential, nonrandomized). B2 = IPI‐refractory patients with melanoma randomized to pembrolizumab at either 2 or 10 mg/kg Q3W. B3 = IPI‐naive, IPI‐treated, or IPI‐refractory patients with melanoma randomized to 10 mg/kg either Q2W or Q3W. D = IPI‐naive patients with melanoma randomized to either 2 or 10 mg/kg Q3W.
Tumor size measurements
Individual lesions were measured using computed tomography or magnetic resonance imaging. Tumor size was recorded as the sum of longest dimensions (diameter) of target lesions, using RECIST version 1.1.
Linking pembrolizumab exposure to response
Area under the concentration‐time curve (AUC) estimates for connecting pembrolizumab serum concentrations to tumor size reduction in both the mixture model and the consolidated exposure‐response model were derived from separate population pharmacokinetic analyses (Ahamadi et al., manuscript submitted).22 The AUC over 6 weeks (AUCss‐6weeks) was chosen as the most appropriate exposure metric to account for a common time interval, given the investigation of both the Q2W and Q3W dosing (see Supplementary Methods).
Covariate exploration
The covariates tested in each analysis depended on clinical interest and on the list available in the clinical dataset at the time of analysis. Covariates to be tested were predefined in modeling analyses plans prior to analyses. For additional details on covariate methodology and model parameterization, please refer to the Supplementary Materials. The covariates explored for the early mixture model included age, body weight, gender, baseline tumor size, number of target lesions, number of lymph nodes affected, B‐type Raf (BRAF) mutation status, disease stage, IPI pretreatment status, study part, randomization status, and regimen. In the later consolidated model, the following covariates were tested for inclusion in a stepwise fashion: programmed death ligand 1 (PD‐L1) expression, Eastern Cooperative Oncology Group (ECOG) status, demographics (age, gender, and weight), baseline tumor size, IPI pretreatment status, and BRAF mutation status.
Initial exposure‐response tumor size (mixture) model
Initial examination of tumor size‐time profiles in KEYNOTE‐001 suggested significant tumor reduction for many patients; however, the overall population exhibited marked heterogeneity in patterns of individual response. Patients responding to treatment typically displayed early declines in tumor burden at either fast or slow rates, whereas those who progressed tended to do so early and to discontinue treatment sooner. To characterize the population heterogeneity in this initial model, the mixture subroutine in NONMEM was utilized to capture the proportion of patients who belonged to one of four distinct subpopulations (“escape” subpopulation for fast progressors, “monophasic slow” for slow responders, “monophasic fast” for fast responders, and “biphasic” for fast responders whose tumors did not change size after an initial drop). One of the motivations behind implementing this initial structure was the need to capture exposure‐response patterns for all patients, including those who dropped out without a postbaseline scan due to fast disease progression. Conveniently, the mixture model parameterization allowed such patients to be retained in the model and allocated to the escape group.
Within each mixture group, tumor growth/shrinkage parameters were estimated in a manner similar to the model originally described by Claret et al.6 in 2009 (see Supplementary Table S1). However, in many patients, tumor size was stable for long durations after an initial drop in tumor size, a pattern that is uncharacteristic for conventional chemotherapies with which relapse would be more common.9, 11 The resistance term in the original Claret model was therefore fixed to zero and the model was further modified as follows to better capture the observed durable pembrolizumab response patterns:
where “Baseline1” represents a dynamic portion of the tumor undergoing growth/shrinkage and “Baseline2” represents time‐invariant tumor mass; KL and KD are the rate constants of tumor growth and tumor shrinkage, respectively (see Supplementary Table S2a).
An automated stepwise forward inclusion/backward elimination procedure was applied to test for significant covariates on the model parameters using the stepwise covariate modeling routine in PsN (Perl speaks NONMEM).23 This automated covariate search was split into two parts: (1) stepwise covariate modeling was performed on the structural parameters of the tumor size model only; and (2) the estimates of mixture categories were used as input to a multinomial logistic regression model, and stepwise covariate modeling was performed on the logarithm of the odds of the mixture probabilities. AUCss‐6weeks was included as a parameter on KD and, irrespective of the statistical significance of the estimated exposure‐slope, was retained in the final model (together with other statistically significant relationships) so that simulations could be used to evaluate the magnitude of the potential exposure‐response relationship. Simulations with the mixture model were conducted as outlined in the Supplementary Methods.
