

Abstract
Nod-like receptor family card containing 4 (NLRC4)/Ipaf is involved in recognition of pathogen-associated molecular patterns leading to caspase-1 activation and cytokine release, which mediate protective innate immune response. Point mutations in NLRC4 cause autoinflammatory syndromes. Although all the mutations result in constitutive caspase-1 activation, their phenotypic presentations are different, implying that these mutations cause different alterations in properties of NLRC4. NLRC4 interacts with SUG1 and induces caspase-8-mediated cell death. Here, we show that one of the autoinflammatory syndrome-causing mutants of NLRC4, H443P, but not T337A and V341A, constitutively activates caspase-8 and induces apoptotic cell death in human lung epithelial cells. Compared with wild type NLRC4, the H443P mutant shows stronger interaction with SUG1 and with ubiquitinated cellular proteins. Phosphorylation of NLRC4 at Ser533 plays a crucial role in caspase-8 activation and cell death. However, H443P mutant does not require Ser533 phosphorylation for caspase-8 activation and cell death. Caspase-8 activation by NLRC4 and its H443P mutant are dependent on the adaptor protein FADD. A phosphomimicking mutant of NLRC4, S533D does not require SUG1 activity for inducing cell death. Ubiquitin-tagged NLRC4 could induce cell death and activate caspase-8 independent of Ser533 phosphorylation. Our work suggests that SUG1-mediated signaling results in enhanced ubiquitination and regulates FADD-dependent caspase-8 activation by NLRC4. We show that the autoinflammation-associated H443P mutant is altered in interaction with SUG1 and ubiquitinated proteins, triggering constitutive caspase-8-mediated cell death dependent on FADD but independent of Ser533 phosphorylation.
Keywords: apoptosis, caspase, cell death, inflammasome, phosphorylation, ubiquitylation (ubiquitination)
Introduction
NOD-like receptor family CARD-containing 4 (NLRC4)4 is a cytoplasmic immune receptor involved in innate immune response. It induces caspase-1-mediated inflammation and pyroptosis in response to bacterial infection or upon detection of bacterial flagellin, type III secretion system rod, or needle proteins, recognition of which is mediated by various NAIP paralogs (1–4). NLRC4 has a distinct N-terminal caspase activation and recruitment domain (CARD), a nucleotide binding domain (NBD), and a leucine-rich repeat (LRR) domain that forms an autoinhibitory loop and is involved in intramolecular interactions with amino acid residues of NBD (5). Detection of an antigen triggers NLRC4 complexation, which results in activation of caspase-1 via structural changes such as reorientation of the autoinhibitory LRR domain away from NBD (5–8); an activated NLRC4 undergoes oligomerization through intermolecular interaction and recruits a number of other proteins including procaspase-1 to form a multiprotein complex known as inflammasome. The multiprotein complex formed by NLRC4 facilitates the proximity-dependent autocatalysis of caspase-1. Studies using murine proteins have shown that other NLR family members, NAIPs determine specificity of NLRC4 toward an antigen and also enable inflammasome formation (2, 4, 9). Protein modification such as phosphorylation at NLRC4-Ser533 has been shown to be crucial in NLRC4 inflammasome activation (10, 11); however, molecular events that follow NLRC4-Ser533 phosphorylation are poorly understood.
More recently, NLRC4 was shown to be expressed in non-hematopoietic gut cells (12, 13) and in lung epithelial cells (14). Given that alveolar epithelium of lung is repeatedly exposed to airborne pathogenic or inflammatory agents, the role of NLRC4 in protection against agents invading lung epithelium needs to be investigated. Although most studies have focused on caspase-1-mediated functions of NLRC4, we have earlier shown that in human alveolar epithelial adenocarcinoma cells, NLRC4 engages caspase-8 to induce apoptotic cell death. In these cells, cell death is independent of caspase-1 activation. The proteasomal component SUG1 directly interacts with NLRC4, leading to its ubiquitination and formation of a caspase-8-containing multiprotein complex resulting in caspase-8 activation and apoptotic cell death. An artificial deletion mutant expressing amino acids 1–561 of NLRC4 (ΔLRR-NLRC4) forms cytoplasmic aggregates and is constitutively active in inducing caspase-8-dependent cell death (15). Importantly, ΔLRR-NLRC4 fails to induce cell death upon knockdown of SUG1 or co-expression of K196M-SUG1, a dominant negative variant that lacks ATPase activity, indicating a crucial role for cellular SUG1 in NLRC4-mediated caspase-8-dependent cell death signaling. Molecular mechanisms involved in multiprotein complex formation by NLRC4 for caspase-8 activation may differ from those involved in caspase-1 activation and must be investigated.
Mutations in NLRC4 have been implicated in various autoimmune genetic disorders; a missense mutation T337S is known to cause recurrent macrophage activation syndrome and autoinflammation (16). Another mutation V341A results in enterocolitis and nearly fatal episodes of autoinflammation (17). More recently, the missense mutation H443P has been shown to result in hypersensitivity of the heterozygotes toward cold exposure and has been implicated to cause familial cold autoinflammatory syndrome (FCAS) in a non-consanguineous Japanese family (18). These three mutants cause constitutive caspase-1 activation and IL-1β production. However, it is likely that these mutants are altered in some other properties that lead to different disease phenotypes.
