

Abstract
Bacterial microcompartments are bacterial analogs of eukaryotic organelles in that they spatially segregate aspects of cellular metabolism, but they do so by building not a lipid membrane but a thin polyhedral protein shell. Although multiple shell protein structures are known for several microcompartment types, additional uncharacterized components complicate systematic investigations of shell architecture. We report here the structures of all four proteins proposed to form the shell of an uncharacterized microcompartment designated the Rhodococcus and Mycobacterium microcompartment (RMM), which, along with crystal interactions and docking studies, suggests possible models for the particle's vertex and edge organization. MSM0272 is a typical hexameric β-sandwich shell protein thought to form the bulk of the facet. MSM0273 is a pentameric β-barrel shell protein that likely plugs the vertex of the particle. MSM0271 is an unusual double-ringed bacterial microcompartment shell protein whose rings are organized in an offset position relative to all known related proteins. MSM0275 is related to MSM0271 but self-organizes as linear strips that may line the facet edge; here, the presence of a novel extendable loop may help ameliorate poor packing geometry of the rigid main particle at the angled edges. In contrast to previously characterized homologs, both of these proteins show closed pores at both ends. This suggests a model where key interactions at the vertex and edges are mediated at the inner layer of the shell by MSM0271 (encircling MSM0273) and MSM0275, and the facet is built from MSM0272 hexamers tiling in the outer layer of the shell.
Keywords: bacterial metabolism, carboxysome, molecular docking, structural biology, transport, X-ray crystallography, bacterial microcompartment
Introduction
Bacteria differ fundamentally from eukaryotes in that they lack the ability to form membrane-bound organelles to segregate aspects of their metabolism. Some bacteria are, however, able to bypass this limitation by encapsulating enzymes catalyzing critical steps of a problematic metabolic pathway in a protein shell; the resulting 100-nm scale bodies are known as bacterial microcompartments (BMCs).2 Recent surveys suggest 23 phyla of bacteria contain operons harboring candidate genes for construction of these microcompartments (1). Ecologically, the most important BMCs are the anabolic carboxysomes, which facilitate carbon fixation in cyanobacteria and chemoautotrophs (2, 3); a variety of BMCs in heterotrophic bacteria also support the catabolism of a variety of small organic compounds (4). Well characterized examples of microcompartments include the two varieties of carboxysomes (α and β) that fix carbon dioxide using Rubisco, as well as the propanediol utilization (pdu), and ethanolamine utilization (eut) microcompartments. Available evidence for less well characterized microcompartments suggests roles in the catabolism of ethanol, choline, 6-deoxyhexoses, and other small molecules (5, 6), while bioinformatics analysis suggests 37 distinct microcompartment gene patterns, some of which are highly likely to encode novel pathways (1). The emergent pattern is that microcompartments typically catalyze successive steps in pathways where the intermediates are toxic and/or volatile (7). Encapsulation of the enzymes within a large (∼100 nm diameter) selectively permeable polyhedral proteinaceous shell therefore serves to bring sequentially acting active sites into spatial proximity, while the selectivity and limited porosity of the shell helps impede the escape of problematic intermediates (8, 9).
The organization of the microcompartment shell has been of considerable research interest; assembly appears to echo patterns previously identified in the organization of icosahedral viruses, but the constituent proteins are not homologous. The facets of the shell are built from members of a single protein family (pfam00936) with a core structure composed of a four-stranded anti-parallel β-sheet flanked by three α-helices in a thioredoxin-like topology; six copies of this domain then form a basic hexagonal tile that can be packed into extended sheets (Fig. 1) (10–13). At least one (and generally several) such protein has been identified in every candidate microcompartment identified to date, and, in systems where the shell composition has been quantified, these bacterial microcompartment-hexameric shell proteins (BMC-H) are the most abundant components of the shell. More recently, a second deeply divergent branch of this family was identified in Cso1D in α-carboxysomes (14). This protein has only very weak sequence similarity to BMC-H proteins, and indeed, the topology of the basic structural domain is permuted, with two permuted domains linked in a head-to-tail fusion. These proteins are here termed BMC-FP (BMC fused, permuted) proteins (note that we use BMC-FP to denote only fused, permuted, and double-ringed proteins, such as CcmP and Cso1D). Only three copies of the protein are required to form a pseudohexagonal ring, but in both known examples two such rings oligomerize face to face forming a double-ring that has been suggested to form a locally double-layered shell (14, 15). BMC-FPs are only associated with a subset of microcompartments and have only been previously characterized in carboxysomes. Other variants on the pfam00936 family are also found, including permuted hexameric proteins (e.g. EutS and PduU) (16, 17) and double-domain non-permuted proteins (PduB, EutL, and CcmO) (16, 18, 19), although these are generally associated with only a subset of microcompartment types and likely play more specialized functional roles.
FIGURE 1.
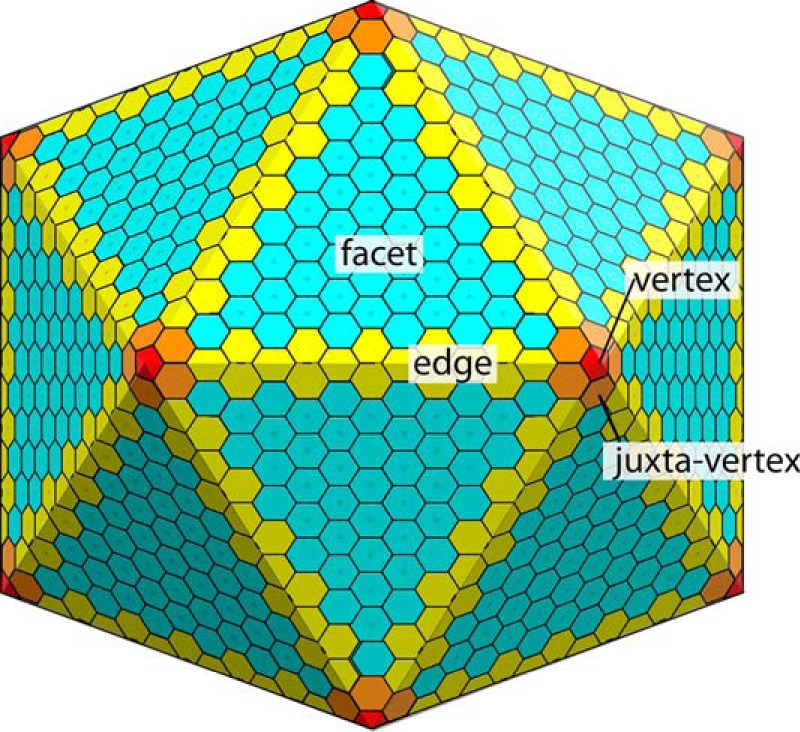
Architecture of bacterial microcompartments. The bulk facet is formed by tiling of BMC-H/FP proteins. The organization around the vertex, and especially the edge, are much more poorly understood.
In addition to BMC-H proteins, the other protein family that seems absolutely required for forming a functional microcompartment shell is pfam03319. These small proteins are built as a five-stranded anti-parallel β-barrel that oligomerizes with four other chains to form a truncated pentameric pyramid (20). Available evidence suggests that voids left in the 5-fold symmetric vertices of the shell are capped by these bacterial microcompartment-pentameric shell (BMC-P) proteins (20, 21). Interestingly, many microcompartments contain several paralogs of these BMC-P proteins (up to seven), suggesting that they may have roles beyond acting as simple plugs.
In this work, we investigate a member of a family of functionally uncharacterized microcompartments, previously identified in members of the Actinomycetales genus (22); in the absence of an experimentally confirmed function, we adopt the terminology of Axen et al. (1), terming these RMM (Rhodococcus and Mycobacterium microcompartment) microcompartments. We have focused on Mycobacterium smegmatis MC2 155 for this work; M. smegmatis is a close saprophytic relative of Mycobacterium tuberculosis, and being non-pathogenic and fast growing, a variety of tools (including transformation, protein expression and genome-wide knockouts) have been developed in M. smegmatis to facilitate use of this organism as a model. The operon associated with the RMM in M. smegmatis contains open reading frames for four shell protein homologs and four enzymes, as well as a possible additional structural protein, a transcription regulator, and an amino acid permease-like transporter. The only enzyme from an RMM operon that has been enzymatically characterized is a short chain alcohol dehydrogenase that has been shown to possess l-1-amino-2-propanol dehydrogenase activity (23). Encoded enzymes that have not been catalytically characterized, but from sequence homology, the operon includes a class III aminotransferase, an aldehyde-alcohol dehydrogenase, and a protein with distant homology to aminoglycoside phosphotransferases. This operon can be induced in Rhodococcus by the addition of 1-amino-2-propanol (24). Together, this suggests that the initial substrate is either 1-amino-2-propanol, and/or a small amino alcohol, or a closely related metabolite. The products that would ultimately enter general metabolism remain unclear. The shell proteins include a BMC-H protein, a BMC-P protein, and two BMC-FP proteins. Intriguingly, this is the smallest number of shell proteins associated with any known microcompartment (tied with the α-carboxysome in Prochlorococcus MED4, which has two BMC-P proteins); this suggests that that the RMM may be a usefully tractable system for characterizing and modeling microcompartment shell assembly. We report here for the first time X-ray structures for all shell components of a single microcompartment (all obtained from a single organism), revealing intriguing new insights into how these shell proteins might interact and function.