Consolidated exposure‐response tumor size model
For exposure‐response analysis conducted with a subsequent, larger dataset from the KEYNOTE‐001, ‐002, and ‐006 studies (data cutoff April 2015), a simpler model structure was used to facilitate model stability, ease of covariate search, and conduct of simulations. This consolidated model structure parameterization is depicted visually in Figure 1 a,b, whereas Table 1 describes the number of patients in the consolidated tumor size modeling dataset with available pharmacokinetic data, categorized by treatment and protocol/cohort (N = 1,366). Here, the labels “f” and “1‐f” represent proportions of target tumor tissue that are accessible to treatment and undergoing unimpeded exponential growth, respectively. Thus, tumor size at a particular time point is described by:
where “tumor size” is the sum of longest diameter (in mm), “baseline” is the tumor size at screening, “kgrowth” is the tumor growth rate, “kdeath” is the tumor shrinkage rate that captures the kinetics of net tumor removal in the responding portion of the tumor, and delay represents the lag in onset of drug activity for tumor shrinkage, interpreted as the time required for immune system activation. The term “f” was logit normally distributed and thus constrained between zero and one, whereas delay, kdeath, and kgrowth were constrained to be positive, with individual parameter logs normally distributed. Also shown in Figure 1 a,b are representative individual tumor size profiles, demonstrating that such parameterization has the flexibility to capture a diverse array of tumor growth patterns.
Figure 1.
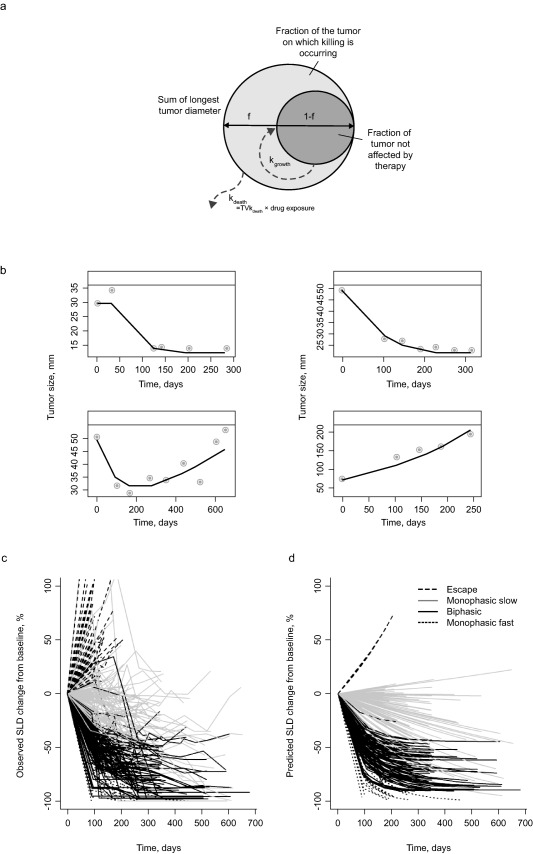
(a) Schematic representation of the structural components of the consolidated tumor size model used for describing melanoma tumor dynamics, where f is the fraction of the tumor in which removal is occurring and represents the proportion of target tumor tissue that is accessible to treatment; 1‐f represents the proportion of target tumor tissue undergoing unimpeded exponential growth; kgrowth represents tumor growth rate; and kdeath represents tumor shrinkage rate. (b) Representative individual profiles from the consolidated melanoma model parameterization. These tumor‐size time plots help demonstrate that the model successfully captures a wide variety of tumor growth patterns (decline in tumor size following an initial delay before drug administration, instantaneous drug effect, tumor growth relapse after an initial decline, and exponential tumor growth). Observed tumor‐size time data (•) and individual predictions (‐). (c) Observed and (d) model‐predicted percentage change in tumor size from baseline for each of the four tumor growth patterns (base mixture model). Tumor size was recorded as the sum of the longest diameters of target lesions.
Unlike the mixture model, parameterization that required a latent categorical covariate (mixture state) to be estimated for each patient, interindividual variations in kdeath, kgrowth, and f captured a variety of tumor size patterns observed across the melanoma population. This structure was found to be the most parsimonious that adequately described the combined data.
To account for the effect of drug exposure in this exposure‐response analysis, AUCss‐6weeks was incorporated into the structural model parameterization on the tumor size shrinkage rate. However, at the time of analysis, an imbalance in the integrated dataset required estimation of two distinct exposure‐response relationships. Specifically, IPI‐naive patients (who responded better to pembrolizumab than did IPI‐experienced patients) made up a higher proportion of the 10 mg/kg dose groups and a lower proportion of the 2‐mg/kg dose groups. This imbalance could manifest inappropriately as an exposure‐dependency in tumor size patterns in a pooled exposure‐response analysis; separate exposure‐response parameters were therefore estimated for IPI‐naive and IPI‐experienced patients.
The estimated confidence interval (CI) of the slope parameters (θIPI‐naive and θIPI‐treated) determined the extent of the pembrolizumab exposure response. Statistical significance was assessed by testing for a difference in the estimated slopes from the value zero.