To study function of NLRC4 and consequences of its mutations on caspase-8 activation, we used the human alveolar epithelial adenocarcinoma cell line (A549), which represents a model for studies on lung biology. We provide evidence for the role of FADD in caspase-8 activation by an NLR family cytosolic receptor NLRC4. A FCAS causing mutant of NLRC4, H443P, otherwise known to mediate caspase-1-dependent inflammation, also induces caspase-8-dependent and caspase-1-independent apoptosis in A549 cells. This mutant shows enhanced interaction with SUG1, which mediates caspase-8 activation and cell death. We also identify the requirement of NLRC4-Ser533 phosphorylation for caspase-8-mediated apoptotic cell death and show that NLRC4-H443P is not dependent on this phosphorylation for caspase-8 activation and induction of cell death. Thus our results show that the H443P mutant is altered in several properties in addition to constitutive caspase-1 activation reported previously.
Results
NLRC4-H443P Constitutively Activates Caspase-8-dependent Cell Death Signaling
Missense mutations H443P, T337S, and V341A in NLRC4 result in constitutive activation of NLRC4 triggering caspase-1-mediated inflammatory response and have been implicated in various sterile autoinflammatory diseases (16–18). Positions of these mutations are indicated in Fig. 1A. NLRC4 mediates caspase-8-dependent apoptotic cell death independent of caspase-1 in lung alveolar epithelial cells (15). To test whether the missense mutations H443P, T337S, and V341A in NLRC4 result in caspase-8 activation, GFP fusion proteins of WT-NLRC4, NLRC4-H443P, NLRC4-T337S, and NLRC4-V341A were transiently expressed in A549 cells for 20 h, and the cells were examined for subcellular localization of overexpressed protein and features of cell death such as nuclear fragmentation, cytoplasmic shrinkage, and membrane blebbing. We observed that all the mutants show diffused cytoplasmic distribution (supplemental Fig. S1A), a pattern similar to that of WT-NLRC4. NLRC4-H443P induced apoptotic cell death in a significantly greater number of expressing cells compared with WT-NLRC4, NLRC4-T337S, and NLRC4-V341A (Fig. 1B). Cell death induced by H443P mutant was further confirmed using an apoptosis assay kit, which detects phosphatidylserine and membrane integrity as apoptotic markers. We observed that H443P mutant indeed induced apoptosis in a significantly higher number of expressing cells compared with WT-NLRC4 or T337S or V341A mutants (Fig. 1C). Western blotting analysis of whole cell lysates of A549 cells expressing NLRC4 and NLRC4-H443P showed significantly higher caspase-8 activation in NLRC4-H443P-expressing cells compared with cells expressing WT-NLRC4 (Fig. 1D) as detected by abundance of p43/p41 cleaved fragment of pro-caspase-8, an indicator of active caspase-8. We also confirmed that expression of all three mutants constitutively activates caspase-1 (Fig. 1E) as described earlier (18). To determine the roles of caspase-8 and caspase-1 in cell death induced by NLRC4-H443P, we co-expressed catalytically inactive C360A-caspase-8 (mCaspase-8) or catalytically inactive mCaspase-1 along with NLRC4-H443P in A549 cells for 20 h and quantitated cell death. Co-expression of mCaspase-8 showed a significant reduction in cell death induced by NLRC4-H443P, whereas co-expression of mCaspase-1 had no significant effect (Fig. 1F). Levels of overexpressed proteins indicated in the Western blot showed that reduced cell death was not due to reduced expression of NLRC4-H443P in mCaspase-8-expressing cells. These observations suggest that NLRC4-H443P induces apoptotic cell death mediated by caspase-8 and independent of caspase-1. We have earlier shown that caspase-8 activation by NLRC4 is not restricted to only one cell line and that ΔLRR-NLRC4 could activate caspase-8 in MCF-7 cells, as well as in human macrophage THP-1 cells (15). Unlike THP1 cells, A549 or HEK293 cells do not show detectable levels of NLRC4 and caspase-1 (supplemental Fig. S1C). To further exclude that effects of NLRC4-H443P mutant are restricted to A549 cells, we examined levels of apoptosis in HEK293 cells expressing WT-NLRC4 or H443P mutant by morphological criteria, as well as annexin-V/7-AAD staining. Indeed, NLRC4-H443P induced significantly higher apoptotic cell death compared with WT-NLRC4 (Fig. 1G).
FIGURE 1.