Results
Structure of MSM0273
The pentameric shell protein (BMC-P) MSM0273 crystallized in space group P3121 and diffracted to a resolution of 1.6 Å. The asymmetric unit contained five protomers arranged into a pentamer. Of the 87 native amino acids, one chain is missing only the last residue, while the last 9–10 residues are disordered in the other four. Each protomer consists of a single pfam03319 domain, organized around a five-stranded antiparallel β-barrel (1, −2, 3, 5, −4 topology) (Fig. 2A). The axis of this barrel lies within the plane of the pentamer, radiating out from the central pore. The long β1-β2 loop (β1a) extends out toward an adjacent protomer, which in turn pairs with β5 and an extended region of the C terminus. In the one chain where ordered, the extended C terminus forms a β-strand along the outside edge of β1 extending outward from the β-barrel. A short six-residue α-helix is inserted into the β4-β5 loop; the N-terminal end of this helix lines the central pore of the pentamer.
FIGURE 2.

Overall structures of RMM shell proteins. A, MSM0273, the BMC-P. B, MSM0272, the BMC-H. C, MSM0271, and D, MSM0275, the BMC-FP proteins. The top sub-panels show the protomeric structure (colored blue to red N to C terminus) with secondary structure elements labeled. The 2nd sub-panel shows the oligomer (pentamer in A, otherwise a hexamer) shown looking down the axis of maximal symmetry and in the same orientation as the protomer in the top panel. Additional chains are shown in white, except for the BMF-FP proteins, where one protomer in the opposite ring is shown in beige to show the relative orientations of the two rings. Small triangles indicate the position the 3-fold symmetry axis would be found should the top (black) or bottom (white) trimer be tiled in an arrangement analogous to classical BMC-H proteins. The 3rd panel shows the oligomer from a viewpoint orthogonal to that in the 2nd panel.
Searching with DALI reveals that EutN (PDB code 2Z9H), GrpN (PDB code 4I7A), CcmL (PDB code 2QW7), and CsoS4 (HnCsoS4, PDB code 2RCF) are the closest structural homologs, with Z-scores in the range of 12–13 (25). Sequence identity to EutN is closest at 35%. These structures all generally resemble one another, with MSM0273 having a shorter β2-β3 loop and a shorter C terminus than most homologs. Similar to homologous structures, a pore is formed at the 5-fold axis and is lined by the N-terminal end of α1; the hydroxyl group and amide nitrogen of Ser-56 lines an electropositive hydrophilic pore ∼5 Å in diameter (Fig. 3, A–C).
FIGURE 3.

Details of potential transport routes through RMM shell proteins. A, electrostatic surface of MSM0273. B, organization of the MSM0273 pore, seen from the concave side. Similar to other BMC-P proteins, MSM0273 is restricted to a narrow ∼3.5 Å pore with 5-fold symmetry lined by the side chain of Ser-56. C, cross-section of the MSM0273 pore. Note that the pore is noticeably electropositive. D, electrostatic surface of MSM0272; note that the central pore area is highly electronegative. E, details of the more open MSM0272 pore. F, same pore as in E but shown in the plane of the pore. G, details of the pore of MSM0272 in the more closed ring, where Asp-40 from one protomer inserts into the pore, partially occluding it. H, MSM0275 electrostatic surface, sliced along the 3-fold symmetry axis. Note that the hexamer is closed at both the upper and lower surface. However, there are narrow pores open to the side in protomers where the β1-α1 loop is in an extended conformation. I, details of the interactions at the 3-fold axis of MSM0275. The interactions between Gln-70 and Arg-69 possibly add additional stability to this interface. J, electrostatic surface for MSM0271, sliced along the 3-fold symmetry axis. Note that both top and bottom surfaces are sealed, and there are no lateral passages. K, details of the interaction at the 3-fold axis in MSM0271.
Structure of MSM0272
MSM0272 is a BMC-H protein that is 68% identical to Clostridium EutM and 59% identical to Salmonella PduA. The structure of MSM0272 was determined at a resolution of 2.2 Å, with an Rfree of 23.3%. Crystals were in the space group P43 with two complete hexamers in the asymmetric unit. At minimum, all protomers have residues 4–88 (of 93) ordered, while chain A (the most complete chain with the lowest atomic displacement parameters) is missing only residue 1, which is likely removed by methionine amino peptidase. MSM0272, as expected for a BMC-H protein, is built as a hexamer of single pfam00936 domains, with a four-stranded antiparallel β-sheet (with a 2, −3, 1, −4 topology) that has two α-helices packed onto one face (Fig. 2B). The C terminus forms a third helix-like structure that packs onto the equivalent sheet face on an adjacent protomer. Six protomers are organized into a hexameric ring that has a regular hexagonal outline, with near straight edges and distinct convex and concave (where α3 is located) faces.
Although α3 is well defined with up to three hydrogen bonds in a 310 pattern in one protomer, in others these residues only form a single hydrogen bond and more resemble two adjacent turns. The structural flexibility of this region, especially the disorder of residues 89–93 in many protomers is interesting, as this region in the close homolog PduA is proposed to mediate interactions with terminal helical peptides from encapsulated residues (26, 27). In PduA, this helix is required to recruit PduP to the microcompartment, with His-81, Val-84, and Leu-88 (here His-81, Leu-84, and Phe-88) being the critical residues. The relative mobility of these residues, with Phe-88 disordered in 7 of the 12 protomers, suggests that this helix may be able to reorganize to maximize interactions with the targeting peptide.
In common with other BMC-H proteins, a small pore is formed where all six protomers meet at the 6-fold rotational axis and is lined by a β-hairpin motif (between β2 and β3). In common with most BMC-H proteins, this turn has a sequence ΦGZGX, where Φ is a small hydrophobic residue; X is any residue; and Z is the residue that immediately lines the pore (note that the full diversity of BMC-H proteins includes more radical variants, including significant insertions and deletions into this loop (1); however, these have not been structurally characterized, and an understanding of the characteristics of the pore they form is lacking). This pore-lining residue is important as its ability to interact with adjacent copies of the motif helps define pore diameter, conformation, and flexibility, while its properties define the interactions any ligands will be able to form as they pass through the pore. Most commonly, this pore-lining residue is small and polar, generally serine (in CcmK1, CcmK2, and CcmK4 from the β-carboxysome) or glycine (CsoS1A and CsoS1C from the α-carboxysome). In MSM0272, the pore-lining residue is an aspartate, Asp-40, which by default carries a negative charge, resulting in a pore environment with crowded carboxylate groups that should strongly repel one another. In the structure, the side chains of Asp-40 protrude on the convex face of the protein, forming a ring of carboxylates that face the solvent (Fig. 3, D–G). The carboxylate generally makes hydrogen bonds only to water molecules, although the nearby placement of the Lys-12 ζ-amine from the same or an adjacent protomer (generally around 3.5 Å, but not hydrogen bonded) helps stabilize the excess negative charge while adopting several alternative conformations; in some orthologs, Arg-12 is found to substitute. Two additional nearby acidic groups, Glu-10 and Glu-70, ensure that the convex face of the hexamer is highly acidic. Pore diameter is 5 Å (i.e. the diameter of the largest object that could readily pass through) at the Asp-40 amide nitrogen (which is relatively fixed), and a slightly narrower 4.5 Å at the level of the carboxylate group, which is more mobile. Interestingly, the second independent hexamer in the crystal displays an asymmetric conformation. Here Asp-40 of one chain is inserted into the pore, making a hydrogen bond to the amide backbone of an adjacent Asp-40; this conformation constricts the pore to 3.0 Å diameter (Fig. 3G).
Structures of MSM0271 and MSM0275
MSM0271 crystallized in space group P6322, diffracted to a resolution of 2.7 Å, and was refined to an Rfree of 22.4%. The asymmetric unit contains a trimer of protomers arranged in a ring, with each copy of the chain being largely identical (r.m.s.d. values of 0.2 Å or less). A second trimer related by crystallographic symmetry completes the hexameric double ring structure. Residues 10 to the C terminus (218) are ordered in all three protomers; however, residues 31–36 in the β1-α1 loop are disordered in all protomers (with 29 and 30 also disordered in one chain).
