

The regenerative capacity of peripheral nerve is significantly different from injury responses in the central nervous system. Regeneration of injured axons, re-myelination and functional recovery are readily observed after injury to peripheral nerve. Much of this capacity can be attributed to the unique injury program of Schwann cells, which survive injury and release diverse signaling molecules that are vital to the nerve regeneration process. Previous reviews have highlighted the ability of Schwann cells of injured nerve to downregulate myelin genes, clear myelin debris, and signal to macrophages (Jessen and Mirsky, 2016). In addition, denervated Schwann cells initiate an autophagy process (termed myelinophagy) and transform into an elongated bipolar morphology and align in columns, termed Bands of Bungner. These structures provide substrates for regenerating axons to reach their target tissues. Several signaling pathways are involved in Schwann cell responses, including activation of Jnk, Notch and Erk pathways. Interestingly, the age-dependent impairment of peripheral nerve regeneration appears to be due to deficient Schwann cell responses rather than intrinsic neuronal properties (Painter et al., 2014).
Several lines of evidence indicate that Schwann cells in injured nerve do not merely dedifferentiate, but that they establish a novel differentiated state. An elegant demonstration of this concept is that the c-Jun transcription factor is not required for Schwann cell development, but is required for many Schwann cell responses after nerve injury. In the absence of c-Jun in Schwann cells, a number of processes are impaired in injured nerve, such as myelin debris clearance, signaling to macrophages and formation of Bands of Bungner (Arthur-Farraj et al., 2012). In addition, Schwann cells in injured nerves begin to express proteins that are not normally expressed in Schwann cell development such as sonic hedgehog and neuregulin Type I (Stassart et al., 2013; Lin et al., 2015). Therefore, Schwann cells in injured nerve are profoundly reprogrammed, involving dynamic changes in gene expression of more than 1,000 genes, to enable nerve repair. Because the roles of Schwann cells in nerve regeneration differ substantially from those in axon ensheathment and myelination, they have been termed repair Schwann cells (Arthur-Farraj et al., 2012; Jessen and Mirsky, 2016).
Similar to the reprogramming mechanisms characterized in stem cell studies, the transformation of Schwann cells to repair cells requires dynamic changes in several transcription factors. For example, analysis of c-Jun mutants has identified ~100 genes that require c-Jun for induction after nerve injury (Arthur-Farraj et al., 2012). Some injury-induced genes are expressed in embryonic Schwann cells and then become repressed during the myelination phase of Schwann cell development. Many of these genes are repressed by the zinc finger transcription factor EGR2/KROX20, which is required for the terminal differentiation program and maintenance of the differentiated state of Schwann cells. EGR2 positively regulates several mature myelin genes, such as peripheral myelin protein 22 and myelin protein zero, but also represses genes coding for negative regulators of myelination (Le et al., 2005; Srinivasan et al., 2012), including c-Jun and Sox2. Therefore, some of these genes become activated due to the injury-associated decrease of the EGR2 transcription factor. EGR2 achieves transcriptional repression through the association with the Nab corepressors that form a bridge to the Chd4-containing nucleosome remodeling and deacetylase (NuRD) complex. Deletion of Nab1/Nab2 and Schwann cell-specific deletion of Chd4 resulted in derepression of c-Jun, Sox2 and other genes that are normally repressed in mature Schwann cells (Le et al., 2005; Hung et al., 2012).
Cell reprogramming involves not only transcriptional pathways but also modification of chromatin structure. Chromatin transitions are presumably involved in reprogramming Schwann cells, and we have focused on exposing epigenomic pathways that are implicated in the regulation of Schwann cell plasticity between terminally differentiated myelinating glia and reprogramed repair cells. We recently characterized changes in a histone mark that denotes active enhancers (histone H3 Lys27 acetylation, H3K27ac) and found that approximately 4,000 new sites appear after nerve injury in vivo, whereas a similar number of active enhancers lose this mark in injured nerve (Hung et al., 2015). H3K27ac is made by the CBP/p300 acetylases that work with a broad range of transcription factors. The dynamically regulated sites are linked to nearby genes that change expression after nerve injury, and motif analysis revealed that injury-induced enhancers are highly enriched in binding motifs for the c-Jun transcription factor. Based on some stem cell studies, we had anticipated that injury-induced enhancers may be pre-marked, or poised, for activation after injury. However, profiling the hallmarks of poised enhancers (open chromatin, H3K27me3) revealed that injury-induced enhancers were not pre-marked, but rather appear to be activated de novo from repressed chromatin (Ma et al., 2016). This suggests that at least some injury-induced transcription factors must act as pioneer factors to open up repressed chromatin.