NLRC4-H443P induces caspase-8-dependent apoptotic cell death. A, schematic showing domain organization of NLRC4 and sites of missense mutations H443P, T337S, and V341A. B, effect of expression of NLRC4 mutants on survival of A549 cells. WT-NLRC4 and indicated mutants were expressed in A549 cells plated on coverslips. B and C, cell death was quantitated by morphological criteria (B) and annexin V/7-AAD staining (C) after 20 h in expressing and non-expressing cells. The data represent means ± S.D. (n = 4). ***, p < 0.0005. D, lysates of untransfected (UT) A549 cells or those expressing indicated constructs were analyzed for caspase-8 activation using Cl.Casp-8 antibody. Lysates of TNF/cycloheximide (CHX)-treated A549 cells were used as positive control for Cl.Casp-8. GAPDH was used as loading control. Representative Western blot and quantitation from four independent experiments is shown in the bar diagram. *, p < 0.05. E, effect of NLRC4 and its mutants H443P, T337S, and V341A on activation of caspase-1. Whole cell lysates of HEK cells co-expressing caspase-1 and WT-NLRC4 or the indicated mutants were subjected to Western blotting analysis to detect levels of cleaved caspase-1. F, effect of co-expressing catalytically inactive mutants of caspase-1 and caspase-8 on NLRC4-H443P-induced apoptosis (n = 4). **, p < 0.005. Lysates of untransfected A549 cells or those expressing the indicated plasmids were analyzed by Western blotting to check the levels of overexpressed proteins. G, effect of NLRC4-H443P mutant on survival of HEK293 cells was examined by morphological criteria and annexin V/7-AAD staining. Bar diagrams indicate means ± S.D. (n = 4). **, p < 0.005. The expression levels of the indicated proteins is shown in blot. Exp, expressing.
SUG1 Mediates NLRC4-H443P-induced Cell Death and Caspase-8 Activation
SUG1 is a 26S proteasomal component involved in cellular homeostasis and other functions like transcriptional regulation (19–21). Previous work from our laboratory has shown that SUG1 physically interacts with NLRC4 and enables it to induce apoptotic cell death, suggesting a possible role for SUG1 in innate immune response. To test whether NLRC4-H443P-mediated cell death signaling involves SUG1, we co-expressed catalytically inactive K196M-SUG1, which dominantly inhibits endogenous SUG1 activity, and NLRC4-H443P (Fig. 2A) in A549 cells and examined cell death. Co-expressing K196M-SUG1 with NLRC4-H443P significantly inhibited NLRC4-H443P-induced cell death, as well as caspase-8 activation (Fig. 2B). Expression of NLRC4 constructs or K196M-SUG1 did not alter levels of endogenous SUG1 (supplemental Fig. S1D). Caspase-8 activation was detected by immunostaining using cleaved caspase-8 antibody, which specifically detects only the cleaved fragments of human caspase-8 that indicate caspase-8 activation. We observed that fewer cells co-expressing NLRC4-H443P and K196M-SUG1 show the presence of Cl.Casp-8 compared with cells expressing NLRC4-H443P alone (Fig. 2B). Western blotting analysis of whole cell lysates of A549 cells co-expressing NLRC4-H443P and K196M-SUG1 for 20 h also showed reduction in procaspase-8 cleavage, indicating its reduced activation (Fig. 2C). In addition, shRNA-mediated knockdown of endogenous SUG1 resulted in reduced NLRC4-H443P-mediated cell death (Fig. 2D). SUG1 knockdown was confirmed by Western blotting analysis (Fig. 2E).
FIGURE 2.

NLRC4-H443P induces SUG1-mediated cell death. A, schematic showing domain organization of SUG1 and regions of NLRC4 involved in SUG1 and LRR binding. * shows site of phosphorylation at Ser-533. B, bar diagram shows quantitation of apoptosis or Cl.Casp-8 positivity in A549 cells expressing NLRC4-H443P with or without catalytically inactive K196M-SUG1 for 20 h (n = 4). *** p < 0.0005. C, Western blotting analysis of lysates of A549 cells expressing indicated plasmids showing caspase-8 activation. Lysates of TNF/cycloheximide (CHX)-treated A549 cells were used as positive control for Cl.Casp-8. Actin was used as loading control. D, knockdown of endogenous SUG1 reduces NLRC4-H443P-induced cell death. Quantitation of apoptosis in A549 cells upon expression of NLRC4-H443P and co-transfection of indicated plasmids for 24 h is shown (n = 4). **, p < 0.005. E, Western blot shows efficacy of SUG1 knockdown by shRNA. Con, control.
Because SUG1 interacts with NBD and H443P mutation is in the winged helix domain (WHD) of NLRC4 (supplemental Fig. S2A), we therefore wished to determine the domain requirement for caspase-8 activation by NLRC4. Using various deletion constructs of NLRC4, we observed that CARD, as well as NBD (including WHD) are required for caspase-8 activation and induction of cell death (supplemental Fig. S2, B and C). Truncated variants having CARD alone, NBD and WHD alone, or the SUG1-interacting region alone do not induce cell death.