A stepwise covariate search was used to find covariates predictive of variability on kdeath, kgrowth, and f. The magnitude of drug exposure‐tumor size response was depicted visually using simulations from the final model that took into account uncertainty in the model parameters, interindividual variability, and residual variability, as described in the Supplementary Methods.
RESULTS
Initial mixture model to characterize melanoma tumor growth and pembrolizumab exposure dependency
Parameter estimates of the base mixture model were conducted with good precision. Additionally, standard diagnostics (observations vs. population and individual predictions, as well as residual‐based diagnostics) indicated good model fits (Supplementary Table S2a and Supplementary Figure S1a). Individual observed (Figure 1 c) and model‐predicted changes (Figure 1 d) from baseline are shown for each of the four (estimated) mixture model categories, demonstrating the ability of the model to adequately describe observed data patterns.
During the covariate searches, the number of target lesions (P < 0.0001) and baseline ECOG‐performance status (P = 0.0004) were found to be covariates predictive of baseline tumor size, whereas baseline tumor size and the number of lymph nodes were found to be predictive of mixture category classification (Supplementary Table S3). Importantly, AUCss‐6weeks was not a statistically significant covariate. However, the AUCss‐6weeks relationship was retained in the final model so that further simulations could be used to evaluate the magnitude of the potential exposure relationship.
Parameter estimates of the final mixture model were estimated precisely, as indicated by the relatively narrow bootstrap CIs (Table 2 (mixture model) and Supplementary Table S2b (multinomial regression)). Goodness‐of‐fit plots demonstrated that the model adequately fit the data (Supplementary Figure S1b). A visual predictive check for the final model demonstrated that the model describes the central tendency (median) of the data well in all mixture groups (Figure 2 a–d).
Table 2.
Final mixture model
| Bootstrap estimate a | |||||
|---|---|---|---|---|---|
| Parameter, units | Description | Estimate | Median | 90% CI | Shrinkage, % |
| Fixed‐effects parameters | |||||
| KL, 10− 3/day b | Growth rate constant | 2.76 | 2.77 | 2.26–3.9 | |
| KD, 10− 3/day b | Kill rate constant | 3.57 | 3.61 | 2.96–4.74 | |
| BASEL1, mm | Baseline size of shallow tumor compartment | 56.3 | 56.4 | 49.8–63 | |
| BASEL1∼NTARGET | Exponent of relationship between number of target lesions and BASEL1 | 0.654 | 0.661 | 0.555–0.765 | |
| BASEL1∼BECOGN | Fractional change in BASEL1 for ECOG status 1 compared to ECOG status 0 | 0.344 | 0.344 | 0.151–0.577 | |
| BASEL2, mm | Baseline size of deep tumor compartment | 25.1 | 25.8 | 18–37.9 | |
| BASEL Rel. Diff. | Difference of BASEL1 in escape group relative to all other groups | 2 | 2 | 1.66–2.35 | |
| KD Rel. Diff. | Difference in KD in biphasic and fast monophasic groups relative to slow monophasic group | 4.17 | 4.12 | 3.42–4.85 | |
| LGT2 | Logit of probability of being in slow monophasic subpopulation | 0.903 | 0.916 | 0.513–1.47 | |
| LGT3 | Logit of probability of being in slow biphasic subpopulation | 0.48 | 0.488 | 0.0969–0.934 | |
| LGT4 | Logit of probability of being in fast monophasic subpopulation | −0.92 | −0.926 | −1.61 to −0.269 | |
| P1 c | Probability of being in escape subpopulation | 0.294 | 0.248 | 0.196–0.298 | |
| P2 c | Probability of being in slow monophasic subpopulation | 0.389 | 0.415 | 0.34–0.506 | |
| P3 c | Probability of being biphasic subpopulation | 0.255 | 0.268 | 0.197–0.335 | |
| P4 c | Probability of being in fast monophasic subpopulation | 0.0628 | 0.064 | 0.0353–0.104 | |
| Random‐effects parameters d | |||||
| ETA_EPS | Interindividual variability of residual error (variance) | 26.1 | 26 | 15.9–33.1 | 28.9 |
| IIV KD, %CV | Interindividual variability of KD | 34 | 32.9 | 25.4–42.2 | 26.7 |
| IIV BL1, %CV | Interindividual variability of BASEL1 | 79.6 | 79.1 | 71.4–88.3 | 2.8 |
| IIV BL2, %CV | Interindividual variability of BASEL2 | 225 | 213 | 116–357 | 8.1 |
| Corr, BL1∼BL2 | Correlation coefficient BASEL1∼BASEL2 | 0.863 | 0.87 | 0.743–0.941 | |
| Residual error | |||||
| Proportional, %CV | 10.3 | 10.2 | 8.84–11.8 | ||
| Additive, mm | 3.29 | 3.24 | 2.65–4.1 | 2.9e | |
BECOGN, baseline Eastern Cooperative Oncology Group numeric; CI, confidence interval; CV, coefficient of variation; ECOG, Eastern Cooperative Oncology Group.
aObtained from 695 successfully minimized replicate runs of 1,000. bLog‐transformed parameter estimated and back‐transformed for reporting. cDerived from estimates of the logits and corrected for the frequency of patients with missing post‐baseline scans. dCV% was calculated as: ; eepsilon‐shrinkage refers to the combined error model, not only the additive part.
