

Abstract
Chemical cross-linking coupled with mass spectroscopy (CXMS) provides proximity information for the cross-linked residues and is used increasingly for modeling protein structures. However, experimentally identified cross-links are sometimes incompatible with the known structure of a protein, as the distance calculated between the cross-linked residues far exceeds the maximum length of the cross-linker. The discrepancies may persist even after eliminating potentially false cross-links and excluding intermolecular ones. Thus the “over-length” cross-links may arise from alternative excited-state conformation of the protein. Here we present a method and associated software DynaXL for visualizing the ensemble structures of multidomain proteins based on intramolecular cross-links identified by mass spectrometry with high confidence. Representing the cross-linkers and cross-linking reactions explicitly, we show that the protein excited-state structure can be modeled with as few as two over-length cross-links. We demonstrate the generality of our method with three systems: calmodulin, enzyme I, and glutamine-binding protein, and we show that these proteins alternate between different conformations for interacting with other proteins and ligands. Taken together, the over-length chemical cross-links contain valuable information about protein dynamics, and our findings here illustrate the relationship between dynamic domain movement and protein function.
Keywords: calmodulin (CaM), mass spectrometry (MS), molecular dynamics, protein cross-linking, protein domain, structural model, protein dynamics, protein excited state
Introduction
Present-day structural biology largely focuses on the predominant ground-state structures of proteins, which are most populated and readily detectable. Nevertheless, it is becoming clear that a protein can transiently adopt alternative, often lowly populated excited-state conformations. Dynamic inter-conversion between protein ground and excited states, together constituting the ensemble structures of a protein, enables the protein to perform its function (1, 2). For a multidomain protein, the dynamics usually involve the rearrangement between the domains, which can be essential for ligand recognition, catalytic activity, and allosteric modulation of the protein (3, 4). Despite technical advances in X-ray crystallography (5), nuclear magnetic resonance (NMR) spectroscopy (6), and cryo-electron microscopy (7), the excited states of proteins remain difficult to characterize. Among the existing techniques, NMR spectroscopy is known for identifying the excited states and for elucidating protein dynamics (6). Yet NMR requires a large quantity of purified, isotopically enriched recombinant proteins and is mainly applicable to proteins <50,000 Da.
Chemical cross-linking of proteins coupled with mass spectrometry (CXMS)4 is an emerging technique in structural biology and has been increasingly used for modeling protein structures (8, 9). The cross-linked residues that are identified by high resolution mass spectroscopy should be close to each other, within the reach of the cross-linker used. However, it has been shown that the straight-line distance between the Cα atoms of cross-linked residues calculated from the known structure sometimes exceed the maximum length of the cross-linker (10, 11). The discrepancy between experimental cross-links and protein structures may be due to false identifications of the cross-links. Yet, even after eliminating erroneous assignments with stringent criteria (12), the discrepancy persists (13–16). As a common practice in protein structure modeling, the distance restraints derived from the over-length cross-links were often relaxed (17, 18) or even discarded (19).
Recently it has been shown that for protein-protein complexes, intermolecular cross-links incompatible with a unique complex structure may arise from some alternative arrangement(s) of the complex (11, 20). We thus reason that over-length intramolecular cross-links can be explained by the dynamic movement between different parts of the protein. Simple straight-line distance restraints derived from experimental cross-links has been used for modeling protein structures. However, as the cross-linker cannot go through the protein and can only go around the protein, intramolecular cross-links are better assessed with solvent-accessible surface (SAS) distances (21). The SAS distance for an intramolecular cross-link is usually longer than the straight-line Euclidean Cα-Cα distance for the two cross-linked residues, and therefore, the discrepancy between the calculated distance and maximum length of the cross-linker is even larger.
To account for all intramolecular cross-links identified with high confidence, here we present a method for assessing the conformational fluctuations of multidomain proteins (the associated software DynaXL is freely available from the author upon request). In this method we represent the cross-linkers with atomic details, and we model the protein alternative conformations with conjoined rigid body/torsion angle simulated annealing. We illustrate our method by characterizing the dynamics of three multidomain proteins, and we show how the domain arrangements of these proteins can enable their specific functions.
Results
The Workflow of DynaXL Modeling
A multidomain protein may dynamically interconvert between the ground state and the excited state, in which the domains have different arrangements. As illustrated in Fig. 1, the open conformation corresponds to the ground state, whereas the closed conformation corresponds to the excited state. For a protein existing only in an open conformation, the residues located in the opposite domains are too far away to be cross-linked despite that a mono-linked product can be readily formed. On the other hand, if the protein can transiently switch from the open conformation to the closed conformation, even for a short period of the time, the alternative conformation may permit the cross-linking reaction to complete. The residues that can react with the cross-linking reagent have to be appropriately located in the opposite domains. Thus the alternative conformation may not be efficiently captured. Conversely, if one or more over-length interdomain cross-links were identified by mass spectrometry with high confidence, it is likely that the predominant open state transiently fluctuates to some alternative conformation, as discussed below.
FIGURE 1.

Protein dynamics can be manifested from over-length cross-links, as illustrated here with the open-closed domain movement for a two-domain protein. A cross-linking reagent (e.g. BS3 or BS2G) first reacts with a lysine residue to form a mono-linked intermediate. The second conjugation reaction may only take place when the protein fluctuates to the alternative closed conformation.
We implemented this concept into our modeling approach. The workflow for the associated software DynaXL is illustrated in Fig. 2 and explained in detail with the analysis of calmodulin followed by two other examples (a snapshot of the software interface is shown in supplemental Fig. S1). DynaXL takes as input the known PDB structure of a protein and a list of high confidence intramolecular cross-links obtained from CXMS analysis on a 1:1 mixture of the 14N- and 15N-labeled proteins. Collecting CXMS data on this mixed sample, we were able to differentiate intramolecular versus intermolecular cross-links and also to eliminate false positives (see “Experimental Procedures” for details). Domain boundaries in multidomain proteins are delineated by multithreading alignment (22, 23) and validated with molecular dynamics (MD) simulations, which allows the definition of rigid bodies and the classification of intra- versus interdomain cross-link. The intradomain cross-links are examined against the known protein structures for compatibility, thus to confirm the rigidity of each domain. All interdomain cross-links involving rigid residues are used for characterizing protein ensemble structure that comprises the ground-state conformation and any additional excited-state conformation. If a two-conformer ensemble structure cannot account for all interdomain cross-links, DynaXL attempts a three-conformer ensemble structure and so on until all cross-links are satisfied.
FIGURE 2.