NLRC4-H443P Shows Altered Interaction with SUG1 and Ubiquitinated Proteins
SUG1 has been shown to physically interact with NLRC4, and a region spanning amino acids 91–253 in NBD of NLRC4 (Fig. 2A) is sufficient for SUG1 interaction (15). Given that NLRC4-H443P-induced cell death is mediated by SUG1, we tested whether NLRC4-H443P shows any altered interaction with endogenous SUG1. GFP-tagged NLRC4-H443P and WT-NLRC4 were overexpressed in HEK293T cells and whole cell lysates subjected to immunoprecipitation using GFP antibody. Western blotting analysis of the precipitates showed that higher levels of endogenous SUG1 coprecipitated with NLRC4-H443P than with WT-NLRC4 (Fig. 3, A and B). We also examined the level of ubiquitinated proteins in the immunoprecipitates and found that compared with WT-NLRC4, the H443P mutant immunoprecipitates showed a significantly higher level of cellular ubiquitinated proteins (Fig. 3, A and C). These results suggest that compared with WT-NLRC4, the H443P mutant shows stronger interaction with endogenous SUG1 and ubiquitinated proteins. The ubiquitination of the H443P mutant was also significantly higher compared with WT-NLRC4, as determined by quantitation of the corresponding band in the ubiquitin blots (Fig. 3, A and D). We also observed that, unlike NLRC4-H443P, the V341A mutant neither showed enhanced interaction with SUG1 nor higher ubiquitination of coprecipitated cellular proteins (Fig. 3E).
FIGURE 3.

NLRC4-H443P shows stronger interaction with SUG1 and cellular ubiquitinated proteins relative to WT-NLRC4. A, immunoprecipitation assay showing co-precipitation of endogenous SUG1 with NLRC4 and NLRC4-H443P. HEK293T cells were transfected with the indicated plasmids for 16 h. GFP-tagged proteins were pulled down using agarose-conjugated GFP antibody (GFPTrap), and the complexes were analyzed by Western blot using SUG1, ubiquitin, and GFP antibodies. The arrow indicates position of GFP-NLRC4. B and C, bar diagrams show relative amounts of SUG1 (B) and ubiquitinated proteins (C) co-precipitated with H443P mutant compared with NLRC4, averaged from four independent assays normalized with GFP signal. D, relative levels of ubiquitination on H443P mutant compared with NLRC4 is shown. *, p < 0.05; **, p < 0.005. E, blot showing co-precipitation of endogenous SUG1 with NLRC4 and NLRC4-V341A mutant. F, bar diagram showing effect of co-expressed K48R and K63R mutants of ubiquitin on cell death induced by NLRC4-H443P (n = 4). ***, p < 0.0005. Western blot shows expression of indicated proteins. Con, control; Exp, expressing; IP, immunoprecipitation; WB, Western blot; WCL, whole cell lysate.
Ubiquitination Is Required for NLRC4-H443P-induced Cell Death
Because H443P mutant-induced cell death is dependent on SUG1 and SUG1 is known to mediate ubiquitination of proteins including NLRC4 (15), it is likely that NLRC4-H443P-induced cell death is dependent on ubiquitination. We examined this possibility by using K48R and K63R mutants of ubiquitin that block polyubiquitination by inhibiting Lys48-linked and Lys63-linked chain elongation, respectively (22). Co-expression of K48R-ubiquitin strongly inhibited NLRC4-H443P-induced cell death, whereas wild type ubiquitin had no significant effect (Fig. 3F). Co-expression of K63R-ubiquitin also inhibited cell death, although it was less effective than K48R mutant (Fig. 3F). These results suggest that Lys48-linked polyubiquitination and to a lesser extent Lys63-linked polyubiquitination mediates NLRC4-H443P-induced cell death.
FADD Is Required for NLRC4-mediated Cell Death and Caspase-8 Activation
FADD is well known for its role in Fas-mediated cell death signaling (23). FADD and caspase-8 have been shown to play crucial role in many cell death and inflammation pathways (11, 24–28). We tested whether FADD is involved in cell death signaling mediated by NLRC4. dnFADD is a deletion construct that lacks death effector domain (DED) and acts as a dominant negative for functional FADD (Fig. 4A). A truncated variant of NLRC4 which lacks the LRR domain (Fig. 4A) is constitutively active in inducing caspase-8 activation and cell death (15). We overexpressed the two constitutively active forms, ΔLRR-NLRC4 and NLRC4-H443P, with or without dnFADD in A549 cells and examined cells for apoptotic features. We found that dnFADD significantly inhibited cell death induced by NLRC4-H443P (Fig. 4B) and ΔLRR-NLRC4 (Fig. 4E). Expression level of proteins were checked by Western blotting (Fig. 4, D and F) to confirm that the difference in extent of cell death was not due to differences in expression of NLRC4-H443P or ΔLRR-NLRC4. As expected, co-expression of dnFADD with NLRC4-H443P reduced the number of Cl.Casp-8-positive cells (Fig. 4C). Reduced Cl.Casp-8 was also seen in the lysates of cells co-expressing ΔLRR-NLRC4 with dnFADD (Fig. 4F). Earlier, our work has shown that co-expression of SUG1 with WT-NLRC4 enables it to initiate caspase-8-dependent apoptotic cell death. We found that FADD is also required for NLRC4-SUG1-mediated cell death because co-expressed dnFADD could rescue apoptotic cell death (Fig. 4G) induced by co-expressed NLRC4 and SUG1.
FIGURE 4.