MSM0275 crystallized in space group C2221 and diffracted to a resolution of 2.1 Å. The asymmetric unit contains six protomers arranged as a dimer of trimers. Of the 202 amino acids in the native protein 189 (chain E) to 201 (chain A), amino acids could be traced. Up to seven additional amino acids derived from the hexahistidine tag were ordered in chains A and B, forming a short antiparallel four α-helical bundle with tags from adjacent hexamers; this motif mediates the main packing interactions in the c direction between layers of molecules arranged in the a/b plane (not depicted).
MSM0271 and MSM0275 are tandem domain shell proteins. The protomer is built from a pair of fused, permuted pfam00936 domains, each with a four-stranded antiparallel β-sheet (1, −2, 4, −3 and 5, −6, 8, −7 topology for the N- and C-terminal repeats respectively). Two α-helices α2 and α3 (α5 and α6 in the C-terminal domain) pack on one face of the β-sheet, while α1 (α4) is part of a long loop-helix-loop motif inserted between β1 and β2 (β4 and β5) packs on the opposite face and mediates interaction between adjacent rings.
Although the protomeric organizations of MSM0271 and MSM0275 closely resemble one another (see Fig. 2, C and D; the chains can be superimposed with an r.m.s.d. of 0.69 Å), MSM0275 and MSM0271 hexamers show emergent differences. MSM0275 rings associate in a manner similar to CsoS1D and CcmP, where interactions are mediated largely by α1 packing on its C-terminal domain equivalent helix, α4. The 2-fold symmetry axis run through the diagonals of the hexagon. MSM0271 uniquely has its domains offset by 60°, so α1 packs onto α1 and α4 packs onto α4; the internal symmetry axis therefore runs through the centers of the facets of the hexagon (Fig. 2, C and D). This unique arrangement requires two distinct sets of interactions to hold the rings together.
It is worth noting that the two rings in MSM0271 and MSM0275 are not properly oriented to form a double-layered sheet with canonical BMC-H-like interactions. This is because, as noted previously for CcmP (15), the two trimeric rings are not appropriately aligned but rather are offset by about 10°. One way to envision this is to map the locations of the 3-fold axes at the corners of the (pseudo)hexamers were these hexamers to partake in canonical facet packing (as seen in PduA; Fig. 4A). These axes are offset in the two layers (triangles in Fig. 2, C and D), meaning that BMC-FP proteins cannot pack into facet-like sheets with both rings simultaneously.
FIGURE 4.

Crystal packing and potential interactions mediated by RMM shell proteins. All are shown from the concave side of the respective (pseudo)hexamer, unless indicated otherwise. A, crystal packing in PduA from Salmonella enterica typhimurium (PDB code 3ngk), a hexameric shell protein, as a well established model for the canonical interactions mediating facet tiling. Individual hexamers interact along their edge, forming a seal. Key residues include Lys-26, Asn-29, and Arg-79, which reaches across the interface to interact with to the C-terminal end of α1. B, corresponding facet of MSM0272. MSM0272 is 59% identical to PduA and conserves all of the important interface residues. C, ID interaction interface of MSM0271. Note that key residues are not conserved, including Arg-79 and Lys-26 (here Arg-171). D, IC interaction interface of MSM0275. Here again, Arg-67 is substituted for the usual Lys-26. E, crystal packing in MSM0275 in the a/b plane; chains A–F are colored green, red, magenta, yellow, pink, white, respectively. Note that interactions between the A and B protomers follows the canonical tight facet interaction shown in A. This results in a zigzag pattern of rings linked in a continuous line (bright versus faded). The interactions between the magenta protomers involves the β1-β2 loop adopting an extended conformation, and packing between molecules (F). F, details of the conformation change in the β1-β2 loop. This region shows large changes in conformation between protomers, with individual residues moving up to 15 Å. The loop adopts a fully compacted conformation (cyan) where close contacts exist with adjacent molecules, and an extended conformation (orange) where there is available space between molecules. The blue conformation appears to be an intermediate conformation where most of the loop is disordered. Note that the structural transition also involves substantive rearrangement of α1. G, interactions mediated by the tight (ID-ID) interface of MSM0275. Lys-158 and Arg-12 are equivalent to Lys-26 and Arg-79 in PduA, and mediate similar interactions. The interface is gapless, and the interactions appear at least as strong as in PduA. H, interactions mediated by the second pseudohexamer at the same interface as in Fig. 2G. Note that these interactions are very different from the canonical facet interactions. Although Lys-56 appears in the equivalent position, the geometry of the two rings of MSM0275 (and by extension all structurally characterized BMC-FP proteins) means making the tight interactions in C preclude these lysine residues from interacting similarly. Instead they are displaced laterally, so the two rings only interact across two-thirds of their length. This arrangement precludes a tight seal forming between these rings, although additional stabilization is afforded by the interface formed by the opposite rings. Near identical copies of the interactions shown in C and D recur at a second interface in the crystal, offset by 120°. I, alternative, long range ID-ID interaction in MSM0275. This interaction positions the domains much further apart. The gap between protomers is instead filled by the β1-α1 loop, which extends into this space and interacts with the opposite ring. Note that this interface is skewed, with the interfaces angled about 10° apart. This panel shows both pseudohexameric rings, with the ID-ID interface shown from the convex side of the ring.
The concave-to-concave interactions between rings in MSM0271 and MSM0275 create a substantive enclosed volume of space. Similar to what has previously been seen with the CsoS1D and CcmP structures (14, 15), access to this space is blocked by the meeting of symmetry related copies of Arg-81 (Arg-69 in MSM0275) meeting at the 3-fold axis with Arg-81 hydrogen bonding with Glu-80 (Glu-68) from an adjacent protomer (Fig. 3, I and K). In MSM0271 and MSM0275, this interaction seems especially strong, as it is stabilized by an additional inter-chain hydrogen bond contributed by Gln-82 (Gln-70). Arg-81 also packs on Gly-171 and Ala-172 of the topologically equivalent loop from the C-terminal repeat. In CsoS1D and CcmP, this conformation is present in only one ring, while the second ring adopts an open conformation that reveals a substantial (∼13 Å wide) pore. In MSM0271 and MSM0275, the rings on both protomers are closed, blocking access to the central cavity from outside. Atomic displacement parameters for these residues are similar to the rest of the structure, giving little indication of a tendency to opening by order-to-disorder transitions.
Structural Plasticity in the MSM0275 β1-α1 Loop
All six copies of MSM0275 in the asymmetric unit can be superimposed with an r.m.s.d. 0.29–0.55 Å. The only region where the protomeric structures diverge significantly is in the α1-helix and the loops connecting this motif to β1-β2 in the N-terminal domain (Fig. 4F). Residues Trp-24–Asn-33 are disordered in two of the six protomers. In one protomer, chain F, all residues are ordered and very closely match the conformation of the equivalent residues in CsoS1D and CcmP, in a conformation that we will refer to as the retracted conformation. Two additional chains (A and B) closely match this conformation, but residues 29–32 are displaced up to 2 Å from the site of chain F. In chain D, the C-terminal end of the helix has become slightly unwound, with Trp-24 sitting in the position usually occupied by Ile-25, with residues 25–32 being disordered. The largest change is seen in chains C and E. Note that in these chains density is weak with high atomic displacement parameters; for chain C, the central residues of this region are disordered, while for chain E, residues 26–33 are partially stabilized by interactions with an adjacent molecule. In these chains, helix α1 partly unwinds and shifts 3.8 Å. This results in Thr-23 occupying the approximate pocket occupied by Pro-30 in the other chains. This rearrangement leaves residues 26–33 protruding from the surface of the hexamer, with Ala-28 of chain E shifted 21 Å from the position it occupies in chain B. This conformation also leaves a narrow channel open from the central pore cavity to the surface of the facet. We refer to this loop as the extensible loop and conformations similar to that in chain E as the extended conformation.
In the MSM0271 structure, all chains are similar. Residues 29–35 (equivalent to residues 20–24 in MSM0275) are disordered in all three chains of the MSM0271 structure, but the positioning of these residues implies that the disordered residues are located in the central cavity, rather than exposed on the surface.