Schematic for characterizing protein ensemble structures based on CXMS data as implemented in DynaXL. The workflow of DynaXL includes four steps: CXMS analysis, preparation of the structure file, structure refinement with explicit modeling, and validation. Domain boundaries in multidomain proteins are delineated by multithreading alignment and validated with MD simulations, allowing the classification of intra- versus interdomain cross-link. The intradomain cross-links are examined against the known structures of the protein for compatibility so as to confirm the rigidity of each domain. Based on interdomain cross-links, protein ensemble structure was refined using conjoined rigid-body/torsion angle-simulated annealing. If a two-conformer ensemble structure cannot account for all interdomain cross-links, DynaXL attempts a three-conformer ensemble structure and so on until all cross-links are explained. Lastly, the alternative conformational states are subjected to further assessment and validation.
To account for intramolecular cross-links, it is better to use SAS distances (21). To this end, DynaXL treats the geometry of the cross-linker and the proximity between the reactive side chains with atomic details. With explicit modeling, the cross-linkers can only be located at protein surface. Due to the curvature of a protein, SAS distance restraints imposes a more stringent restraint than does a straight-line Cα-Cα distance restraint. To our knowledge, all previous modeling of protein structures from CXMS data used only straight-line Euclidean distance restraints applied to the Cα atoms (or Cβ atoms) of cross-linked residues (11, 13–16, 24, 25).
Identifying Intramolecular Cross-links with High Confidence
For proof of principle, we first visualized the conformational fluctuation of calcium-loaded calmodulin (Ca2+-CaM). CaM comprises an N-terminal domain and a C-terminal domain. In the absence of a ligand, Ca2+-CaM was found to exist in an open, extended conformation in crystal (26), with the two domains well separated (Fig. 3A).
FIGURE 3.
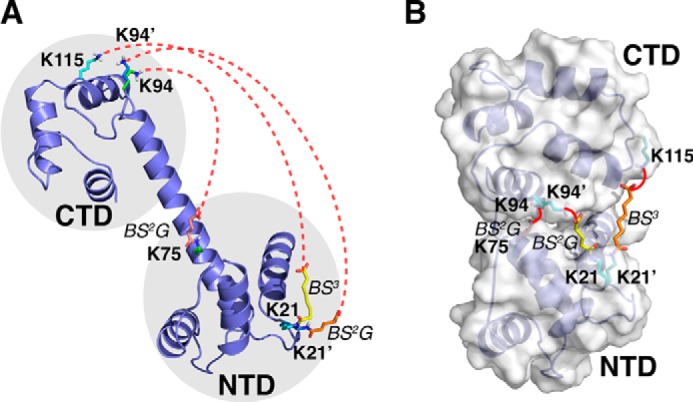
Ligand-free Ca2+-CaM can fluctuate between open and closed states. A, the interdomain cross-links (indicated by red dotted lines) involve residues too far away to be cross-linked in the open state. Note that Lys21 and Lys94 are each cross-linked with two different residues. For clarity, the N-hydroxysuccinimide ester at one end of the cross-linker and the Ca2+ ions bound to the protein are not shown. B, ensemble refinement with explicit modeling of the cross-linkers using the DynaXL approach revealed the closed-state structure of Ca2+-CaM, which would allow the cross-linking reaction to take place (indicated by red lines).
We performed CXMS experiments on an equimolar mixture of 14N-labeled (natural isotope abundance) and 15N-labeled Ca2+-CaM to exclude cross-links that may arise from protein homo-oligomers (20, 27). As illustrated in supplemental Fig. S2, an intramolecular cross-link should yield just two cross-link isoforms of equal abundance, one between two 14N-labeled light peptides (L1-L2) and one between two 15N-labeled heavy peptides (H1-H2). An intermolecular cross-link yields two additional isoforms each consisting of a 14N-labeled peptide and a 15N-labeled peptide (L1-H2 and H1-L2). Thus cross-links with a significant intermolecular component were eliminated, leaving only the intramolecular cross-links.
We performed CXMS experiments using amine-specific bis-sulfosuccinimidyl suberate (BS3) and a shorter cross-linking reagent bis-sulfosuccinimidyl glutarate (BS2G, supplemental Fig. S3) on different Ca2+-CaM protein samples. Filtering the data with stringent criteria to eliminate potentially false identifications (see “Experimental Procedures” for details), we identified eight intramolecular cross-links for ligand-free Ca2+-CaM with high confidence (supplemental Fig. S4 and Table S1). Some of the cross-links were only observed with BS3, indicating that the protein amine groups are likely too far away to react with the shorter BS2G (supplemental Table S1). The four intradomain cross-links agree with the respective domain structure (supplemental Fig. S5 and Table S1), confirming that the dynamics within each domain are small if present. The rigidity within each domain was further confirmed with MD simulations; except for N- and C-terminal residues and linker residues, most residues display backbone root mean square fluctuations of <2 Å (supplemental Fig. S6). In contrast, none of the four interdomain cross-links agree with the open structure of Ca2+-CaM, even with the flexibility of the explicitly modeled lysine side chains and attached cross-linker considered (Fig. 3A).
Modeling the Closed State of Ligand-free Ca2+-CaM
To account for the over-length interdomain cross-links, we introduced a second conformer of Ca2+-CaM and refined the ensemble structure based on the three interdomain cross-links involving rigid lysine residues. The interdomain cross-link Ala1-Lys94 was not used in the modeling due to the flexibility of the N terminus. We grouped the N-terminal domain (NTD, residues 4–77) and the C-terminal domain (CTD, residues 82–147) as rigid bodies connected by a polypeptide linker (residues 78–81). The arrangement between NTD and CTD was optimized using the DynaXL approach, thus allowing the interdomain cross-linking reactions to take place. Based on the cross-links identified with high confidence, we obtained the alternative closed conformation of ligand-free Ca2+-CaM (Fig. 3B and supplemental Table S2). The closed-state structures are highly converged, with an overall r.m.s. deviation of 1.08 ± 0.35 Å for backbone heavy atoms (supplemental Fig. S7). The closed-state structures obtained can also be assessed with spherical coordinates. With respect to the open structure, the vector from the center of mass of the NTD to the center of mass of the CTD rotates by 58.5 ± 5.1° and shortens from 37.5 Å to 23.8 ± 0.7 Å (supplemental Fig. S8, A and C).
To ensure complete sampling of the conformational space, we also started the refinement with the domains in random relative positions. The resulting structures for the closed state are almost identical to those calculated from the open-state structure of Ca2+-CaM (supplemental Fig. S8, B and D), consistent with the expectation that the correct structure should converge irrespective of the starting conformation. For the cross-linking reaction, a cross-linker can be first attached to either domain to form a mono-linked intermediate before being cross-linked to another domain. Accordingly, we modeled the cross-linkers to either the NTD or CTD, which yielded almost identical structures (supplemental Fig. S9). Because a two-conformer ensemble already satisfied all cross-links, introducing an additional conformer brought no improvement. To illustrate how DynaXL can be used to model larger ensembles, we added two artificial cross-links. The new restraint list can no longer be satisfied with a two-conformer ensemble but can be accounted for with a three-conformer ensemble in which the two conformers adopt closed states with distinct domain arrangements (supplemental Fig. S10).