FADD mediates caspase-8 activation and cell death induced by NLRC4-H443P or NLRC4 co-expressed with SUG1. A, schematic showing domain organization of NLRC4 and FADD, and deletion constructs. * shows site of phosphorylation at Ser-533. B and C, quantitation of apoptosis in A549 cells upon transfection of indicated plasmids for 20 h by morphological criteria (B) and Cl.Casp-8 positivity (C) (n = 4). **, p < 0.005. D, Western blot shows expression of indicated proteins. E, effect of dnFADD on cell death induced by ΔLRR-NLRC4 (n = 6). ***, p < 0.0005. E and F, indicated plasmids were transfected in A549 cells for 24 h and quantitated for apoptosis (E) and analyzed by SDS-PAGE for levels of Cl.Casp-8 (F). G, FADD is required for cell death induced by NLRC4 co-expressed with SUG1. The bar diagram shows quantitation of apoptosis induced by NLRC4 co-expressed with SUG1 in presence or absence of dnFADD (n = 4). ***, p < 0.0005. H, immunoprecipitation assay showing complex formation of NLRC4 and ΔLRR-NLRC4 with dnFADD. GFP-NLRC4 and GFP-ΔLRR-NLRC4 were co-expressed with HA-tagged dnFADD in HEK293T cells. Lysates were immunoprecipitated using HA antibody and blotted with GFP and HA antibodies. The arrows indicate GFP-NLRC4 and GFP-ΔLRR-NLRC4. Con, control; Exp, expressing; IP, immunoprecipitation; WB, Western blot; WCL, whole cell lysate.
To further investigate the role of FADD in cell death signaling induced by NLRC4, we examined whether FADD is part of the multimolecular complex formed by NLRC4 or ΔLRR-NLRC4. Because overexpression of FADD induced extensive cell death, HA-tagged dnFADD was overexpressed in HEK293T cells along with GFP fusion proteins of NLRC4 or ΔLRR-NLRC4 for 24 h; whole cell lysates were subjected to immunoprecipitation using HA antibody and analyzed for the presence of NLRC4 or ΔLRR-NLRC4 using GFP antibody. We observed that WT-NLRC4, as well as the constitutively active ΔLRR-NLRC4, was present in immunoprecipitates of dnFADD (Fig. 4H).
Ser533 Phosphorylation Is Required for NLRC4-mediated Cell Death Signaling
Ser533 of NLRC4 is an evolutionarily conserved residue, and its phosphorylation has been shown to be critical for NLRC4 inflammasome activation (10). The role of Ser533 phosphorylation in NLRC4-induced cell death signaling remains uncharacterized to date. We generated GFP-tagged phosphodefective mutants NLRC4-S533A and ΔLRR-NLRC4-S533A, as well as phosphomimetic mutants NLRC4-S533D and ΔLRR-NLRC4-S533D. The phosphomimetic mutant GFP-NLRC4-S533D showed diffused cytoplasmic distribution like WT-NLRC4 (supplemental Fig. S1B) and induced a significantly higher percentage of cell death and caspase-8 activation in expressing cells compared with GFP-NLRC4-S533A or GFP-NLRC4 (Fig. 5, A and B), indicating a crucial role for Ser533 phosphorylation in apoptotic signaling. As expected, Western blotting analysis confirmed increased Cl.Casp-8 levels in GFP-NLRC4-S533D-expressing cells (Fig. 5C). We also observed that transient expression of NLRC4-S533D resulted in significantly higher number of cleaved caspase-3-positive cells compared with WT-NLRC4, as detected by immunostaining using a cleaved caspase-3 antibody which specifically detects only the active fragment, further confirming apoptotic cell death (data not shown). As described earlier (10), we also observed that S533D mutant constitutively activates caspase-1 (Fig. 5D). These results suggested that the phosphomimetic mutant of NLRC4 was constitutively active with respect to caspase-1, as well as caspase-8 activation. Overexpressed GFP-ΔLRR-NLRC4-S533A in A549 cells formed cytoplasmic aggregates much like ΔLRR-NLRC4 but induced significantly lower cell death compared with the WT form (Fig. 5E). Interestingly, the phosphomimetic GFP-ΔLRR-NLRC4-S533D did not show enhanced cell death compared with GFP-ΔLRR-NLRC4 (Fig. 5E). Overexpression of phosphodefective mutant GFP-ΔLRR-NLRC4-S533A shows a significantly lower percentage of Cl.Casp-8-positive cells compared with GFP-ΔLRR-NLRC4 or GFP-ΔLRR-NLRC4-S533D (Fig. 5F). SUG1 co-expression enables NLRC4 to induce caspase-8-mediated cell death; however, A549 cells co-expressing SUG1 and GFP-NLRC4-S533A did not show enhanced apoptosis (Fig. 5G).
FIGURE 5.