Interaction Faces and Packing Interactions
BMC-H proteins preferentially adopt a specific edge-to-edge interaction geometry; the interface in PduA, for example, shows the key conserved residue Lys-26 packing antiparallel to its symmetry-related mate, while Arg-79 reaches across the interface and makes a hydrogen bond with the C-terminal end of α1 (Fig. 4A); this interface straddles a 2-fold symmetry axis, so these interactions are repeated on the other side of the interface. Lys-26, Asn-29, and Arg-79 have all been shown to be required to form a stable functional shell (13). In crystal structures of several BMC-H proteins, this interface is repeated at each face, resulting in a continuous gapless sheet of molecules that is believed to serve as a close model for the packing in the microcompartment facets. We term this the “canonical” interaction geometry.
The MSM0272 crystal form obtained packs individual hexamers in non-sheet-like orientations; however, MSM0272 is a close homolog of PduA, and all key residues of the canonical interface are conserved in MSM0272 (with identical numbering) (Fig. 4B). Modeling indicates that MSM0272 should be capable of packing in very similar sheet-like arrangements, with essentially identical packing interactions.
Exploring the possible interactions of MSM0275 and MSM0271 in the microcompartment shell requires awareness of two complicating factors. First, because of the fused nature of the dimers, there are two distinct potential canonical interfaces in each protein ring; one face is characterized by the meeting of two domains of one chain (inter-domain or ID facet), and the other is characterized by the meeting of adjacent chains (inter-chain or IC facet). In both proteins, these facets differ in the amino acids presented at the interface. Second, because these proteins form rigid back-to-back double-ringed hexamers, interactions in one ring force a specific interaction geometry in the second ring. Therefore, interactions likely require optimizing both interfaces simultaneously.
MSM0271 fails to form any meaningful lateral interactions in the crystals. Moreover, the potential interaction interface shows some substantive differences from characterized BMC-H and BMC-FP proteins. Lys-26, otherwise near absolutely conserved, is replaced by arginine, Arg-67/171. The arginine that usually interacts with the adjacent ring is replaced by an aspartate, Asp-21/127 (Fig. 4, C and D). Despite the lack of the usual interacting motifs, the facet remains flat, and there is a strong similarity between the potential interaction surfaces formed at the ID and IC faces.
MSM0275 hexamers pack in the crystal as layers of flat sheets of molecules. The hexamer in the asymmetric unit abuts six others related by 2-fold rotational and screw symmetry operations, generating four crystallographically distinct interfaces. Two of these interfaces (where chain A packs on A and chain B on B) mediate a tight interface that places the center-to-center distance between hexamers at 67.6 Å, very close to that seen in crystal structures that are the basis of the canonical single sheet facet model, PduA (3ngk; 67.2 Å) and CcmK2 (3dnc, 67.4 Å) (11, 28). Equivalent side chains also mediate very similar interactions (Fig. 4G); symmetry related copies of Lys-158 are in van der Waals contact, while making hydrogen bonds to Asp-11 in the same chain; Arg-12 reaches across the interface and interacts with the C-terminal end of helix α5. In addition, Gln-187 hydrogen bonds to Asp-11 carbonyl oxygen, and Thr-80 hydrogen bonds with Asp-184, providing additional stabilizing interactions. The packing and specific interactions involved closely match the canonical interaction; we will refer to this as the canonical MSM0275 interaction. Note that at this interface, the extendable β1-α1 loop is fully ordered in the retracted conformation, and indeed, extending it out into the space between hexamers would result in clashes. This tight interaction simultaneously forces a specific geometry on the second trimeric ring of these hexamers, which interact through the IC interface (Fig. 4H). These rings interact more weakly, with the only hydrogen bond occurring between symmetry related copies of Ser-114, and some van der Waals contacts between Ala-183 and the carbonyl oxygen atoms of Lys-56 and His-57. The IC facet lacks the equivalent of Asp-11 and Arg-12 (the equivalent residues are Ser-113 and Ser-114) or Gln-187 (Ala-85). Despite the lack of strong contacts, this surface leaves no large gaps along its interface. However, the subunits at the interface are effectively displaced 13 Å from their more usual packing positions, meaning any attempt to tile a surface with these interactions will leave large gaps at the 3-fold axes. These interactions therefore appear to further stabilize the ID-ID interface but do not themselves form a well sealed IC-IC interface.
Unlike what has been observed for BMC-H proteins, the crystal does not display canonical packing at each face of the hexamer, so uniform two-dimensional tiling is absent. Rather, the placement of these canonical interfaces results in strips of molecules being tightly connected in extended lines, with the 120° offset of connections resulting in a zigzag pattern (Fig. 4E). Note that CcmP form 1 crystals pack in a very similar pattern of zigzagged linear strips, although with an interface that is displaced from the canonical packing interaction; electron microscopy suggests that this protein prefers this organization even when unconstrained in a crystal lattice (15). It should be noted that these canonical ID-ID MSM0275 interactions cannot readily be extended into a model for packing MSM0275 into a continuous facet, as the ID facet edge is found alternatively in the top and bottom trimers of the hexamer as one proceeds around the ring. Forming the canonical interface alternatively through the upper and lower rings results in severe clashes, as the off-centered placement of the neighboring IC-IC interface causes it to protrude into the space required for the ID-ID interaction.
A third independent interface also features ID-ID packing, between chain C and chain E (pink in Fig. 4E). This packing interface is looser, reflecting a significantly longer center-to-center distance between hexamers (72.4 Å versus 67.6 Å), and the facets are angled about 10° apart in plane, so packing along one face is closer than the other (possibly because the constraints of crystal packing preclude optimal interactions). This interface is characterized by interactions mediated by the α1-β1 loop that is in the fully extended conformation from both chains, although only fully ordered in chain E, where interactions are closer (Fig. 4I). Leu-29 from this loop forms van der Waals interactions with Pro-15C, Ile-25C, and Pro-134F, while Arg-31E hydrogen bonds with Asp-161C. Additional interactions are formed by Glu-79E hydrogen bonding with Gln-187C, and the guanidinium groups of Arg-12E/C being in van der Waals contact. Residues 29–32 of chain C are disordered, but if they were to adopt a conformation similar to those of chain E, a sealed interface would seem at least possible. In the opposing ring, chains A and F interact through van der Waals interactions over a reasonably wide interface, but chains B and D (the second half of this IC interface) are too far apart to interact.
The final independent interface in the crystal is again different. This interface pairs an ID with an IC interface, with centers of hexamers 69.2 Å apart. The resulting interface is weakly stabilized by van der Waals packing, but it appears loosely packed and porous.
Modeling the Vertex by Docking
Tanaka et al. (20), in the original study investigating the organization of the vertex by computational modeling, suggest that the BMC-Ps have an appropriate size and shape to plug a pentameric defect where five facets of BMC-H proteins meet. However, this pioneering study was limited by trying only one of the six candidate hexameric shell proteins for modeling and the use of rigid body docking. In addition, the authors modeled the pentameric defect between CcmK2 and CcmL by simply bending the canonical interface to the required 141° angle. However, the acute angle between BMC-H/FP proteins at the vertex introduces the possibility that they pack substantively differently, as the vertex is not an extension of the interactions in the facet but rather of those at the edge. We instead systematically investigated the local organization of the RMM vertex by docking (with side chain flexibility) each of the three candidate BMC-H/FP proteins in a geometry appropriate for forming the juxta-vertex ring, followed by docking of the BMC-P into the resulting pentameric defect (Fig. 5A). Docking results were compared on the basis of their interface score (I_sc; the difference in energy between the complex and its component structures: typically an I_sc of −5 to −10 is considered “good”) as well as total buried surface area.
FIGURE 5.

Modeling the RMM vertex by docking. A, overall logic of the docking. The formation of the vertex requires forming two distinct protein-protein interfaces; these are investigated sequentially using Rosetta dock. First ➀, the interface between BMC-H (MSM0272) or BMC-FP (MSM0271 or MSM0275) was investigated. Two copies of a given protein (blue and cyan) were positioned with the appropriate geometry for a vertex and allowed to rotate and translate along their rotational symmetry axis. The interface was then repacked and evaluated using Rosetta dock. For the BMC-H protein, this was done separately with the concave side facing outward (as shown) or inward. For the BMC-FP proteins, the ID and IC faces were evaluated separately. The highest scoring pose for each protein was then fixed and used to dock MSM0273 ➁, the BMC-P, again systematically investigating rotation and translations around the 5-fold symmetry axis. B, table summarizing results of docking of each BMC-H/FP shell protein to itself, then to MSM0273. N.D. indicates that the docking was not done, due to poor docking results in the first stage. C, I_sc versus r.m.s.d. (versus top scoring pose) for docking of MSM0271 on MSM0271. The presence of a funnel, where all of the lowest energy solutions closely resemble the best solution, is indicative of a successful docking run. D, I_sc versus r.m.s.d. for docking MSM0273 (concave outward) into the previously docked MSM0271 ID-ID complex. E, final model of the complex. MSM0271 is in cyan, and MSM0273 in orange. The second ring of each interacting MSM0271 hexamer is shown as a schematic. F, details of the MSM0271-MSM0271 interface. Inset on right shows the same interaction looking along the interface. Note that the interface occurs over a wide surface. G, same MSM0271-MSM0271 interface, shown looking down the interface from the side. H, details of the MSM0271-MSM0273 interface.