As such, we have shown that the DynaXL approach allows the modeling of the alternative closed state of ligand-free Ca2+-CaM. The structural convergence is due to the explicit modeling of the cross-linker, to the conjoined rigid-body/torsion angle refinement approach, and to the self-consistency of CXMS restraints. Indeed, refining against any one of the interdomain CXMS restraints can already lead to a closed-state structure for the ligand-free Ca2+-CaM. Although dispersed, the centers of mass of the resulting structures are similar to each other and to those refined with all three interdomain cross-links (supplemental Fig. S11, A–C). The structures become more converged when refining with two interdomain cross-links (supplemental Fig. S11, D–F) because the alternative structure has to satisfy both restraints at the same time. Importantly, the CXMS restraints can be cross-validated (the free reactive end of the mono-linked intermediate is found in close proximity to the target lysine side chain in the opposite domain provided that the other two cross-links have been formed (supplemental Table S2). Moreover, the ensemble structure can also account for the Ala1-Lys94 cross-link, which was not used in the refinement (supplemental Fig. S12). Taken together, the DynaXL approach reveals that the ligand-free Ca2+-CaM transiently adopts a closed state, and the cross-linking captures and stabilizes this preexisting excited-state conformation.
Validating the Closed-state Structure of Ligand-free Ca2+-CaM
The ensemble structure of Ca2+-CaM can also be validated with other techniques. Previous NMR studies have shown that ligand-free Ca2+-CaM transiently adopts a closed conformation, and the excited state is present for only ∼5% of the time (28–30). Using paramagnetic relaxation enhancement (PRE) data reported with a probe attached at S17C site (30), we calculated the closed-state structure for ligand-free Ca2+-CaM following the established protocol (supplemental Fig. S13A) (31). The PRE and CXMS structures are similar to each other (supplemental Fig. S13B), and the backbone r.m.s. difference was as small as 1.66 Å (Fig. 4A). The structural similarity further attests that CXMS can detect and visualize the lowly populated excited states of proteins, a task that has been mostly reserved for NMR (6).
FIGURE 4.
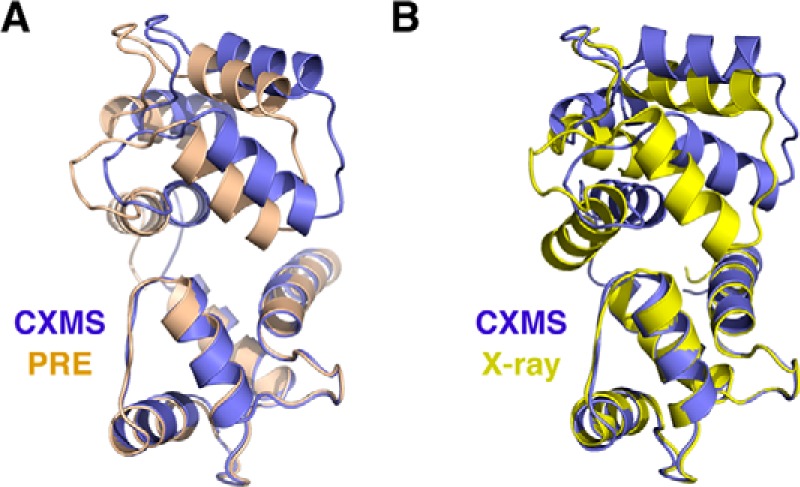
Assessment of the closed-state structure of ligand-free Ca2+-CaM. A, comparison to the closed-state structure obtained by refining against the PRE NMR data. B, comparison to the closed-state structure of ligand-bound Ca2+-CaM, which was previously determined using X-ray crystallography (PDB code 2BE6).
There are a number of structures determined for ligand-bound Ca-CaM2+, and the exact domain arrangement varies depending on the bound ligand (supplemental Fig. S14A). The closed-state structures calculated for ligand-free Ca2+-CaM based on the CXMS data and the closed-state structures for ligand-bound Ca2+-CaM are similar (supplemental Fig. S14A), and the smallest r.m.s. difference is 1.93 Å as referenced to the complex structure bound to an IQ-domain from voltage-gated calcium channel (Fig. 4B) (16). Thus, a cognate ligand of Ca2+-CaM can stabilize the closed-state structure already present for ligand-free Ca2+-CaM, which is characteristic of a conformational selection mechanism (supplemental Fig. S15).
Visualizing the Phosphoryl-receiving Conformation of an Enzyme I Domain
To further evaluate our DynaXL method for modeling protein excited states, we assessed the dynamics of the N-terminal domain of enzyme I. Enzyme I (EI), a bacterial phosphotransferase, comprises an N-terminal domain (EIN) and a C-terminal domain (EIC) (32). In the presence of phosphoenol pyruvate (PEP), EIN is autophosphorylated by EIC (32, 33). In turn, EIN transfers the phosphoryl group to the downstream partner HPr (34). EIN itself comprises two domains, the α/β domain (residues 1–20 and residues 148–231) and the α domain (residues 24–142), which can be defined from computational analysis (22) and by inspecting the known structures.
Using high resolution mass spectrometry, we identified 13 intramolecular cross-links with high confidence for EIN (supplemental Table S3). All seven intradomain cross-links agree with the ground-state structure of either EIN (PDB code 1ZYM) by itself or EIN in complex with HPr (PDB code 3EZA), which confirms the rigidity of each domain. In contrast, for two of the six interdomain cross-links, Lys49-Lys20 and Lys49-Lys175 (supplemental Fig. S16), the theoretical distances calculated from the ground-state structure of EIN far exceed the maximum lengths of the corresponding cross-linkers, even with the flexibility of the cross-linkers considered (Fig. 5A). Therefore, these interdomain cross-links may arise from an excited state of EIN in which the α/β domain and the α domain are arranged differently.
FIGURE 5.
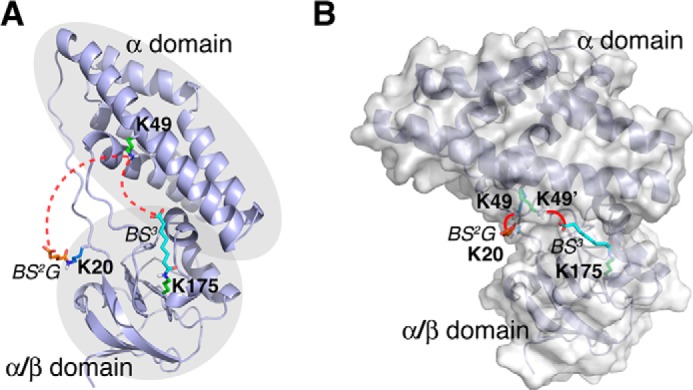
Visualizing the conformational fluctuations of EIN from over-length cross-links. A, the structure of EIN either by itself or in complex with HPr cannot explain the two cross-links between α-domain and α/β-domain (indicated by red dotted lines). B, modeled from interdomain cross-links, EIN is found to exist in an alternative conformation, likely responsible for phosphoryl receiving. This conformational state allows BS2G cross-link between Lys20 and Lys49 and the BS3 cross-link between Lys175 and Lys49 (indicated with red lines) to be formed.