NLRC4-mediated cell death and caspase-8 activation is dependent on Ser533 phosphorylation. WT-NLRC4, phosphomimetic mutant NLRC4-S533D, and phosphodefective mutant NLRC4-S533A were overexpressed in A549 cells for 24 h. Bar diagrams show quantitation of apoptosis by morphological criteria (A) (n = 5) and the percentage of Cl.Casp-8 positivity (B) (n = 3) in A549 cells expressing indicated plasmids. C, lysates of A549 cells expressing the indicated plasmids were analyzed by Western blotting for caspase-8 activation. Representative blot and bar diagram indicating relative caspase-8 activation induced by NLRC4-S533D, compared with NLRC4 from three independent experiments is shown. *, p < 0.05. D, Western blot showing activation of caspase-1 by NLRC4 mutants. E, ΔLRR-NLRC4 requires Ser533 phosphorylation to induce cell death. The bar diagram shows the percentage of apoptosis (n = 6) (E) and Cl.Casp-8 positivity (n = 4) (F) in A549 cells, induced by ΔLRR-NLRC4 and its mutants. ***, p < 0.0005; **, p < 0.005. Whole cell lysates analyzed by Western blotting to indicate expression levels of various proteins is shown. G, SUG1 co-expression does not enhance cell death induced by NLRC4-S533A. The bar diagram shows the percentage of apoptosis in A549 cells expressing HA-tagged SUG1 along with WT-NLRC4 or NLRC4-S533A (n = 40). ***, p < 0.0005. Western blot shows expression level of overexpressed proteins. H, role of SUG1 and FADD in NLRC4-S533D-induced cell death. GFP-NLRC4-S533D was co-expressed with HA-tagged K196M-SUG1 or dnFADD for 24 h and quantitated for apoptosis (n = 6). ***, p < 0.0005. Expression of proteins was checked by SDS-PAGE using HA and GFP antibodies. GAPDH was used as loading control. I, immunoprecipitation assay showing increased ubiquitination of cellular proteins coprecipitated with NLRC4-S533D compared with WT-NLRC4. Indicated plasmids were overexpressed in HEK293T cells, and lysates were subjected to immunoprecipitation using GFP antibody. Immunoprecipitates were analyzed by Western blotting for abundance of ubiquitinated proteins and endogenous SUG1. Con, control; Exp, expressing; IP, immunoprecipitation; UT, untransfected; WB, Western blot; WCL, whole cell lysate.
To determine the role of SUG1 and FADD in NLRC4-S533D-induced cell death, we co-expressed HA-tagged K196M-SUG1 and dnFADD along with NLRC4-S533D in A549 cells for 24 h. Quantitation of apoptosis revealed that co-expressed dnFADD can reduce NLRC4-S533D-induced cell death, but K196M-SUG1 does not have any effect on NLRC4-S533D-mediated cell death signaling (Fig. 5H). Immunoprecipitation assay showed that, unlike NLRC4-H443P, NLRC4-S533D does not show stronger affinity for cellular SUG1 compared with WT-NLRC4; however, we observed higher ubiquitination of NLRC4-S533D as well as coprecipitated cellular proteins compared with WT-NLRC4 (Fig. 5I).
NLRC4-H443P Induces Apoptosis Independent of Phosphorylation at Ser533
We also checked for the role of Ser533 phosphorylation in NLRC4-H443P-induced cell death. The loss of the Ser533 phosphorylation site in NLRC4-H443P did not have any effect on its ability to constitutively induce caspase-8-mediated cell death because a phosphodefective double mutant NLRC4-H443P/S533A showed apoptosis in 29.52 ± 3.181.90% expressing cells compared with 28.67 ± 1.430.30% cell death induced by NLRC4-H443P (Fig. 6A). Also, Cl.Casp-8 levels were found to be similar in whole cell lysates of cells expressing NLRC4-H443P or NLRC4-H443P/S533A mutants (Fig. 6B), indicating redundancy of Ser533 phosphorylation for NLRC4-H443P-induced apoptotic signaling.
FIGURE 6.
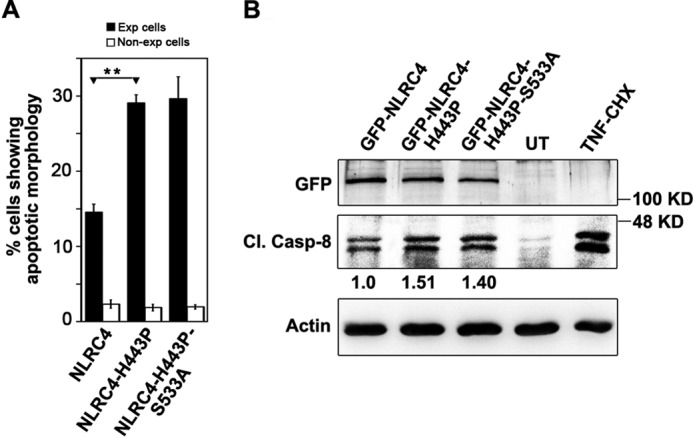
NLRC4-H443P induces caspase-8-mediated cell death independent of Ser533 phosphorylation. A549 cells transfected with indicated plasmids for 20 h were quantitated for apoptosis (A) (n = 4) and caspase-8 activation (B). **, p < 0.005. Actin was used as loading control. CHX, cycloheximide; UT, untransfected.