As Tanaka et al. (20) point out, single ringed BMC-H proteins can plausibly meet at a 5-fold axis with either their concave or convex face on the inner edge of the vertex. For BMC-FP proteins, however, we observe that the presence of a second bulky ring on the concave face implies that these proteins can only interact at the vertex with the convex face inward (i.e. localized to the luminal side of the compartment) so that the second rings diverge from one another rather than collide. We also assume that if BMC-FP proteins line the vertex, they are likely do so consistently with a given face (ID or IC) forming BMC-FP self-interactions and the other face forming BMC-FP to BMC-P interactions; any other organization would force a complex mixture of interfaces, with an asymmetric pentameric defect housing the symmetric MSM0273. To minimize assumptions about vertex organization, we started by modeling the self-interactions of the ring of BMC-H/FP molecules immediately adjacent to the vertex. This necessitates modeling six different interactions (MSM0272 with the convex and concave side inward and MSM0271 and MSM0275 each interacting through their distinct IC and ID faces). The highest scoring model for each protein was then used to model interactions with MSM0273, with MSM0273 in either possible orientation (base in or base out).
BMC-H/FP self-docking yielded solutions with roughly comparable interaction scores for all three proteins; however, none of these interactions generated the canonical-facet-bent model originally proposed for lining the vertex. MSM0272 gave significantly better packing with the convex side facing inward rather than outward (I_sc of −6.99 versus −5.24; see Fig. 5B). Interestingly, the optimal (convex inward) interaction geometry occurs where the edges are offset laterally 5 Å from their positions in the canonical facet interaction; indeed, no good packing arrangement was found for any BMC protein with the distorted canonical-like interactions originally assumed by Tanaka et al. (20). This MSM0272 interaction results in a tight seal along the edge, with no discernible gaps. The interactions extend the interacting surface to include interactions between side chains at the C-terminal end of α2. Docking MSM0273 onto this optimal complex, however, does not yield any convincing solutions; the highest observed I_sc is −18.05, but examination of the model shows that the protruding helices α1 and α2 mediate most of the contacts, and the clefts left between these helices leave large gaps (∼10 Å) between molecules (note that five separate interfaces are being summed in these models, resulting in much higher I_sc scores). In the optimal concave inward packing, α2 from MSM0272 forms a prominent ridge that precludes a tight fit with MSM0273, with docking giving poor packing (I_sc −12.38), resulting in even larger (∼14 Å) gaps between molecules. Similarly, poor packing was observed for MSM0273 in either orientation docking onto the optimal convex-outward MSM0272 complex. Although MSM0272 can form plausible self-interactions at the vertex, these position the hexameric rings in a way that makes productive docking of MSM0273 impossible.
Modeling MSM0275 self-interactions around the vertex suggests that this protein interacts significantly more strongly through the IC face than the ID face (I_sc −5.21 versus −3.31). The optimal packing is displaced about 5 Å laterally from the canonical interface, but in the opposite direction than that seen in MSM0272; this then results in a very different interface. The model has hydrogen bonds between Lys-56 and Ser-114 and between Asp-92 and Arg-193, with a small pore (∼3 Å) at the center of the interface. Docking MSM0273 into the IC complex, however, yields sub-optimal packing, with I_sc of −15.11, and significant gaps in the interface where interacting MSM0275 rings meet the MSM0273 pentamer.
In contrast with MSM0275, MSM0271 packs with noticeably better geometry along its ID face than its IC face (I_sc −6.09 versus −5.22). Optimized packing at the IC face pulled the molecules into configuration with geometry incompatible with a juxta-vertex ring; this solution was therefore not considered further. The interaction along the ID face positions the rings in an orientation analogous to that seen as optimal for MSM0275 (Fig. 5, C, F, and G). Similar to that interaction, Arg-171 hydrogen bonds with Glu-20 at the facet-like interface, and Arg-116 hydrogen bonds with Glu-112, residues provided by the longer helix α6 and the β4-β5 loop (joining domains). Leu-93 also packs against Arg-171 and Val-173, contributing hydrophobic stabilization. Effectively, the ID face in MSM0271 is padded out by these features, extending the interacting area when the proteins are oriented at a 141° angle. The buried interface is considerably larger (1802 Å2) than that observed for any of the other self-interactions modeled. Docking MSM0273 into this model results in a considerably better fit than any of the other models, with an I_sc of −21.38. This arrangement allows favorable electrostatic interactions between Glu-95 (MSM0271) and Arg-80 (MSM0273) and between Arg-98 and Asp-78, as well as various hydrogen bonds and favorable hydrophobic interactions (Fig. 5, D, E, and H). This configuration has no large gaps in the interface, allowing a good seal. Docking therefore suggests the preferred geometry for organizing the RMM vertex is with MSM0271 self-interacting through its ID face, with MSM0273 docking into the resulting pentameric hole with its base facing outward.
Discussion
The four proteins whose structures are described above are presumed to together build the shell of the RMM microcompartment. In particular, the organization of these building blocks must somehow account for all architecturally distinct environments in the polyhedral shell (at minimum bulk facet, edge, vertex, and juxta-vertex) and provide passage for all metabolites that need to enter or leave the microcompartment.
Substrates, Transport, and Selectivity
Bacterial microcompartment shells need to allow passage of all substrates and products required in the reactions of the encapsulated enzymes. However, the need to transport stoichiometric quantities of bulky co-factors is now understood to be bypassed by pairing reactions that generate and consume each co-factor within the shell (29) (for example, the NAD(P)H required for the alcohol dehydrogenase and aldehyde dehydrogenase encoded in the RMM operon is likely to be handled in this way). The presence of an aminotransferase homolog, as well as work indicating that the alcohol dehydrogenase acts on 1-amino-2-propanol, strongly indicates that the initial substrate for the RMM is an amine. MSM0272 has a highly electronegative central pore lined with aspartate residues (Fig. 3, D–G). This motif seems well suited to interact with an amine substrate, and likely it provides the substrate ingress channel. The nature of the product(s) and route of egress are more difficult to discern. The presence of the aldehyde dehydrogenase implies that one of the products is likely anionic (e.g. a carboxylic acid or a phosphate ester), making it a problematic substrate for the highly anionic MSM0272 pore; an amine acceptor and its product (e.g. α-ketoglutarate/glutamate) may also require passage. Possibly the more electropositive central pore in MSM0273 might provide a channel, although there are likely only 12 copies of this complex in the shell (one at each vertex of an icosahedral particle) limiting flux, and there is little that differentiates this pore from all related BMC-P proteins, meaning that there is unlikely to be much specificity associated with this pore. The fact that the number of distinct central pores is likely smaller than the number of substrates passaged is interesting as it implies that either these pores are not the only available route or the shell has limited substrate specificity. An alternative model is that specific metabolite channels form where packing of specific faces of specific shell proteins leaves a gap. This model has the advantage that the pores can, like the substrates, be asymmetric, allowing a greater degree of specificity; using double-shelled BMC-FP proteins in this manner has the advantage that the second pseudo-hexamer can provide an additional set of buttressing interactions (as seen for the MSM0275 canonical interaction) that compensates from the loss of interaction energy where a pore is formed. This idea of specific pores emerging from the interaction between appropriate pairs of shell proteins is interesting but difficult to address experimentally.
Transport Roles of BMC-FP Proteins
Previous work on BMC-FP proteins has led to the proposal that these act as gated channels, where weak binding of a metabolite within the pore opens the exterior gate (15). It is not clear whether this would apply to MSM0271 and MSM0275, especially as stochastic gate opening events would be required to first allow entry of gate triggering metabolites from the microcompartment lumen. We propose instead that the gates at either end may open randomly through thermal fluctuations, allowing any small enough molecule (∼1 kDa) to enter or leave from either the cytoplasmic or luminal face. These permuted double-ring shell proteins would then function as sub-stoichiometric nonspecific transporters for larger metabolites, most importantly co-enzymes. Recent results have demonstrated that bacterial microcompartments maintain co-enzyme pools separate from the general metabolic pools, where NADH consumed in one reaction will be regenerated in another, for example (29). However, regeneration is likely not completely efficient over time. Any escape from the microcompartment of the metabolic intermediates between the two opposed reactions will lead to a gradual accumulation of the co-factor in one state or the other. In addition, NAD(P)H can be oxidized non-enzymatically by oxidizing agents and can also be damaged in side reactions (30); without some mechanism to replenish these cofactor pools, microcompartments would gradually lose efficacy as they age. Slow sub-stoichiometric exchange of co-factors with the larger cytoplasmic pool would ensure that these co-factors retain access to repair mechanisms and can be replenished if depleted. The cationic nature of the space between gates may help recruit co-enzymes, which are typically anionic due to their phosphate-rich nucleotide groups. In carboxysomes, this mechanism may be required to allow encapsulated Rubisco activase access to the ATP it requires to function (31, 32).