Using the DynaXL approach, we found a two-conformer representation for EIN that can account for all intramolecular cross-links (supplemental Table S4). In addition to the predominant conformation responsible for phosphoryl transferring, we identified an alternative conformation of EIN and determined its structure to convergence, with backbone r.m.s. deviations of 2.01 ± 1.11 Å (supplemental Fig. S17). In the second conformation, the EIN α domain is tilted to one side of the α/β domain (Fig. 5B). This rearrangement would expose the active-site residue His189 in the α/β domain and possibly allows His189 to be phosphorylated by EIC. Indeed, such an alternative conformation of EIN has been found in the full-length enzyme I protein in crystal bound with an inhibitor (32) or in solution with a point mutation introduced (33). The r.m.s. difference between the alternative conformation of EIN modeled with DynaXL and the structure of EIN in the full-length enzyme I is as small as 1.74 Å (supplemental Fig. S18). As such, EIN is inherently dynamic with or without EIC, and the dynamics can be functionally important for the relaying of the phosphoryl group (supplemental Fig. S19).
Characterizing a Partially Closed Conformation of ApoQBP
Lastly, we assessed the dynamic domain movement of glutamine-binding protein (QBP). QBP, a bacterial periplasmic solute binding protein, comprises two domains, domain I (residues 1–86 and residues 185–226) and domain II (residues 90–180), which are connected by two shorter linkers (35). The domains can be defined by automatic threading approach (22) or by comparing ligand-free and ligand-bound structures of QBP. In the absence of the ligand, QBP adopts an open conformation in crystal (35). Upon glutamine binding, the two domains undergo a hinged rotation for 37.6° (supplemental Fig. S20A) (36).
We identified 35 intramolecular cross-links with high confidence for apoQBP (supplemental Table S5). All intradomain cross-links agree with the crystal structure, which confirms that the domain structures are rigid regardless of the domain movement. Among the interdomain cross-links, however, Lys76-Lys125 and Lys77-Lys125 cannot be accounted for by the open structure of apoQBP (supplemental Fig. S21 and Fig. 6A). The over-length cross-links are unlikely to arise from the contamination of holoQBP for these reasons: first, any bound glutamine would have been washed off after several rounds of denaturation, dialysis, and renaturation during sample preparation; second, if the apoQBP sample had been contaminated with holoQBP, we expected to see two BS2G cross-links that were specific for holoQBP, Lys77–Lys125 and Lys87–Lys218 (supplemental Fig. S22), but we did not.
FIGURE 6.
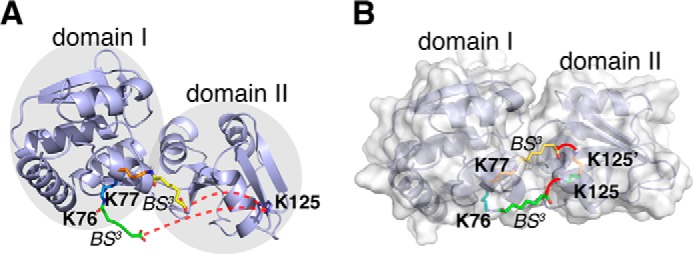
Visualizing the conformational fluctuations of apoQBP from MS-identified cross-links. A, the apoQBP exists in an open conformation and cannot account for the two interdomain cross-links (denoted by red dotted lines). B, with the DynaXL modeling, apoQBP is found to exist in a partially closed conformation, which enables the cross-linking reactions between Lys76 and Lys125 and between Lys77 and Lys125 (indicated with red lines).
Using the DynaXL modeling approach, we obtained a two-conformer ensemble structure for apoQBP that accounts for all 12 high confidence interdomain cross-links. The second conformation of apoQBP can explain the two cross-links incompatible with the open structure of apoQBP, and the cross-links can in part be cross-validated with the new structure (supplemental Table S6). Interestingly, the second conformation of apoQBP displays a rotation of ∼26.8° for the two domains (Fig. 6B) and, therefore, is only partially closed with respect to the holoQBP structure (supplemental Fig. S20B). Such a partially closed state can be corroborated with small angle X-ray scattering (SAXS) analysis, as the ensemble comprising the open and partially closed structures but not the ensemble comprising the open and fully closed structures can best account for the experimental data (supplemental Fig. S23). Being partially closed yet still accessible to a ligand, the alternative structure of apoQBP may facilitate the transition to the fully closed structure and enable specific ligand recognition, as have been demonstrated for other periplasmic binding proteins (3, 30).
Discussion
Cross-linking coupled with mass spectrometry has been increasingly used for protein structure modeling. Quite often it has been found that a subset of the highly reliable cross-links cannot be explained by the known structure of the protein or protein complex, or these cross-links are inconsistent with other cross-links or other experimental data (12, 13, 17, 18). In those cases, the over-length cross-links were often discarded or the corresponding distance restraints were relaxed. Here we show that the over-length cross-links are treasure, not trash, containing valuable information about protein dynamics.
Demonstrated with three multidomain proteins, Ca2+-CaM, QBP, and EIN, we have shown that the otherwise incompatible cross-links manifest protein-excited states. The over-length cross-links allowed us to model the transiently closed conformation of ligand-free Ca2+-CaM with a population of only ∼5% (30). The CXMS also enables the depiction of a partially closed conformation of apoQBP. For EIN, the alternative conformation should have a low occupancy and could not be observed without the addition of an inhibitor or introduction of a point mutation in the past. Thus we show that the CXMS is highly sensitive to the lowly populated conformational states of proteins.
CXMS-based structural modeling has been explored using various software tools, yet a lot of these studies aimed for a single structure that had the lowest energy and best satisfied the experimental data (11, 13, 24, 25). To fully account for the intramolecular cross-links, we developed the DynaXL method. In this method we invoke ensemble representation for the protein, and we explicitly model the cross-linkers and cross-linking reactions. The explicit modeling approach not only imposes a realistic distance limit for each cross-link, but also takes into account the flexibility of the cross-linker and the van der Waals interactions between the cross-linker and the protein.