FADD Is Required, but Ser533 Phosphorylation Is Dispensable for NLRC4-ubiquitin-induced Cell Death and Caspase-8 Activation
NLRC4 modified by addition of two ubiquitin molecules at its C-terminal end (Fig. 7A) can recruit caspase-8 in to a multimolecular complex, resulting in caspase-8 activation and cell death (15). We tested involvement of FADD in NLRC4-Ub2-induced caspase-8 activation. A549 cells expressing NLRC4-Ub2 with or without dnFADD were examined for apoptotic features. We observed that dnFADD co-expression significantly inhibits NLRC4-Ub2-induced cell death (Fig. 7B). As expected, Western blotting analysis of Cl.Casp-8 levels in whole cell lysates also revealed compromised caspase-8 activation upon dnFADD co-expression (Fig. 7C). These results indicated that FADD has a crucial role in NLRC4-Ub2-mediated cell death signaling. We also tested dependence of NLRC4-Ub2-mediated apoptotic signaling on Ser533 phosphorylation. A549 cells were examined for apoptotic features as well as caspase-8 activation upon transient expression of NLRC4-Ub2 and phosphodefective mutant NLRC4-Ub2-S533A for 24 h. It was observed that NLRC4-Ub2 remains constitutively active despite the loss of the Ser533 phosphorylation site (Fig. 7, D and E). To test whether caspase-8 forms a molecular complex with NLRC4 or NLRC4 modified with two ubiquitin moieties, mCaspase-8 was transiently expressed along with WT-NLRC4 or NLRC4-Ub2 in HEK293 cells, and whole cell lysates were subjected to immunoprecipitation using caspase-8 antibody. Western blotting analysis of the immunoprecipitates showed that both WT-NLRC4 and NLRC4-Ub2 form a complex with mCaspase-8. Interaction of mCaspase-8 with GFP-NLRC4-Ub2 was stronger than with NLRC4 because relatively more protein coprecipitated (Fig. 7F).
FIGURE 7.

Role of FADD and Ser533 phosphorylation in ubiquitin-tagged NLRC4-induced caspase-8 dependent cell death. A, schematic showing construct of NLRC4 modified with two ubiquitin moieties and site of Ser533 phosphorylation. * shows site of phosphorylation at Ser-533. B, bar diagram shows quantitation of NLRC4-Ub2 induced apoptosis in A549 cells in the presence or absence of dnFADD (n = 4). **, p < 0.005. C, Western blotting analysis of whole cell lysates of A549 cells transfected with the indicated plasmids for 24 h to showcaspase-8 activation; the levels of actin are shown as loading control. D, A549 cells expressing NLRC4-Ub2 or NLRC4-Ub2-S533A for 24 h were observed for apoptotic features. The bar diagram shows quantitation of apoptosis (n = 4). E, Western blotting analysis of whole cell lysates showing Cl.Casp-8 levels. F, lysates of HEK293 cells transiently expressing GFP-NLRC4 or GFP-NLRC4-Ub2 along with mCaspase-8 were subjected to immunoprecipitation using caspase-8 antibody and Western blotting. The presence of GFP-tagged NLRC4 or NLRC4-Ub2 was detected using GFP antibody and is indicated by arrows. CHX, cycloheximide; Con, control; Exp, expressing; IP, immunoprecipitation; UT, untransfected; WB, Western blot; WCL, whole cell lysate.
Discussion
In this study, we have used disease associated mutants of NLRC4 (H443P, T337S, and V341A) to examine their effect on apoptotic signaling in human lung epithelial cells. We identify a novel function for NLRC4-H443P to induce constitutive caspase-8 activation and cell death, in addition to its ability to mediate caspase-1 activation during inflammatory response. NLRC4, a component of inflammasome, has primarily been shown to aid in sensing of intracellular bacterial components and enable caspase-1 activation to trigger release of cytokines. NLRC4 is a direct transcriptional target of p53 and contributes to p53-mediated cell death (29, 30). Recent evidence shows that NLRC4 also contributes to cell death and brain injury in mice (31). Here, we show that NLRC4-H443P engages SUG1 and FADD to induce caspase-8-dependent cell death independent of caspase-1 activation. Interestingly, our results show that two other disease-associated mutations, T337S and V341A, known to induce caspase-1 activation, do not activate caspase-8, nor do they induce cell death. NLRC4 is an ATPase and is known to remain bound to an ADP molecule in its inactive state. The ADP molecule is stabilized via extensive hydrogen bonding with His443 and residues Pro119, Pro128, Thr135, Gly172, Gly174, Lys175, Ser176, and Thr177 of NBD (amino acids 95–298), which is also the SUG1-binding region (amino acids 91–253) of NLRC4. In case of the mutation, H443P, it is likely that the ADP binding interactions are disrupted, triggering intramolecular reorganization and resulting in enhanced affinity of NBD for binding partners like SUG1, thus enabling NLRC4-H443P to induce SUG1 mediated caspase-8 activation and cell death (Fig. 8).
FIGURE 8.