Organization of the RMM Shell
The RMM microcompartment investigated here is the first BMC for which the structures of all candidate shell proteins are known. These structures together strongly constrain the possible organization of the shell, given the requirement that a largely sealed capsule needs to be generated from interactions between these four proteins. In keeping with what has previously been discovered with other microcompartments, MSM0272 is a straightforward hexameric BMC closely related to PduA that has canonical edge interaction motifs that should allow it to readily tile in flat sheets to form the shell facets (28). MSM0273 seems a typical BMC-P protein; the role of these proteins in plugging pentameric defects in the icosahedral shells that are created where five facets meet is generally accepted in the field (20, 21, 33). The role of the two BMC-FP proteins is more difficult to ascertain, as there are no published models that clearly suggest how they contribute to shell organization. We argue that, at least in the RMM microcompartments, these proteins play a critical role in forming the edges and vertex, a conclusion that has interesting geometric implications.
Incorporating BMC-FP protein(s) into a BMC shell introduces an important inner/outer layer distinction into descriptions of the shell. In particular, assuming that BMC-H/FP proteins can only interact strongly along their edges when arrayed in parallel (i.e. with their convex face facing the same direction), then the outer layer has its concave side, and the inner layer its convex side facing the lumen. Because the inter-ring interaction geometry of BMC-FP proteins seems to preclude them from readily organizing in continuous sheets, and the BMC-H protein should occupy only a single layer, the bulk of the facets probably show a mostly single-layered BMC-H protein-dominated organization, possibly with isolated BMC-FP proteins embedded (although this organization may not be true of β-carboxysomes, where abundant CcmK2 can form double-ringed dodecamers with geometry appropriate for extended tiling (34)). One critical constraint on shell organization is that, in common with many BMC-H proteins, MSM0272 is likely required to interact with helix-forming peptides fused onto the termini of encapsulated enzymes. Because this occurs through the α3-helix on the BMC-H concave face (27), MSM0272 proteins should strongly prefer to be localized in the outer shell layer. However, modeling of possible interactions between BMC-H/FP proteins and the BMC-P at the vertex seems to strongly favor models with concave-outward interactions in the inner layer. Docking between MSM0272 rings does not produce favorable packing in vertex models where these hexamers are assumed to be localized to the outer layer; placing MSM0272 at the inner layer of the vertex also seems unlikely, given that it does not seem to form favorable packing interactions with MSM0273, and interactions with the encapsulated enzyme targeting peptides will in any case tend to pre-orient it with its concave side inward. Docking studies suggest that the vertex is built from MSM0271 interacting with MSM0273 through its inner ring, with ID faces mediating MSM0271 self-interactions, and the IC faces mediating interactions with MSM0273. However, this model is only possible if the edges of the particle also meet in the inner layer of the shell.
To date, there has been relatively little discussion in the literature of the organization of the microcompartment edge. The edge of the facet truncates the regular pattern of horizontal tiling, and therefore it leaves a serrated edge that must somehow be welded to a similar edge. It is worth noting that the particle vertex is formed by the terminal members of this edge pattern. The only detailed model for the organization of the edge of a microcompartment proposed to date is for the Eut microcompartment, where EutS forms a bent wedge-shaped hexamer that could join two facets at the appropriate angle (16); however, it should be noted that both the Clostridium EutS homolog and the closely related protein PduU form conventional symmetric hexameric rings, raising questions as to the wider applicability of this model (17, 35). The RMM seems to lack a similar protein, meaning that one or more of the other shell proteins is required to stabilize the edge through a different mechanism. In MSM0275, tight canonical interfaces are found at alternating facets, organizing the molecules into linear strips in a zigzagged fashion (Figs. 4E and 6). The close lattice match between MSM0275 and PduA (a close homolog of MSM0272) suggests that these strips could be seamlessly added to the edge of a facet made of MSM0272 hexamers. Two such edging strips would then meet at a 141° angle to form an edge. Although the interaction geometry likely does not allow tight interactions between the bodies of the rings in all positions, the extensible β1-α1 loop, unrestricted by tight packing, could extend and organize so as to maximize interactions with the abutting strip, sealing the gap. The emergence of this motif from the equator of the hexamer is useful in this regard as these motifs would be exposed from both rings where molecules are bent back at the edge. Parallel lines of MSM0275 molecules that meet in a hinge-like arrangement and are sealed by this adaptable loop would therefore provide a mechanism for mediating sealing the edges where two facets meet (see Fig. 6).
FIGURE 6.
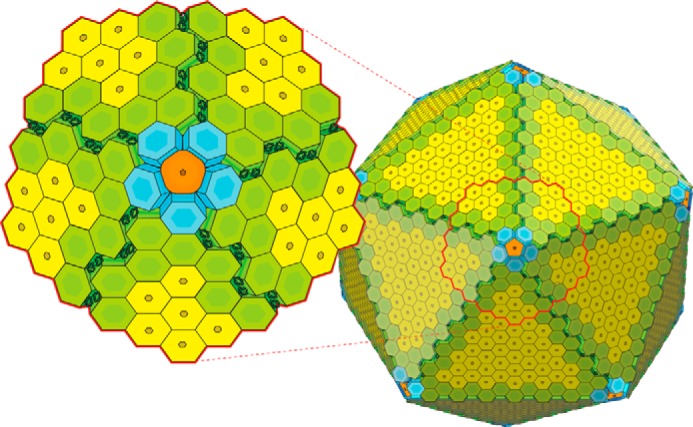
Overall model for the organization of the RMM shell. MSM0273 (orange), the BMC-P protein occupies the vertices, surrounded by a ring of the BMC-FP protein MSM0271 (blue) at each vertex. Interactions at the vertex occur through only the inner ring of MSM0271. The other BMC-FP protein MSM0275 (green) is organized in long strips that line the edge of the facets. The bulk of the facet is made of MSM0272 (yellow) the BMC-H protein. Two lines of MSM0275 proteins meet at the edge. Here, an imperfect geometric match between the MSM0275 strips is compensated for by packing the extendable loops between the adjacent rings.
In conclusion, we present for the first time the structures of a complete set of microcompartment shell proteins. These strongly constrain the possible architecture and transport strategies of the RMM microcompartment. The BMC-FP proteins in particular present several novel features that may point to an important role in shell architecture, including the ability to form well sealed edges in the icosahedral particle.
Experimental Procedures
DNA Methods
Mycobacterium smegmatis mc2 155 was grown in LB and after heat killing and centrifugation, genomic DNA was purified following the Invitrogen mini-prep kit procedure. For cloning, genes were amplified from genomic DNA using PfuX7 (a generous gift from Dr. D. Christendat) and ligated into vectors so as to leave either an N-terminal (pET28a) or a C-terminal (pET22b) hexa-histidine tag. MSM0271 (UniProt accession no. A0QP48_MYCS20) was amplified using primers GGTTCGCATATGGTCGCACCGGAAACCGAGAGGATCCGTACC and GGTTCGCTCGAGTCAGTGTTCCTGCCCTTCGATCGCGCTCAGC and ligated into the NdeI and XhoI sites of pET28a. MSM0272 (UniProt accession no. A0QP49_MYCS2) was amplified using primers GAGCTACATATGTCCAGCAACGCAATCGGATTGATC and GATGCTCTCGAGCTTGCTGGACACCGAGAAGTGC and ligated into the NdeI and XhoI sites of pET22b. MSM0273 (UniProt accession no. A0QP50_MYCS2) was amplified using primers ACGTATCTCGAGCTCGGCGGGGTTGCTATCTGATCTG and TAGCAACATATGTTGAGAGCGACCGTCACCGGCAATG and ligated into the NdeI and XhoI sites of pET22b. MSM0275 (UniProt accession no. A0QP52_MYCS2) was amplified using primers TCGGGTCATATGGCAGAACTACGTTCCTTCATCTTCATCG and ATTTCAAAGCTTCGCACCCTGCAGGACGGCGAGC and ligated into the NdeI and HindIII sites of pET22b. Plasmids were propagated in Escherichia coli DH5α cells and sequenced at the University of Guelph Agriculture and Food Laboratory Services facility. Plasmids containing the correct sequences were used to transform E. coli BL21 cells for overexpression.