The success of the DynaXL method is due to several factors. First, the conjoined rigid-body/torsion angle refinement approach narrows the conformational search space and permits efficient convergence to the structure that best accounts for all experimental data. Second, with the cross-linkers and cross-linked residues explicitly modeled and with flexible linker residues opting for certain rotamers (37), the conformational space possibly adopted by the protein-excited state is further narrowed. Third, the cross-links identified with high confidence are partially redundant. For cross-validation, therefore, it is better to have at least two over-length, but highly reliable interdomain cross-links between every two domains in a protein.
DynaXL method is not limited to the characterization of the more compact state over the ground state. As we show with the example of EIN, any alternative conformation with different domain arrangement can in theory be detected as long as the cross-links involve residues incompatible with the ground-state structure. In addition, DynaXL may also be used to visualize local protein dynamics, as we show for the N-terminal tail of Ca2+-CaM (supplemental Fig. S9). Nevertheless, depending on the accessibility and reactivity of the side chains, not all the reactive residues that are within the reach the cross-linkers are experimentally identified, and the cross-linking restraints are sparse. As a result, certain excited state(s) may not be captured with CXMS.
Besides CXMS, other MS techniques such as hydrogen/deuterium exchange and ion mobility mass spectrometry have also been used to characterize dynamic protein-protein interactions (38) and protein domain movement (39, 40). As CXMS uncovers lowly populated conformers that only have been captured with sensitive NMR techniques, CXMS is probably more sensitive than other MS techniques in depicting protein excited-state structure. Unlike NMR, CXMS is not limited by the size of the protein. Thus the DynaXL method can be used to depict the ensemble structures of large multidomain proteins. Together, we envision that DynaXL will be used as a general tool either by itself or in conjunction with other techniques for visualizing protein dynamics.
Experimental Procedures
Sample Preparation
The ligand-free human Ca2+-CaM, the N-terminal domain of Escherichia coli enzyme I (EIN, residues 1–249), and E. coli periplasmic QBP were prepared as described (41–43). The 15N-labeled recombinant proteins were purified from bacterial cells grown in M9 minimum medium with U-15NH4Cl (Cambridge Isotope Laboratory, Tewksbury, MA) as the sole nitrogen source. To remove bound glutamine, 2 m guanidine hydrochloride was added to purified QBP, and the partially denatured protein was desalted in the presence of 2 m guanidine, which was later removed with a second desalting step to allow the apoQBP to refold.
Cross-linking reactions were performed for 0.6 μg/μl protein at room temperature for 1 h in 20 mm HEPES, pH 7.2, buffer containing 0.5 mm BS3 or BS2G (Thermo Scientific, Waltham, MA) and were quenched after 1 h with 20 mm NH4HCO3. The cross-linked proteins were precipitated with ice-cold acetone, air-dried, and resuspended in 100 mm Tris, pH 8.5, buffer containing 8 m urea.
CXMS Experiments
Subsequent to trypsin digestion, LC-MS/MS analysis was performed on an Easy-nLC 1000 UPLC (Thermo Scientific) coupled with a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific). Peptides were loaded on a precolumn (75-μm inner diameter, 8 cm long, packed with ODS-AQ 12 nm–10-μm beads from YMC Co., Kyoto, Japan) and were separated on an analytical column (75-mm inner diameter, 11 cm long, packed with Luna C18 100 Å, 1.8-μm resin from Welch Materials, Austin, TX) using an acetonitrile gradient from 0 to 28% in 60 min at a flow rate of 200 nl/min. The top 10 most intense precursor ions from each full scan (resolution 70,000) were isolated for HCD MS/MS scans (resolution 17,500; normalized collision energy 27) with a dynamic exclusion time of 20 s. Precursors with 1+, 2+, >6+, or unassigned charge states were excluded. Each cross-linking reaction was performed twice (two biological repeats), and each CXMS sample was analyzed twice on the LC-MS/MS (two technical repeats). A database search using Prolucid (44) against E. coli (BL21) protein sequences was performed. Spectra of mono-links as well as peptides not modified by cross-linkers were removed.
Identification of High Confidence Intramolecular Cross-links
We performed CXMS experiments on samples containing equal molar amounts of unlabeled (i.e. 14N-labeled) and 15N-labeled proteins in order to exclude intermolecular cross-links, based on the strategy previously described (20, 27, 45). The MS/MS spectra of L/L cross-links, in which both peptides were 14N-labeled, were identified using pLink (46). CXMS analysis of a 1:1 mixture of the 14N- and 15N-labeled protein allowed us to establish a set of criteria to eliminate false positives. False positives were recognized because they lacked the characteristic peak pair of L1-L2 and H1-H2 in MS1 spectra. In the case of Ca2+-CaM, we found 4 such falsely identified cross-links after filtering the results with a false discovery rate of 0.05 at the spectrum level followed by an E-value cutoff of 10−3. Their best E-values were in the range of 10−8–10−7 (not shown). Therefore, we updated the pLink filtering criteria as follows: false discovery rate <0.05, E-value <10−3, spectral count ≥2, and the best E-value <10−8 for each cross-link identification. Furthermore, for each intramolecular cross-link, we required the precursor intensity ratios of the L/H (or H/L) isoform over the L/L (or H/H) isoform to be <0.14, which were computed using pQuant (47). At this ratio, the intramolecular contribution is at least three times as much as the intermolecular contribution. This approach is superior to CXMS analysis on the monomeric bands from SDS-PAGE, as we obtained many more high quality intramolecular cross-links.
Domain Boundaries Definition
Domain boundaries are automatically defined with multiple threading alignments using Threadom (22). The domain definition can also be achieved by superimposing the available structures for the protein crystallized under different conditions; the segments with smallest r.m.s. differences should belong to the same domain. The domain definitions used are: NTD, (the CaM comprises a N-terminal domain, residues 4–77), CTD (C-terminal domain, residues 82–147), EIN (comprises the α/β domain (including discontinuous segments of residues 3–20 and residues 148–249) and the α domain (residues 24–142)), and QBP, comprises domain I (including two discontinuous segments of residues 5–86 and residues 185–224) and domain II (residues 90–180)). Note that the terminal residues are missing and are likely highly dynamic in the crystal structures of ligand-free Ca2+-CaM (residues 1–3 and 148), EIN (residues 1–2), and apoQBP (residues 1–4 and 225–226), with the PDB codes 1CLL (26), 1ZYM (48), and 1GGG (35), respectively.
The domain definitions were validated with molecular dynamics simulations using AMBER software package with ff12SB force field in the Amber14 package (49); the r.m.s. fluctuations are small within each domain and are much larger between the domains. The starting structures for ligand-free Ca2+-CaM, apoQBP, and EIN are known PDB structures. To illustrate local flexibility and to calculate theoretical Cα-Cα distances, residues missing in the crystal structures were patched up. The protein was solvated with a cube containing TIP3P water molecules with a 10 Å padding in each direction For CaM, the Ca2+ ions were patched using the force field parameters in AMBER. Long range electrostatics were treated with the Particle Mesh Ewald method (50), and van der Waals term was truncated at 10 Å with energy shift. The temperature was kept in 298 K, and the time step was 2 fs. The simulations were run for 100 ns.