Figure 2.
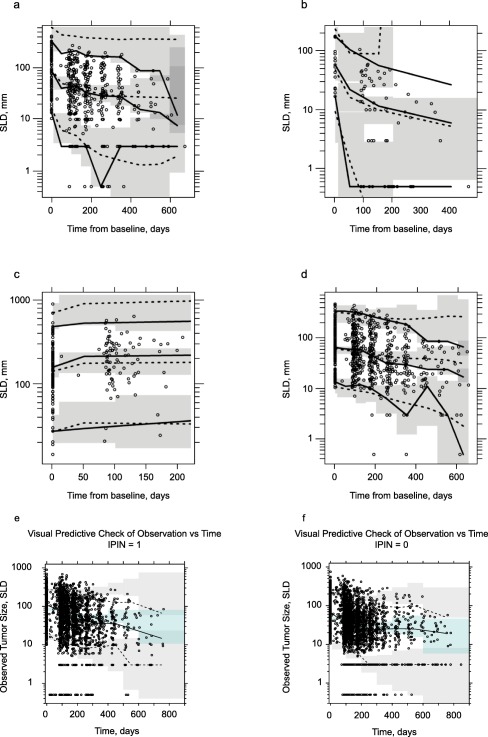
isual predictive check for each subpopulation of the final tumor size reduction model (initial mixture modeling approach): (a) biphasic, (b) monophasic fast, (c) escape, and (d) monophasic slow. Visual predictive check for the final tumor size (consolidated) model for (e) ipilimumab‐naive and (f) ipilimumab‐experienced patients. For a‐d, solid lines are the 5th, 50th, and 95th percentiles of the observations; dotted lines connect the respective percentiles of the predictions for each time bin; shaded gray areas represent the 95% confidence interval (CI) around the percentiles of the predictions. Tumor size was recorded as the sum of longest diameter (SLD) of target lesions. For e and f, the solid line represents the 50th percentile of the observations, whereas dashed lines represent the 10th and 90th percentiles. The shaded areas connect the respective percentiles of the predictions for each time bin, with the uppermost and lower shaded areas representing the 95% CI around the percentiles of the predictions, and the middle shaded area representing the upper and lower. IPIN, ipilimumab‐naive. IPIN = 0, ipilimumab‐naive patients; IPIN = 1, ipilimumab‐experienced patients.
Finally, the mixture model and the multinomial regression model were combined together to simulate response rates across doses (Figure 3 a). Specifically, the median objective response rate (i.e., percentage of patients with a complete or partial response) at week 28 was 32.9% (90% CI = 28.2–37.7) in the 2 mg/kg Q3W group and 35.9% (90% CI = 31.1–40.4) in the 10 mg/kg Q3W group. This small, nonsignificant difference suggests that, within the dose range studied, the exposure‐response relationship is relatively flat and is likely close to the plateau of maximal efficacy.
Figure 3.
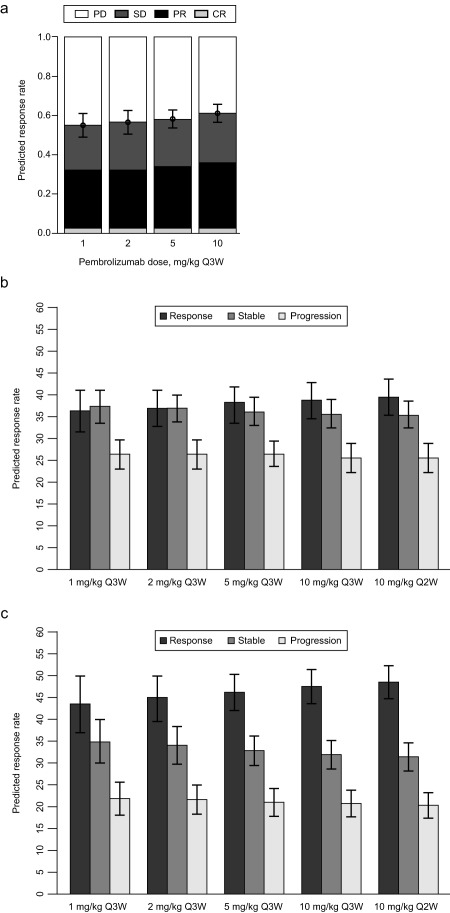
(a) Median response rates at week 28 for the different response categories of 1,000 simulated trials, each with 10,000 patients (mixture model). Median simulated response rates for patients whose tumor is positive for programmed death receptor 1 ligand 1 (PD‐L1) at week 24 for dose schedules ranging from 1 mg/kg every 3 weeks (Q3W) to 10 mg/kg every 2 weeks (Q2W), using the consolidated model structure; (b) ipilimumab‐pretreated patients; and (c) ipilimumab‐naive patients. Error bars represent the 90% confidence intervals around the estimates. Response is defined as change from baseline (CFB) ≤−30%; stable is defined as CFB −30 to −20%; progression is defined as CFB ≥20%. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Simulations of 1 mg/kg Q3W were included as an aid to inform the therapeutic window around the clinical dose range. Consistent with the aforementioned results suggesting that 2 mg/kg Q3W falls near the plateau of the dose‐response curve, only a modest reduction in efficacy was predicted at 1 mg/kg Q3W. These results further suggest that individuals with reduced pembrolizumab exposures (due to intrinsic/extrinsic factors) by as much as 50% will maintain meaningful efficacy.