One example of this is the Sonic Hedgehog gene, which is normally silenced in embryonic and postnatal Schwann cells. However, Shh is activated in repair Schwann cells and therefore is considered a unique marker of repair Schwann cells (Arthur-Farraj et al., 2012; Lin et al., 2015; Jessen and Mirsky, 2016). While many Shh enhancers have been identified in limb bud and CNS development, a unique set of enhancers appear in peripheral nerve after injury (Hung et al., 2015), which are distinct from all previously characterized enhancers.
More recently, we have obtained additional insight into the activation mechanism of repair genes by in vivo profiling of repressive histone modifications of mature intact nerves. These studies revealed that trimethylation at Lys27 of histone H3 tail (H3K27me3) is often associated with promoters and/or gene bodies that become activated after injury. H3K27 methylation is catalyzed by polycomb repressive complex 2 (PRC2), consisting of protein methyltransferase EZH2 and the nonredundant core subunits, suppressor of zeste 12 (SUZ12) and embryonic ectoderm development (EED). H3K27 methylation represses transcription by recruiting other factors such as polycomb repressive complex 1 (PRC1) that in turn catalyzes ubiquitination of Lys119 of histone H2A tail (H2AK119Ub1), blocking transcriptional elongation. To investigate the function of PRC2 in peripheral nerves, we used a mouse model with a Schwann cell-specific deletion of Eed (Eed conditional knockout, Eed cKO). The deletion greatly reduced H3K27me3 of Schwann cells, but Schwann cell development and myelination proceeded normally with no apparent defects. At later timepoints, loss of Eed eventually disrupts the homeostasis of adult peripheral nerves and defects include hypermyelination of small diameter axons and structural abnormalities of myelinated fibers (Ma et al., 2015). Gene expression profiling of the Eed cKO nerves also led to significant dysregulation in gene expression, including derepression of a subset of injury-activated genes at young age when no apparent ultrastructural phenotype was observed. These data revealed that a subset of the injury program could be activated simply by removing polycomb repression. Consistent with these studies, ChIP assays revealed that injury causes demethylation of H3K27 and an increase of trimethylation of H3K4 (H3K4me3), a histone mark generally associated with active promoters (Ma et al., 2016). In addition, the reduced level of H3K27me3 at promoters and gene bodies of injury-induced genes in Eed cKO nerves led to premature and heightened activation of several well-known repair genes such as Shh, Gdnf, Vegfa and Bdnf after injury, suggesting that demethylation of H3K27 is a rate-limiting step for gene induction.
Histone methylation is reversible, and demethylases specific for H3K27, JMJD3/KDM6B and UTX/KDM6A, were previously identified. Interestingly, peripheral nerve injury increases the protein expression of JMJD3/KDM6B (Gomez-Sanchez et al., 2013). To test the role of demethylase activity in repair gene activation, we found that pharmacological inhibition of H3K27 demethylases prevented full activation of repair genes using a nerve explant model (Ma et al., 2016). Collectively, these data suggest that H3K27 demethylation is sufficient to activate some injury genes, but that it is also required for a broader cross-section of injury genes.
Identification of mechanisms involved in gene activation has been a critical part of the study of nerve regeneration. Our findings illustrate how Schwann cells become repair-promoting cells by altering chromatin structure in response to injury (Figure 1). However, these studies are still preliminary and we are testing if the premature and enhanced activation of polycomb-target genes eventually leads to earlier and improved functional recovery. In addition, there remain a number of open questions regarding the switch that leads to reversal of polycomb repression, and how it is coordinated with other Schwann cell changes. In addition, mechanisms of targeting of H3K27 demethylation remain to be elucidated, since studies in other systems have shown that Jmjd3 can associate with specific transcription factors. Ultimately, c-Jun controls a number of injury processes in Schwann cells through regulation of a relatively small subset of injury-induced genes (Arthur-Farraj et al., 2012), and it remains to be established which phenotypic aspects of the repair Schwann cell are controlled by regulation of H3K27 methylation and other chromatin remodeling pathways. Understanding the chromatin reprogramming of Schwann cells will hopefully contribute to optimized therapeutic management for sustainable expression of the repair genes that effectively promote peripheral nerve regeneration.