Explicit Modeling of the Cross-linker
The amino-specific cross-linkers BS2G and BS3 cross-link two lysine residues spaced <20.3 Å and <24.0 Å apart (21, 51) or a protein N terminus and a lysine residue spaced <14.9 Å and <18.8 Å apart, respectively, as measured with a straight line between two Cα atoms. BS2G and BS3 cross-linkers were drawn in PyMOL (52) and were energy-minimized for bond lengths and bond angles. Note that protein N-terminal residue is usually dynamic, and therefore, the local flexibility should also be accounted for in addition to the flexibility of the cross-linker.
The cross-linker was first patched to one of the lysine residues (to form mono-linked intermediate) via isopeptide bond. The lysine side chains and the cross-linkers were given full torsion angle freedom. To allow the isopeptide formation between the free reactive end of the cross-linker in the mono-linked intermediate and the side chain of another lysine residue, i.e. to form the cross-linked product, the carboxylate group and the amine group should be in close proximity to allow the nucleophilic attack to occur, 5.0 Å for the upper limit (1.2× that of the van der Waals radii of nitrogen and carbon atoms, a distance at which the Leonard-Jones potential remains in the negative regime) and 1.3 Å for the lower distance limit (i.e. the C-N covalent bond length). No energy penalty is given if the distance between the carboxylate carbon and the amine nitrogen is within 1.3–5.0 Å.
If a lysine residue is highly reactive and is found cross-linked to multiple lysine residues, its side chain as well as the patched cross-linker is replicated. The different copies of the lysine residue and the attached cross-linker are allowed to overlap, thus to representing the different possibilities. The compatibility between intradomain cross-links and the structure of each domain was evaluated to make sure that domains were correctly defined.
Ensemble Structure Refinement
Structural refinement was performed with conjoined rigid body/torsion angle-simulated annealing invoking Xplor-NIH (53). The PDB structures 1CLL (26), 1ZYM (48), and 1GGG (35) were used as the starting coordinates for Ca2+-CaM, EIN, and QBP, respectively. The residues within the domain were grouped as rigid bodies in the refinement, and complete torsion angle freedom was given to the polypeptide linker residues between the rigid domains, the mono-linked lysine residues, the covalently conjugated cross-linkers, and the lysine residues to be cross-linked. In theory, our method could also permit local flexibility to be introduced, but that has to be supported by MD simulations, by detailed knowledge of the protein, and by other experimental evidence.
Besides the interdomain CXMS restraints for modeling the cross-linking reactions, the restraint list also included covalent energy terms (including bond, angle and impropers), van der Waals repulsive term (to avoid unfavorable steric clashes) applied to the entire protein and to the cross-linkers, and a torsion angle database potential term (37) applied to the polypeptide linkers between rigid domains and to the lysine side chains. The CXMS energy term was defined as kΔ2, in which Δ was the deviation from the upper or lower distance limits (mostly applied when the distance >5Å, as the lower bound is unlikely to be reached before van der Waals repulsive term builds up), and k was the force constant ramped from 1 to 100 kcal·mol−1·Å−2 as the temperature gradually decreased from 3000 K to 25 K. If both BS2G and BS3 cross-links were identified, only the cross-linking reaction of BS2G was modeled.
The ensemble structure comprises two or more conformers that collectively account for the CXMS data. A CXMS restraint is satisfied as long as it is accounted for by one member structure. Using ligand-free Ca2+-CaM as an example, the ensemble structure comprises an open conformation (PDB code 1CLL, fixed during the refinement) and an alternative conformation (to be refined against the CXMS and other restraints). In the refinement, the NTD of Ca2+-CaM was fixed, whereas the CTD was grouped as a rigid body connected to NTD via a polypeptide linker (residues 78–81, excluding the flexible termini and the rigid domains), and was allowed to reorient relative to the NTD. The refinement started either from the open structure or from a randomized structure, with random translational and rotational operations applied to the CTD as well as the linker residues. Alternatively, the CTD of Ca2+-CaM could be fixed, and relative arrangement of the mono-linked NTD was optimized. The refinement was repeated many times with different random seeds. Multiple conformers would be included into the ensemble structure if necessary, until all interdomain cross-links were accounted for. Structures with neither steric clashes nor violation against any CXMS or covalent terms were selected for further analysis. For cross-validation of the CXMS restraints, the refinement was performed using a subset of the CXMS restraints.
The same refinement protocol was used to characterize the ensemble structures of apoQBP and EIN. The ground-state structure was fixed during the refinement, and the excited-state structure with alternative domain arrangement was determined, which accounts for the interdomain cross-links otherwise inexplicable by the ground-state structure. The members of the ensemble structure were allowed to overlap, and the CXMS restraints were to be satisfied by any conformer in the ensemble. The ensemble refinement method based on the CXMS data was implemented in DynaXL software written specifically developed for this purpose (freely available online).
Assessment of the Ensemble Structures
The ensemble structures obtained from experimental cross-links could be cross-validated using a subset of the cross-links, assessed against alternative structures of the protein with either ligand bound or mutation introduced, or validated with other orthogonal techniques, such as PRE and SAXS. The interdomain PRE data for ligand-free Ca2+-loaded CaM with a paramagnetic probe (nitroxide spin radical) conjugated at the S17C site was obtained from the literature (30). With an effective correlation time of 9.9 ns (30), the intradomain PRE data were used to refine the distribution of the paramagnetic probe using a three-conformer representation, whereas the interdomain PRE data were used for the refinement of the closed structure at different occupancies (from 1% to 10% in 1% increment). The domains were grouped as rigid bodies, and the linker residues were subjected to torsion angle dynamics. The refinement was performed with the established protocol (3), and the ensemble structures for ligand-free Ca2+-CaM could be reproduced from the published work (30). It was found that a 5% occupancy and a 2-conformer ensemble could best account for the PRE data, which is in good agreement with the literature (30).
The ensemble structure of apoQBP was also assessed with SAXS. The apoQBP protein was prepared in 20 mm, pH 7.2, HEPES buffer with 100 mm NaCl. The data were collected under two different temperatures (40 °C and 50 °C) at the National Centre for Protein Science Shanghai with the BL19U2 beamline. A total of 20 consecutive frames of 1-s exposure time for each were recorded and were averaged, with no difference seen between consecutive frames. Similarly, the scattering profiles of the buffer were recorded for background subtraction. Protein radius of gyration (Rg) was calculated using software PRIMUS (54) with Guinier approximation at the low scattering angle with q × Rg ≤1. The paired-distance P(r) distribution profiles were obtained by indirect Fourier transformation of the I(q) scattering profiles.