Consolidated exposure‐response tumor size model using integrated data from KEYNOTE‐001, ‐002, and ‐006
For the integrated analysis across KEYNOTE‐001, ‐002, and ‐006, a consolidated model was developed that utilized variability in three parameters (a logit normally distributed fraction of tumor that responds to therapy, and log normally distributed first‐order growth and shrinkage rates) to capture diverse patterns. Prior to modeling the data, exploratory plots were generated to investigate trends of response to treatment vs. exposure. The visual exploration was supported by the results of a simple linear regression, where appropriate. These analyses suggested that separate exposure response slopes were required for IPI‐naive and IPI‐experienced patients (Supplementary Figure S2).
Parameter value estimates of the base structural model along with the parameter uncertainty estimates are shown in Supplementary Table S4a. Generally, all model parameters were estimated with good precision. Basic goodness‐of‐fit plots (Supplementary Figure S3a) demonstrate that the model adequately described the data.
An overview of patient‐ and study‐specific factors pertinent to the covariate analysis is shown in Supplementary Tables S5a,b. The final results of the covariate search suggest that expression of PD‐L1, baseline tumor size on kdeath, prior IPI treatment history, baseline tumor size on f, and BRAF mutation status on kgrowth were predictive of the interindividual variability in these parameters (Supplementary Table S4b and Supplementary Figure S4). Despite these relationships, there was still overlap in parameter values across these covariate groupings. Therefore, the covariates did not necessarily predetermine outcome at an individual patient level. These results suggest that all patients, regardless of baseline tumor size, PD‐L1 expression status, BRAF mutation status, and prior IPI treatment status may have the potential to benefit from therapy.
Exposure (AUCss‐6weeks) was not a significant predictor of tumor shrinkage, with P values of 0.20 and 0.25 for IPI‐naive and IPI‐experienced patients, respectively. Therefore, this statistically insignificant exposure‐response parameter suggested at most a small influence of exposure over the fivefold dose range studied across KEYNOTE‐001, ‐002, and ‐006. The estimated exposure dependence and its associated uncertainty were retained in the model for visualization purposes so that further simulations could be used to assess the magnitude of any potential relationship.
Similar to the base model, parameter estimates of the final tumor size model were also estimated with good precision (Supplementary Table S4c). Goodness‐of‐fit plots and additional supportive plots demonstrate that the model adequately captured the data (Supplementary Figure S3b). A visual predictive check for the final model (stratified by IPI treatment history) is shown in Figure 2 e,f. As noted in the methods, the model describes tumor growth by an unconstrained exponential term; however, physiological tumor growth cannot proceed unimpeded in reality without the patient progressing and eventually coming off the study. Furthermore, observed melanoma tumor size responses suggest that most patients whose tumor size has increased twofold from baseline tend to drop out from the study. Because of these reasons and to mimic the limited duration of data from patients prior to study withdrawal, study discontinuation was accounted for during construction of the visual predictive check by censoring the simulated patients whose tumors had grown more than threefold above baseline and who had been on treatment for at least 6 months. Considering these nuances in capturing longitudinal tumor size data from trial data, the model describes the central tendency of the data relatively well for both IPI‐naive and IPI‐experienced patients, suggesting that the observed data could arise from the proposed model structure and estimated parameters. Still, a modest overprediction of the 90th percentile of the observed data is present. This could be explained in part by the fact that the prespecified censoring rules might be too stringent. In reality, it is likely that nonresponsive patients discontinued from the study when their tumor sizes doubled, not tripled. Improvements in the prediction of the upper percentile of data were obtained using a less stringent censoring criterion (not shown). However, in reporting the visual predictive check results, it was decided that only the prespecified criteria would be used, and, despite these slight perceived discrepancies, the model generally described the data well.