Figure 1.
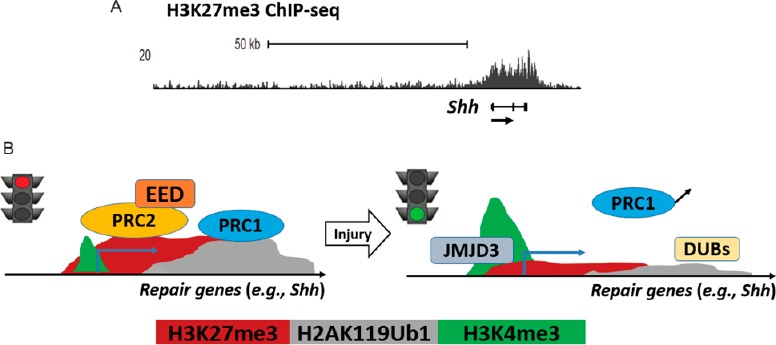
The polycomb pathway regulates repair genes of peripheral nerve injury.
(A) ChIP-seq mapping was performed in intact rat sciatic nerves and identified the H3K27me3-occupancy at repair genes such as sonic hedgehog (Shh). (B) A proposed model of epigenomic regulation after nerve injury. Repression of repair genes in Schwann cells is mediated by H3K27me3 and H2AK119Ub1 of the polycomb pathway and is reversed by the H3K27-demethylase JMJD3 and deubiquitinases (DUBs) after injury, followed by the induction of an active promoter mark H3K4me3.
References
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron. 2012;75:633–647. doi: 10.1016/j.neuron.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Gomis-Coloma C, Morenilla-Palao C, Peiro G, Serra E, Serrano M, Cabedo H. Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain. 2013;136:2262–2278. doi: 10.1093/brain/awt130. [DOI] [PubMed] [Google Scholar]
- Hung H, Kohnken R, Svaren J. The nucleosome remodeling and deacetylase chromatin remodeling (nurd) complex is required for peripheral nerve myelination. J Neurosci. 2012;32:1517–1527. doi: 10.1523/JNEUROSCI.2895-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung HA, Sun G, Keles S, Svaren J. Dynamic regulation of schwann cell enhancers after peripheral nerve injury. J Biol Chem. 2015;290:6937–6950. doi: 10.1074/jbc.M114.622878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The repair Schwann cell and its function in regenerating nerves. J Physiol. 2016;594:3521–3531. doi: 10.1113/JP270874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Svaren J, LaPash C, Araki T, Schmidt RE, Milbrandt J. Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci. 2005;8:932–940. doi: 10.1038/nn1490. [DOI] [PubMed] [Google Scholar]
- Lin HP, Oksuz I, Hurley E, Wrabetz L, Awatramani R. Microprocessor complex subunit DiGeorge syndrome critical region gene 8 (Dgcr8) is required for Schwann cell myelination and myelin maintenance. J Biol Chem. 2015;290:24294–24307. doi: 10.1074/jbc.M115.636407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma KH, Hung HA, Svaren J. Epigenomic regulation of schwann cell reprogramming in peripheral nerve injury. J Neurosci. 2016;36:9135–9147. doi: 10.1523/JNEUROSCI.1370-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma KH, Hung HA, Srinivasan R, Xie H, Orkin SH, Svaren J. Regulation of peripheral nerve myelin maintenance by gene repression through polycomb repressive complex 2. J Neurosci. 2015;35:8640–8652. doi: 10.1523/JNEUROSCI.2257-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MW, Brosius Lutz A, Cheng YC, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T, Woolf CJ. Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron. 2014;83:331–343. doi: 10.1016/j.neuron.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Sun G, Keles S, Jones EA, Jang SW, Krueger C, Moran JJ, Svaren J. Genome-wide analysis of EGR2/SOX10 binding in myelinating peripheral nerve. Nucleic Acids Res. 2012;40:6449–6460. doi: 10.1093/nar/gks313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassart RM, Fledrich R, Velanac V, Brinkmann BG, Schwab MH, Meijer D, Sereda MW, Nave KA. A role for Schwann cell-derived neuregulin-1 in remyelination. Nat Neurosci. 2013;16:48–54. doi: 10.1038/nn.3281. [DOI] [PubMed] [Google Scholar]