Author Contributions
Y.-H. D. conducted the MS experiments. Z. G. and X. D. performed the calculations. K. L. wrote the DynaXL software interface. Z. L. performed the SAXS experiments. C. L. and S.-M. H. modified the pLink program for the analysis of isotopically mixed sample. M.-Q. D. and C. T. conceived the project and analyzed the data. All the authors contributed to the writing of the manuscript.
Acknowledgments
We thank Dr. Na Li and the staff at beamline BL19U2 of Shanghai Synchrotron Radiation Facility for assistance with SAXS data collection and John Hugh Snyder for language editing.
This work was supported by the Chinese Ministry of Science and Technology (2013CB910200, 2016YFA0501200 (to C. T.), 2014CB849800 (to M.-Q. D.), and 2012CB910602 (to C. L.)) and the National Natural Science Foundation of China (31225007 to (to C. T.), 21375010 (to M.-Q. D.), 31400735 (to Z. G.), and 31400644 (to X. D.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S6 and Figs. S1–S23.
- CXMS
- chemical cross-linking coupled with mass spectrometry
- SAXS
- small angle X-ray scattering
- SAS
- solvent-accessible surface
- Ca2+-CaM
- calcium-loaded calmodulin
- PRE
- paramagnetic relaxation enhancement
- EIN
- the N-terminal domain of bacterial phosphotransferase enzyme I
- EIC
- the C-terminal domain of bacterial phosphotransferase enzyme I
- QBP
- bacterial periplasmic glutamine binding protein
- BS3
- bis-sulfosuccinimidyl suberate
- BS2G
- bis-sulfosuccinimidyl glutarate
- MD
- molecular dynamics
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- r.m.s.
- root mean square.
References
- 1. Henzler-Wildman K., and Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]
- 2. Boehr D. D., Nussinov R., and Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tang C., Schwieters C. D., and Clore G. M. (2007) Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 449, 1078–1082 [DOI] [PubMed] [Google Scholar]
- 4. Bouvignies G., Vallurupalli P., Hansen D. F., Correia B. E., Lange O., Bah A., Vernon R. M., Dahlquist F. W., Baker D., and Kay L. E. (2011) Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature 477, 111–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fraser J. S., van den Bedem H., Samelson A. J., Lang P. T., Holton J. M., Echols N., and Alber T. (2011) Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 108, 16247–16252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sekhar A., and Kay L. E. (2013) NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc. Natl. Acad. Sci. U.S.A. 110, 12867–12874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fernandez-Leiro R., and Scheres S. H. (2016) Unravelling biological macromolecules with cryo-electron microscopy. Nature 537, 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jennebach S., Herzog F., Aebersold R., and Cramer P. (2012) Crosslinking-MS analysis reveals RNA polymerase I domain architecture and basis of rRNA cleavage. Nucleic Acids Res. 40, 5591–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Politis A., Stengel F., Hall Z., Hernández H., Leitner A., Walzthoeni T., Robinson C. V., and Aebersold R. (2014) A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods 11, 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merkley E. D., Rysavy S., Kahraman A., Hafen R. P., Daggett V., and Adkins J. N. (2014) Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 23, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferber M., Kosinski J., Ori A., Rashid U. J., Moreno-Morcillo M., Simon B., Bouvier G., Batista P. R., Müller C. W., Beck M., and Nilges M. (2016) Automated structure modeling of large protein assemblies using crosslinks as distance restraints. Nat. Methods 13, 515–520 [DOI] [PubMed] [Google Scholar]
- 12. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Förster F., Beck M., and Aebersold R. (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 9, 901–903 [DOI] [PubMed] [Google Scholar]
- 13. Lössl P., Kölbel K., Tänzler D., Nannemann D., Ihling C. H., Keller M. V., Schneider M., Zaucke F., Meiler J., and Sinz A. (2014) Analysis of nidogen-1/laminin gamma1 interaction by cross-linking, mass spectrometry, and computational modeling reveals multiple binding modes. PLoS ONE 9, e112886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalisman N., Adams C. M., and Levitt M. (2012) Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U.S.A. 109, 2884–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lasker K., Förster F., Bohn S., Walzthoeni T., Villa E., Unverdorben P., Beck F., Aebersold R., Sali A., and Baumeister W. (2012) Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi Y., Fernandez-Martinez J., Tjioe E., Pellarin R., Kim S. J., Williams R., Schneidman-Duhovny D., Sali A., Rout M. P., and Chait B. T. (2014) Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol. Cell. Proteomics 13, 2927–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F. U., Ban N., Malmström L., and Aebersold R. (2012) Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352 [DOI] [PubMed] [Google Scholar]
- 18. Erzberger J. P., Stengel F., Pellarin R., Zhang S., Schaefer T., Aylett C. H., Cimermančič P., Boehringer D., Sali A., Aebersold R., and Ban N. (2014) Molecular architecture of the 40SeIF1eIF3 translation initiation complex. Cell 158, 1123–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plaschka C., Larivière L., Wenzeck L., Seizl M., Hemann M., Tegunov D., Petrotchenko E. V., Borchers C. H., Baumeister W., Herzog F., Villa E., and Cramer P. (2015) Architecture of the RNA polymerase II-Mediator core initiation complex. Nature 518, 376–380 [DOI] [PubMed] [Google Scholar]
- 20. Gong Z., Ding Y.-H., Dong X., Liu N., Zhang E. E., Dong M.-Q., and Tang C. (2015) Visualizing the ensemble structures of protein complexes using chemical cross-linking coupled with mass spectrometry. Biophys. Rep. 1, 127–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kahraman A., Malmström L., and Aebersold R. (2011) Xwalk: computing and visualizing distances in cross-linking experiments. Bioinformatics 27, 2163–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xue Z., Xu D., Wang Y., and Zhang Y. (2013) ThreaDom: extracting protein domain boundary information from multiple threading alignments. Bioinformatics 29, i247–i256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Belsom A., Schneider M., Fischer L., Brock O., and Rappsilber J. (2016) Serum albumin domain structures in human blood serum by mass spectrometry and computational biology. Mol. Cell. Proteomics 15, 1105–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kahraman A., Herzog F., Leitner A., Rosenberger G., Aebersold R., and Malmström L. (2013) Cross-Link Guided Molecular Modeling with ROSETTA. PLoS ONE 8, e73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rappsilber J. (2011) The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol. 173, 530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chattopadhyaya R., Meador W. E., Means A. R., and Quiocho F. A. (1992) Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 228, 1177–1192 [DOI] [PubMed] [Google Scholar]