Finally, trial simulations were conducted, drawing at random with replacement from all patients in the observed dataset but with random reassignment of dose (Figure 3 b,c), as outlined in the Supplementary Methods. All simulations included interindividual and residual variability, and uncertainty in parameters. The percentage of patients in each response group was summarized by dose regimen at week 24 for both IPI‐naive and IPI‐experienced patients. The simulated median (90% CI) response rate at week 24 for IPI‐naive patients was 44.7% (38.8–49.8%) at 2 mg/kg Q3W and 47.4% (43.7–51.3%) at 10 mg/kg Q3W. For prior IPI‐experienced patients, the response rate was 36.9% (90% CI 32.8–41.3%) at 2 mg/kg Q3W and 38.8% (90% CI = 35.2–42.7%) at 10 mg/kg Q3W. These simulations show that although both IPI‐naive and IPI‐experienced patients responded well to treatment, there was a tendency toward a higher response rate among the IPI‐naive group. In addition, across both populations, the substantial overlap in the CIs of median response across the wide range of exposures studied indicates there would be little meaningful difference in response with increasing dose/frequency. Furthermore, the similarity of response rates at 1 mg/kg Q3W compared to 2 mg/kg Q3W suggests that patients with decreased exposure at 2 mg/kg will likely experience the same benefit from pembrolizumab as those with higher exposures at the same dose.
Note that tumor size outputs from model simulations were categorized into three response categories (progressive disease, stable disease, and responders), reflecting the standard RECIST version 1.1. Aside from target tumor size, other clinical factors can influence RECIST‐based clinical response assessments (e.g., shrinkage in nontarget tumors, such as pathologic lymph nodes) and appearance of malignant lesions indicative of progression. Because the model only accounted for tumor size measured based on target lesions without the distinction between lymph node or other tissue/metastases, such nuances in the RECIST could not be fully accounted for in the simulations. Therefore, caution is urged in the interpretation of results and drawing direct comparisons with actual clinical response categories. Another limitation in extrapolation of simulations to the clinical data is that the model does not account for dropouts when allocating patients to response categories, and this could potentially impact the ratios of category assignments at specific timepoints.
DISCUSSION
To our knowledge, the exposure‐response analyses described in this report represent the first comprehensive pharmacokinetic/pharmacodynamic tumor size models for melanoma. In both the initial mixture model and consolidated tumor size model, longitudinal tumor kinetics were successfully described via a nonlinear mixed‐effects framework. The results of these model‐based analyses indicate a durable response in many patients, but with wide interpatient variability in the time course of tumor burden. Covariate searches for both models demonstrated that baseline disease severity (i.e., tumor size during screening) was related to the magnitude of tumor regression, whereas the relationship between changes in tumor size and exposure across a fivefold dose range was not statistically significant (P > 0.05). Further simulations from both models revealed the lack of a clinically meaningful impact of pembrolizumab exposure on response rates. This finding is consistent with earlier translational exposure‐response modeling work, which indicated that doses of 1–2 mg/kg and higher were likely to be associated with meaningful efficacy (Lindauer et al., manuscript e‐Published).24
Both tumor size models described here were fit‐for‐purpose at sequential steps along the pembrolizumab clinical development path for advanced melanoma. The initial mixture model supported an early dose of 2 mg/kg in the 2014 regulatory filing for advanced melanoma in patients with disease progression following treatment with IPI (and a BRAF inhibitor, if BRAF V600 is mutation‐positive) based on a smaller dataset from KEYNOTE‐001. The subsequent consolidated model structure included data from KEYNOTE‐002 and ‐006 in addition to KEYNOTE‐001, and supported dosing decisions for advanced melanoma in patients refractory or naive to IPI. Based on patterns of individual response, the initial mixture model used a probabilistic classification of subjects as different categories of responders (“biphasic,” “monophasic slow,” or “monophasic fast”) or as progressors (the so‐called “escape” category) based on patterns of individual response, whereas the consolidated model captured this diversity in tumor kinetics as a reflection of variation in three factors: the proportion of actively dividing cells (the growth fraction, f), a growth parameter (kgrowth), and the rate of cell loss under treatment (kdeath). The consolidated model structure was implemented with the objective to create a simpler, more flexible model for integrating data across protocols and accounting for dose imbalances in the pooled dataset. This easily adaptable model framework has also been extended to support dosing decisions in other indications.25
Methodology and results from both modeling approaches have many similarities, but also some differences. Most importantly, findings from both models confirmed a flat exposure‐response relationship from 2 mg/kg to 10 mg/kg in advanced melanoma. This evidence has been favorably received by regulatory agencies in the United States and European Union, and helped to establish the registration dose and regimen for pembrolizumab breakthrough designation filings in 2014 and 2015.