Protein Expression and Purification
Large volume cultures (1 liter of 2YT media with appropriate antibiotic 30 μg/ml kanamycin for pET28a or 50 μg/ml ampicillin for pET22b) were inoculated with a small volume of overnight cultures and incubated at 37 °C with shaking until an optical density of 0.8 at 600 nm was reached. Cultures were induced for protein expression by addition of 0.4 mm isopropyl β-d-1-thiogalactopyranoside, followed by incubation for 18–20 h at 16 °C with shaking. Cultures were pelleted by centrifugation at 4 °C for 20 min at 4400 × g. Cell pellets were resuspended in 35 ml of lysis buffer (20 mm Tris, pH 8, 500 mm NaCl; lysis buffer for MSM0272 contained added 10% glycerol) prior to freezing at −20 °C.
Pellets were thawed at 4 °C prior to cell disruption by sonication. The insoluble fraction of cell lysate was pelleted by centrifugation (at 4 °C for 30 min at 48,000 × g), and supernatant was filtered through a 0.45-μm filter prior to loading on a 1-ml nickel-nitrilotriacetic acid-agarose column for immobilized metal affinity chromatography purification. Bound proteins were washed with 10 ml of the respective lysis buffer followed by 10 ml of wash buffer (lysis buffer plus 40 mm imidazole). Proteins were eluted with 10 ml of elution buffer (lysis buffer plus 500 mm imidazole).
Crystallization
For crystallization, MSM0271, MSM0273, and MSM0275 were dialyzed into crystallization buffer (20 mm Tris, pH 8.0, 150 mm NaCl), whereas MSM0272 was kept in elution buffer. Proteins were concentrated by centrifugal concentration, and the final concentration was measured using ninhydrin. MSM0271 (10 mg/ml) was crystallized as hexagonal prisms by sitting drop vapor diffusion at room temperature with a protein solution to crystallization solution (0.8 m sodium/potassium tartrate, 0.1 m Tris, pH 8.5, 0.5% (w/v) PEG 2000MME) ratio of 2:1 or 1:1. MSM0272 (7 mg/ml) was crystallized as tetragonal prisms by sitting drop vapor diffusion at room temperature with a protein solution to crystallization solution (0.1 m Tris, pH 7.5, 11% ethylene glycol, 10% PEG 8000) ratio of 2:1 or 1:1. MSM0273 (10 mg/ml) was crystallized as hexagonal prisms by sitting drop vapor diffusion at room temperature with a protein solution to crystallization solution (90 mm sodium acetate, pH 4.5, 1.8 m NaCl, 10 mm tris(2-carboxyethyl)phosphine) ratio of 2:1 or 1:1. MSM0275 (20 mg/ml) was crystallized as needles by sitting drop vapor diffusion at room temperature with a protein solution to crystallization solution (0.5 m sodium citrate, pH 5.6, 20% PEG3350) ratio of 2:1 or 1:1. Crystals were cryoprotected with Paratone® N (MSM0271, MSM0272, and MSM0275) or a 3:5 v/v glycerol/mother liquor mixture (MSM0273) prior to flash-freezing in liquid nitrogen.
Data Collection and Structure Determination
All datasets were collected at the Canadian Light Source beamline 08ID at 100 K using a wavelength of 0.97949 Å. Data were processed using XDS and scaled using XSCALE (36). All structures were phased using molecular replacement by Phaser (37) in Phenix and initially rebuilt using Autosol (Phenix) (38). Structures were iteratively manually rebuilt in Coot (39), followed by refinement in Phenix. MSM0273 was in space group P3121 and diffracted to 1.6 Å; a pentameric polyalanine model of CcmL (PDB code 2QW7) was used for molecular replacement searches. MSM0272 was crystallized in space group P43 and diffracted to 2.2 Å; a EutM hexamer (PDB code 4AXJ) was used as a search model for molecular replacement. MSM0275 was in the space group C2221, diffracted to 2.1 Å, and solved using a hexamer of CcmP (PDB code 4HT5) as a molecular replacement search model. MSM0271 was in the P6322 space group and diffracted to 2.7 Å; a trimer of MSM0275 was used as a search model for molecular replacement. Ramachandran statistics for percent outliers/favored were 0/99.5, 0.2/97.2, 0/97.8, and 0.3/96.6 for MSM0273, MSM0272, MSM0271, and MSM0275, respectively. Structure statistics are reported in Table 1. All structure figures were prepared in PyMOL.
TABLE 1.
Data collection and refinement statistics
| MSM0273 | MSM0272 | MSM0271 | MSM0275 | |
|---|---|---|---|---|
| PDB code | 5L37 | 5L38 | 5SUH | 5L39 |
| Data collectiona | ||||
| Space group | P3121 | P43 | P6322 | C2221 |
| Cell dimensions | ||||
| a, b, c (Å) | 66.97, 66.97, 140.09 | 93.60, 93.60, 192.54 | 139.89, 139.89, 150.12 | 132.14, 143.48, 116.99 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 | 90, 90, 120 | 90, 90, 90 |
| Resolution (Å) | 50–1.6 (1.64–1.6) | 50–2.2 (2.26–2.2) | 50–2.7 (2.77–2.70) | 50–2.1 (2.15–2.10) |
| Rsym | 0.064 (0.875) | 0.063 (0.745) | 0.133 (0.979) | 0.109 (0.897) |
| I /σI | 17.7 (2.3) | 18.4 (2.5) | 19.2 (2.1) | 12.0 (2.4) |
| Completeness (%) | 0.992 (0.984) | 1.00 (1.00) | 0.999 (1.00) | 1.00 (1.00) |
| Redundancy | 7.3 (7.1) | 5.1 (5.1) | 9.6 (11.1) | 7.5 (7.5) |
| Refinement | ||||
| Resolution (Å) | 45.8–1.6 | 48–2.2 | 47.1–2.7 | 48.6–2.1 |
| No. of reflections | 52,789 | 83,642 | 24,385 | 64,967 |
| Rwork/Rfree | 0.154/0.181 | 0.20/0.233 | 0.173/0.224 | 0.182/0.224 |
| No. of atoms | ||||
| Protein | 2890 | 4228 | 4685 | 8826 |
| Ligand/ion | 5 | |||
| Water | 370 | 925 | 26 | 334 |
| B-Factors | ||||
| Protein | 24.3 | 32.5 | 75.8 | 45.8 |
| Ligand/ion | 34.4 | |||
| Water | 37.1 | 43.0 | 57.2 | 46.7 |
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.017 | 0.004 | 0.010 | 0.005 |
| Bond angles (°) | 1.525 | 0.684 | 1.278 | 0.872 |
a All datasets were collected from a single crystal. Values in parentheses are for the highest resolution shell.
Docking
Docking was performed using Rosetta_Dock with flexible side chains in Rosetta 3.6 (40). Interacting molecules were pre-positioned using a customized script and then minimized using local docking refinement. For BMC-H/FP self-docking, poses were iteratively rotated in 0.5° increments and translated along the rotation axis in 0.5-Å increments, with a total of ∼1000 docking poses evaluated per candidate complex. Docking with MSM0273 in both a concave-in and concave-out conformation was conducted into a pentameric ring of previously docked BMC-H/FP proteins, utilizing rotations and translations around the central symmetry axis. Because of the computational expense of docking the large number of atoms involved and larger angular and translational ranges, sampling here was coarser (1°, 1 Å); ∼600 poses were tested per candidate interaction. Buried surface area was assessed using the CCP4 program areaimol (41).
Author Contributions
Both authors contributed to experimental design. E. M. was responsible for experimental work and contributed to writing and visualization. M. S. K. was responsible for modeling work, writing, visualization and acquiring funding.
Acknowledgments
We thank J. Alexiou and M. Connor who initiated studies on these proteins and P. Grochulski and S. Labiuk at Canadian Light Source for assistance in collecting diffraction data.
This work was supported by Natural Sciences and Engineering Research Council of Canada Grant 04045-2015 (to M. S. K.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5L37, 5L38, 5L39, and 5SUH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BMC
- bacterial microcompartment
- Rubisco
- ribulose-bisphosphate carboxylase/oxygenase
- RMM
- Rhodococcus and Mycobacterium microcompartment
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- ID
- inter-domain
- IC
- inter-chain.