- 27. Taverner T., Hall N. E., O'Hair R. A., and Simpson R. J. (2002) Characterization of an antagonist interleukin-6 dimer by stable isotope labeling, cross-linking, and mass spectrometry. J. Biol. Chem. 277, 46487–46492 [DOI] [PubMed] [Google Scholar]
- 28. Bertini I., Del Bianco C., Gelis I., Katsaros N., Luchinat C., Parigi G., Peana M., Provenzani A., and Zoroddu M. A. (2004) Experimentally exploring the conformational space sampled by domain reorientation in calmodulin. Proc. Natl. Acad. Sci. U.S.A. 101, 6841–6846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bertini I., Kursula P., Luchinat C., Parigi G., Vahokoski J., Wilmanns M., and Yuan J. (2009) Accurate solution structures of proteins from X-ray data and a minimal set of NMR data: calmodulin-peptide complexes as examples. J. Am. Chem. Soc. 131, 5134–5144 [DOI] [PubMed] [Google Scholar]
- 30. Anthis N. J., Doucleff M., and Clore G. M. (2011) Transient, sparsely populated compact states of apo and calcium-loaded calmodulin probed by paramagnetic relaxation enhancement: interplay of conformational selection and induced fit. J. Am. Chem. Soc. 133, 18966–18974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iwahara J., Tang C., and Marius Clore G. (2007) Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 184, 185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teplyakov A., Lim K., Zhu P. P., Kapadia G., Chen C. C., Schwartz J., Howard A., Reddy P. T., Peterkofsky A., and Herzberg O. (2006) Structure of phosphorylated enzyme I, the phosphoenolpyruvate:sugar phosphotransferase system sugar translocation signal protein. Proc. Natl. Acad. Sci. U.S.A. 103, 16218–16223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Venditti V., Schwieters C. D., Grishaev A., and Clore G. M. (2015) Dynamic equilibrium between closed and partially closed states of the bacterial enzyme I unveiled by solution NMR and X-ray scattering. Proc. Natl. Acad. Sci. U.S.A. 112, 11565–11570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garrett D. S., Seok Y. J., Peterkofsky A., Gronenborn A. M., and Clore G. M. (1999) Solution structure of the 40,000 Mr phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr. Nat. Struct. Biol. 6, 166–173 [DOI] [PubMed] [Google Scholar]
- 35. Hsiao C. D., Sun Y. J., Rose J., and Wang B. C. (1996) The crystal structure of glutamine-binding protein from Escherichia coli. J. Mol. Biol. 262, 225–242 [DOI] [PubMed] [Google Scholar]
- 36. Sun Y. J., Rose J., Wang B. C., and Hsiao C. D. (1998) The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: comparisons with other amino acid binding proteins. J. Mol. Biol. 278, 219–229 [DOI] [PubMed] [Google Scholar]
- 37. Bermejo G. A., Clore G. M., and Schwieters C. D. (2012) Smooth statistical torsion angle potential derived from a large conformational database via adaptive kernel density estimation improves the quality of NMR protein structures. Protein Sci. 21, 1824–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lanman J., Lam T. T., Emmett M. R., Marshall A. G., Sakalian M., and Prevelige P. E. Jr. (2004) Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 11, 676–677 [DOI] [PubMed] [Google Scholar]
- 39. Jenner M., Ellis J., Huang W. C., Lloyd Raven E., Roberts G. C., and Oldham N. J. (2011) Detection of a protein conformational equilibrium by electrospray ionisation-ion mobility-mass spectrometry. Angew. Chem. Int. Ed. Engl. 50, 8291–8294 [DOI] [PubMed] [Google Scholar]
- 40. López A., Vilaseca M., Madurga S., Varese M., Tarragó T., and Giralt E. (2016) Analyzing slowly exchanging protein conformations by ion mobility mass spectrometry: study of the dynamic equilibrium of prolyl oligopeptidase. J. Mass Spectrom. 51, 504–511 [DOI] [PubMed] [Google Scholar]
- 41. Tang C., Iwahara J., and Clore G. M. (2006) Visualization of transient encounter complexes in protein-protein association. Nature 444, 383–386 [DOI] [PubMed] [Google Scholar]
- 42. Liu Z., Gong Z., Guo D. C., Zhang W. P., and Tang C. (2014) Subtle dynamics of holo glutamine binding protein revealed with a rigid paramagnetic probe. Biochemistry 53, 1403–1409 [DOI] [PubMed] [Google Scholar]
- 43. Zhou Y., Yang W., Lurtz M. M., Ye Y., Huang Y., Lee H. W., Chen Y., Louis C. F., and Yang J. J. (2007) Identification of the calmodulin binding domain of connexin 43. J. Biol. Chem. 282, 35005–35017 [DOI] [PubMed] [Google Scholar]
- 44. Xu T., Park S. K., Venable J. D., Wohlschlegel J. A., Diedrich J. K., Cociorva D., Lu B., Liao L., Hewel J., Han X., Wong C. C., Fonslow B., Delahunty C., Gao Y., Shah H., and Yates J. R. 3rd (2015) ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petrotchenko E. V., Serpa J. J., Makepeace K. A., Brodie N. I., and Borchers C. H. (2014) 14N15N DXMSMS Match program for the automated analysis of LC/ESI-MS/MS crosslinking data from experiments using 15N metabolically labeled proteins. J. Proteomics 109, 104–110 [DOI] [PubMed] [Google Scholar]
- 46. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., and Dong M. Q. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 47. Liu C., Song C. Q., Yuan Z. F., Fu Y., Chi H., Wang L. H., Fan S. B., Zhang K., Zeng W. F., He S. M., Dong M. Q., and Sun R. X. (2014) pQuant improves quantitation by keeping out interfering signals and evaluating the accuracy of calculated ratios. Anal Chem. 86, 5286–5294 [DOI] [PubMed] [Google Scholar]
- 48. Liao D. I., Silverton E., Seok Y. J., Lee B. R., Peterkofsky A., and Davies D. R. (1996) The first step in sugar transport: crystal structure of the amino terminal domain of enzyme I of the E. coli PEP: sugar phosphotransferase system and a model of the phosphotransfer complex with HPr. Structure 4, 861–872 [DOI] [PubMed] [Google Scholar]
- 49. Case D. A., Babin V., Berryman J., Betz R. M., Cai Q., Cerutti D. S., Cheatham T. E. Iii, Darden T. A., Duke R. E., and Gohlke H. (2014) Amber 14, University of California [Google Scholar]
- 50. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 51. Lee Y. J. (2009) Probability-based shotgun cross-linking sites analysis. J. Am. Soc. Mass Spectrom. 20, 1896–1899 [DOI] [PubMed] [Google Scholar]
- 52. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.7, Schrodinger, LLC, New York [Google Scholar]
- 53. Schwieters C. D., Kuszewski J. J., and Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 [Google Scholar]
- 54. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]