One of the key distinctions between the initial mixture model and the subsequent consolidated approach pertains to the way patients who dropped out of the study provided data for parameter estimation. In the consolidated model, patients who left the study contributed no further data for model parameter estimation beyond the timepoint for which the last measurement of tumor size was made, and were thus effectively ignored beyond their last observation. In contrast, the mixture model allowed patients with no postbaseline scans to be described in the “escape” mixture model, and, therefore, contribute to the exposure‐response relationship and covariate search. In the consolidated analysis, the strict requirement of inclusion of the “escape” group was loosened in order to obtain a more parsimonious model while retaining sufficient flexibility to accommodate different patterns through a distribution of tumor growth and regression patterns. However, 10.8% of patients in the dataset had no postbaseline measurements available for analysis.
In addition, in contrast to the mixture modeling approach, covariates in the consolidated model approach were evaluated in a single step. The results of covariate searches for both analyses determined the baseline tumor size to explain a significant portion of response variability, but in the larger dataset, BRAF mutation, degree of tumor PD‐L1 expression, and IPI treatment history were identified as predictive of individual variability. The testing of covariates in the consolidated model occurred with a much larger dataset than was used for the initial mixture model development. Moreover, PD‐L1 status was not available during initial mixture model development. Identification of PD‐L1 expression as a key determinant of kdeath is consistent with the known mechanism of action for pembrolizumab, and confirms that PD‐L1–positive patients are likely to have better responses to treatment.
Although the mixture model structure contains some useful features (e.g., characterization of subjects with missing postbaseline scans as nonresponders), it is difficult to distinguish among multiple mixture categories from very sparse data (several subjects contained only 2 or 3 data points). Moreover, determination of parameter precision from computation of the Fisher information matrix and identification of covariates predictive of response and simulations are more straightforward with the consolidated model structure. In our view, these properties outweigh the additional precision that can be attained with the mixture model. By presenting both models, readers have the opportunity to see that the choice of model structure has no major impact on the conclusions of whether or not there is an exposure effect on tumor growth within the studied dose range.
We have described herein two modeling approaches that were used to analyze exposure‐response relationships and identify predictors of response to pembrolizumab treatment in advanced melanoma; both approaches indicated that a 2 mg/kg Q3W pembrolizumab dose provides a near‐maximal response. Other mixed‐effects models have been similarly utilized to describe changes in tumor size and responses to drug treatment, including applications in non–small cell lung cancer,9 renal cell carcinoma,10 colorectal cancer,11 thyroid cancer,12 breast cancer,3 gastrointestinal stromal tumors,13 and low‐grade glioma.14 However, the analyses described here are the first of this type of model for melanoma. Unlike RECIST‐based characterization of response as a discrete outcome, model‐based analyses of longitudinal tumor imaging data enable investigation of the dynamics of response on a continuous scale. Moreover, the models developed here lay the groundwork for relating tumor dynamics in melanoma to long‐term survival, as demonstrated for other tumors.9, 11
In both models, an empirical approach was chosen because the simple structure of these models was deemed appropriate for summarizing clinical data and normalizing for differences across observed variables in the studied population (i.e., there was no extrapolation beyond the patient factors studied in the actual clinical trials). The goal of these analyses was to support dose justification in the regulatory filings, whereas more detailed mechanistic understanding of tumor growth and response to immunotherapy will be the topic of future explorations. The potential use of tumor size as predictor of survival response for the case of immune‐modulators, like pembrolizumab, needs to be further investigated.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
We thank Scott Ebbinghaus and Roger Dansey of Merck & Co., Inc. (Kenilworth, NJ) for critical review of the manuscript. Medical writing and editorial support in the preparation of this manuscript was provided by Sarah Adai, MS (ApotheCom, Yardley, PA), Melanie Leiby, PhD (ApotheCom, Yardley, PA), and Chris Ontiveros (ApotheCom, New York, NY) and was funded by Merck & Co., Inc. (Kenilworth, NJ).
Author Contributions
M.S.C., D.C.T., A.L., J.E.S., K.M., J.S., R.d.G, A.G.K., and D.P.d.A wrote the manuscript. J.E.S., M.S.C., D.C.T., A.L., J.E.S., A.S., T.F., and M.A. performed the research. K.M., J.S., R.d.G., A.G.K., and D.P.d.A. analyzed the data. M.S.C., J.E.S., A.L., and D.C.T. contributions equally to the work.
Conflict of Interest
All authors are employees of the stated companies.