References
- 1. Axen S. D., Erbilgin O., and Kerfeld C. A. (2014) A taxonomy of bacterial microcompartment loci constructed by a novel scoring method. PLoS Comp. Biol. 10, e1003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Espie G. S., and Kimber M. S. (2011) Carboxysomes: cyanobacterial RubisCO comes in small packages. Photosyn. Res. 109, 7–20 [DOI] [PubMed] [Google Scholar]
- 3. Heinhorst S., Cannon G. C., and Shively J. M. (2014) Carboxysomes and their structural organization in prokaryotes. Nanomicrobiology 10.1007/978-1-4939-1667-2_4 [DOI] [Google Scholar]
- 4. Chowdhury C., Sinha S., Chun S., Yeates T. O., and Bobik T. A. (2014) Diverse bacterial microcompartment organelles. Microbiol. Mol. Biol. Rev. 78, 438–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seedorf H., Fricke W. F., Veith B., Brüggemann H., Liesegang H., Strittmatter A., Miethke M., Buckel W., Hinderberger J., Li F., Hagemeier C., Thauer R. K., and Gottschalk G. (2008) The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc. Natl. Acad. Sci. U.S.A. 105, 2128–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Craciun S., and Balskus E. P. (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 21307–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kerfeld C. A., and Erbilgin O. (2015) Bacterial microcompartments and the modular construction of microbial metabolism. Trends Microbiol. 23, 22–34 [DOI] [PubMed] [Google Scholar]
- 8. Yeates T. O., Jorda J., and Bobik T. A. (2013) The shells of BMC-type microcompartment organelles in bacteria. J. Mol. Microbiol. Biotechnol. 23, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chowdhury C., Chun S., Pang A., Sawaya M. R., Sinha S., Yeates T. O., and Bobik T. A. (2015) Selective molecular transport through the protein shell of a bacterial microcompartment organelle. Proc. Natl. Acad. Sci. U.S.A. 112, 2990–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kerfeld C. A., Sawaya M. R., Tanaka S., Nguyen C. V., Phillips M., Beeby M., and Yeates T. O. (2005) Protein structures forming the shell of primitive bacterial organelles. Science 309, 936–938 [DOI] [PubMed] [Google Scholar]
- 11. Tanaka S., Sawaya M. R., Phillips M., and Yeates T. O. (2009) Insights from multiple structures of the shell proteins from the β-carboxysome. Protein Sci. 18, 108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dryden K. A., Crowley C. S., Tanaka S., Yeates T. O., and Yeager M. (2009) Two-dimensional crystals of carboxysome shell proteins recapitulate the hexagonal packing of three-dimensional crystals. Protein Sci. 18, 2629–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sinha S., Cheng S., Sung Y. W., McNamara D. E., Sawaya M. R., Yeates T. O., and Bobik T. A. (2014) Alanine scanning mutagenesis identifies an asparagine-arginine-lysine triad essential to assembly of the shell of the Pdu microcompartment. J. Mol. Biol. 426, 2328–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klein M. G., Zwart P., Bagby S. C., Cai F., Chisholm S. W., Heinhorst S., Cannon G. C., and Kerfeld C. A. (2009) Identification and structural analysis of a novel carboxysome shell protein with implications for metabolite transport. J. Mol. Biol. 392, 319–333 [DOI] [PubMed] [Google Scholar]
- 15. Cai F., Sutter M., Cameron J. C., Stanley D. N., Kinney J. N., and Kerfeld C. A. (2013) The structure of CcmP, a tandem bacterial microcompartment domain protein from the β-carboxysome, forms a subcompartment within a microcompartment. J. Biol. Chem. 288, 16055–16063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka S., Sawaya M. R., and Yeates T. O. (2010) Structure and mechanisms of a protein-based organelle in Escherichia coli. Science 327, 81–84 [DOI] [PubMed] [Google Scholar]
- 17. Crowley C. S., Sawaya M. R., Bobik T. A., and Yeates T. O. (2008) Structure of the PduU shell protein from the Pdu microcompartment of Salmonella. Structure 16, 1324–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pang A., Liang M., Prentice M. B., and Pickersgill R. W. (2012) Substrate channels revealed in the trimeric Lactobacillus reuteri bacterial microcompartment shell protein PduB. Acta Crystallogr. D Biol. Crystallogr. 68, 1642–1652 [DOI] [PubMed] [Google Scholar]
- 19. Rae B. D., Long B. M., Badger M. R., and Price G. D. (2012) Structural determinants of the outer shell of β-carboxysomes in Synechococcus elongatus PCC 7942: roles for CcmK2, K3-K4, CcmO, and CcmL. PLoS ONE 7, e43871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka S., Kerfeld C. A., Sawaya M. R., Cai F., Heinhorst S., Cannon G. C., and Yeates T. O. (2008) Atomic-level models of the bacterial carboxysome shell. Science 319, 1083–1086 [DOI] [PubMed] [Google Scholar]
- 21. Keeling T. J., Samborska B., Demers R. W., and Kimber M. S. (2014) Interactions and structural variability of β-carboxysomal shell protein CcmL. Photosyn. Res. 121, 125–133 [DOI] [PubMed] [Google Scholar]
- 22. Jorda J., Lopez D., Wheatley N. M., and Yeates T. O. (2013) Using comparative genomics to uncover new kinds of protein-based metabolic organelles in bacteria. Protein Sci. 22, 179–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kataoka M., Nakamura Y., Urano N., Ishige T., Shi G., Kita S., Sakamoto K., and Shimizu S. (2006) A novel NADP+-dependent l-1-amino-2-propanol dehydrogenase from Rhodococcus erythropolis MAK154: a promising enzyme for the production of double chiral aminoalcohols. Lett. Appl. Microbiol. 43, 430–435 [DOI] [PubMed] [Google Scholar]
- 24. Urano N., Kataoka M., Ishige T., Kita S., Sakamoto K., and Shimizu S. (2011) Genetic analysis around aminoalcohol dehydrogenase gene of Rhodococcus erythropolis MAK154: a putative GntR transcription factor in transcriptional regulation. Appl. Microbiol. Biotechnol. 89, 739–746 [DOI] [PubMed] [Google Scholar]
- 25. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fan C., Cheng S., Liu Y., Escobar C. M., Crowley C. S., Jefferson R. E., Yeates T. O., and Bobik T. A. (2010) Short N-terminal sequences package proteins into bacterial microcompartments. Proc. Natl. Acad. Sci. U.S.A. 107, 7509–7514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan C., Cheng S., Sinha S., and Bobik T. A. (2012) Interactions between the termini of lumen enzymes and shell proteins mediate enzyme encapsulation into bacterial microcompartments. Proc. Natl. Acad. Sci. U.S.A. 109, 14995–15000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crowley C. S., Cascio D., Sawaya M. R., Kopstein J. S., Bobik T. A., and Yeates T. O. (2010) Structural insight into the mechanisms of transport across the Salmonella enterica Pdu microcompartment shell. J. Biol. Chem. 285, 37838–37846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huseby D. L., and Roth J. R. (2013) Evidence that a metabolic microcompartment contains and recycles private cofactor pools. J. Bacteriol. 195, 2864–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Linster C. L., Van Schaftingen E., and Hanson A. D. (2013) Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 9, 72–80 [DOI] [PubMed] [Google Scholar]
- 31. Zarzycki J., Axen S. D., Kinney J. N., and Kerfeld C. A. (2013) Cyanobacterial-based approaches to improving photosynthesis in plants. J. Exp. Bot. 64, 787–798 [DOI] [PubMed] [Google Scholar]
- 32. Tsai Y., Sawaya M. R., and Yeates T. O. (2009) Analysis of lattice-translocation disorder in the layered hexagonal structure of carboxysome shell protein CsoS1C. Acta Crystallogr. D Biol. Crystallogr. 65, 980–988 [DOI] [PubMed] [Google Scholar]
- 33. Wheatley N. M., Gidaniyan S. D., Liu Y., Cascio D., and Yeates T. O. (2013) Bacterial microcompartment shells of diverse functional types possess pentameric vertex proteins. Protein Sci. 22, 660–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Samborska B., and Kimber M. S. (2012) A dodecameric CcmK2 structure suggests β-carboxysomal shell facets have a double-layered organization. Structure 20, 1353–1362 [DOI] [PubMed] [Google Scholar]
- 35. Pitts A. C., Tuck L. R., Faulds-Pain A., Lewis R. J., and Marles-Wright J. (2012) Structural insight into the clostridium difficile ethanolamine utilisation microcompartment. PLoS ONE 7, e48360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 39. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40. Wang C., Bradley P., and Baker D. (2007) Protein-protein docking with backbone flexibility. J. Mol. Biol. 373, 503–519 [DOI] [PubMed] [Google Scholar]
- 41. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]