References
- 1. Therasse, P. et al New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 92, 205–216 (2000). [DOI] [PubMed] [Google Scholar]
- 2. Eisenhauer, E.A. et al New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer. 45, 228–247 (2009). [DOI] [PubMed] [Google Scholar]
- 3. Frances, N. , Claret, L. , Bruno, R. & Iliadis, A. Tumor growth modeling from clinical trials reveals synergistic anticancer effect of the capecitabine and docetaxel combination in metastatic breast cancer. Cancer Chemother. Pharmacol. 68, 1413–1419 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Sharma, M.R. , Maitland, M.L. & Ratain, M.J. RECIST: no longer the sharpest tool in the oncology clinical trials toolbox—point. Cancer Res. 72, 5145–5149 (2012); discussion 5150. [DOI] [PubMed] [Google Scholar]
- 5. Maitland, M.L. , Schwartz, L.H. & Ratain, M.J. Time to tumor growth: a model end point and new metric system for oncology clinical trials. J. Clin. Oncol. 31, 2070–2072 (2013). [DOI] [PubMed] [Google Scholar]
- 6. Goldmacher, G.V. & Conklin, J. The use of tumour volumetrics to assess response to therapy in anticancer clinical trials. Br. J. Clin. Pharmacol. 73, 846–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mould, D.R. , Walz, A.C. , Lave, T. , Gibbs, J.P. & Frame, B. Developing exposure/response models for anticancer drug treatment: special considerations. CPT Pharmacometrics Syst. Pharmacol. 4, e00016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venkatakrishnan, K. et al Optimizing oncology therapeutics through quantitative translational and clinical pharmacology: challenges and opportunities. Clin. Pharmacol. Ther. 97, 37–54 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Wang, Y. et al Elucidation of relationship between tumor size and survival in non‐small‐cell lung cancer patients can aid early decision making in clinical drug development. Clin. Pharmacol. Ther. 86, 167–174 (2009). [DOI] [PubMed] [Google Scholar]
- 10. Stein, A. et al Dynamic tumor modeling of the dose‐response relationship for everolimus in metastatic renal cell carcinoma using data from the phase 3 RECORD‐1 trial. BMC Cancer 12, 311 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Claret, L. et al Model‐based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J. Clin. Oncol. 27, 4103–4108 (2009). [DOI] [PubMed] [Google Scholar]
- 12. Claret, L. , Lu, J.F. , Sun, Y.N. & Bruno, R. Development of a modeling framework to simulate efficacy endpoints for motesanib in patients with thyroid cancer. Cancer Chemother. Pharmacol. 66, 1141–1149 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Hansson, E.K. et al PKPD modeling of VEGF, sVEGFR‐2, sVEGFR‐3, and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. CPT Pharmacometrics Syst. Pharmacol. 2, e84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ribba, B. et al A tumor growth inhibition model for low‐grade glioma treated with chemotherapy or radiotherapy. Clin. Cancer Res. 18, 5071–5080 (2012). [DOI] [PubMed] [Google Scholar]
- 15. Patnaik, A. et al Phase I study of pembrolizumab (MK‐3475; anti‐PD‐1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 21, 4286–4293 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Daud, A. et al Long‐term efficacy of pembrolizumab (pembro; MK‐3475) in a pooled analysis of 655 patients (pts) with advanced melanoma (MEL) enrolled in KEYNOTE‐001. J. Clin. Oncol. 33(suppl), Abstract 9005 (2015). [Google Scholar]
- 17. Hamid, O. et al Safety and tumor responses with lambrolizumab (anti‐PD‐1) in melanoma. N. Engl. J. Med. 369, 134–144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robert, C. et al Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: a randomised dose‐comparison cohort of a phase 1 trial. Lancet 384, 1109–1117 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Ribas, A. et al Pembrolizumab versus investigator‐choice chemotherapy for ipilimumab‐refractory melanoma (KEYNOTE‐002): a randomised, controlled, phase 2 trial. Lancet Oncol. 16, 908–918 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robert, C. et al Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 21. Garon, E.B. et al Pembrolizumab for the treatment of non‐small‐cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 22. Ahamadi, M. , et al. Model‐based characterization of the pharmacokinetics of pembrolizumab, a humanized anti‐PD‐1 monoclonal antibody, in advanced solid tumors. CPT Pharmacometrics Syst. Pharmacol. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)–a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 24. Lindauer, A. , et al. Translational pharmacokinetic/pharmacodynamic modeling of tumor growth inhibition supports dose‐range selection of the anti‐PD‐1 antibody pembrolizumab. CPT Pharmacometrics Syst. Pharmacol. 2016;ePub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chatterjee, M. et al Model‐based analysis of the relationship between pembrolizumab exposure and efficacy in patients with melanoma and NSCLC: a cross‐indication comparison 2015 Annual Meeting of the Population Approach Group in Europe 2–5 June 2015; Hersonissos, Greece; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information