Proposed model showing molecular events during NLRC4-H443P-induced caspase-8 activation and cell death. WT-NLRC4-mediated caspase-8 activation is dependent on SUG1 over expression and Ser533 phosphorylation. Mutation of His443 to proline (seen in patients with FCAS) causes enhanced interaction with cellular SUG1 compared with WT-NLRC4; SUG1 mediates ubiquitination of NLRC4-H443P leading to FADD and caspase-8-dependent apoptotic cell death. The presence of this mutation releases dependence of caspase-8 activation on Ser533 phosphorylation.
Unlike His443, the residues Thr337 and Val341 are not known to be involved in intramolecular interactions involving SUG1-binding region, and therefore, mutations of these residues may not alter the affinity of NLRC4 toward SUG1, and these mutants may not show phenotypes similar to H443P with respect to caspase-8 activation. However, the mutations T337S and V341A must be causing some alteration that results in autoactivation of NLRC4 to induce caspase-1-mediated inflammatory response.
Although the mechanism of caspase-1 activation by NLRC4 is evident from the fact that NLRC4 and casapse-1 interact through heterodimerization of their CARDs or via another CARD containing protein ASC (32, 33), the molecular details involved in caspase-8 activation are not known. FADD has been shown to enable caspase-8 recruitment to death-inducing silencing complex and initiate its activation (34, 35). We observed that NLRC4-H443P or ΔLRR-NLRC4 or NLRC4 co-expressed with SUG1 induce caspase-8 activation and cell death dependent on FADD, suggesting that FADD may play a role in forming an intracellular caspase-8 activation platform. Our results show that NLRC4 as well as ΔLRR-NLRC4 can complex with dnFADD. Whether the interaction between NLRC4 and FADD is direct or is aided by SUG1 is presently not known. Interaction of FADD with NLRC4 is independent of the LRR domain of NLRC4 and death effector domain of FADD. Our observation that mCaspase-8 interacts more strongly with ubiquitinated NLRC4 compared with WT-NLRC4 suggests that caspase-8 is brought into the complex through interaction with ubiquitinated NLRC4. It is known that in addition to death-inducing signaling complex recruitment, other molecular events like polyubiquitination, which are not fully understood, are required for full caspase-8 activation (36). Because the H443P mutant of NLRC4 is also dependent on SUG1 for caspase-8 activation just like ΔLRR-NLRC4, it is possible that SUG1 may be an additional player required for caspase-8 activation. In this context it may be mentioned that we have earlier shown that TNF-α and cycloheximide-induced cell death is dependent on SUG1 (15).
Modifications like phosphorylation and ubiquitination enable transient changes in the properties of molecules. Our experiments demonstrated that H443P mutant but not V341A mutant shows increased association with ubiquitinated cellular proteins. Cell death induced by the H443P mutant was inhibited strongly by K48R mutant of ubiquitin and partially by K63R-ubiquitin, suggesting that specific ubiquitin linkages are engaged to trigger cell death signal. The constitutively active S533D mutant also shows enhanced ubiquitination of associated cellular proteins, although it does not interact better with SUG1 compared with WT-NLRC4. Phosphorylation of Ser533 by PKC-δ is required for caspase-1 activation (10). Phosphorylation at Ser533 of NLRC4 keeps it in a primed but inactive state (10, 11). We show that ΔLRR-NLRC4 or co-expression of NLRC4 with SUG1 activates caspase-8 dependent on Ser533 phosphorylation. NLRC4-H443P mutant can activate caspase-8 even when Ser533 is mutated to a non-phosphorylable form. Because His443 interacts with the phosphate of ADP, mutation in this residue may result in constitutive activation and may not require structural reorganization achieved by phosphorylation at Ser533 to activate caspase-8. Salient findings showing the mechanism of caspase-8 activation and cell death induction by NLRC4-H443P are shown as a model in Fig. 8.
The fact that ΔLRR-NLRC4 is also dependent on Ser533 phosphorylation for activation of caspase-8 indicates that phosphorylation at Ser533 is essential even though NLRC4 is in an open configuration. Unlike NLRC4-H443P, the phosphomimetic mutant S533D does not show enhanced affinity for SUG1 and can activate caspase-8 independent of SUG1. However, enhanced ubiquitination and requirement of FADD remain important steps in cell death signaling mediated by NLRC4-S533D. Because addition of ubiquitin molecules to NLRC4 releases dependence on Ser533 phosphorylation, it may be inferred that mimicking Ser533 phosphorylation in NLRC4-S533D or SUG1 interaction results in ubiquitination, which eventually causes caspase-8 activation.
Recent studies have discovered functions of NLRC4 in intestinal epithelial lining and in brain cells (12, 13, 31). Our study prompts further investigation into how caspase-8 dependent function of NLRC4 contributes to immune response or lung pathology like breakdown of epithelial barrier function caused by cell death. Caspase-8 is known to mediate caspase-1 activation and inflammatory response (26, 27). We have presented evidence for a caspase-1-independent cell death function of caspase-8 downstream of NLRC4. More detailed studies are required to understand what determines caspase-8 activation versus caspase-1 activation by NLRC4. In conclusion, our results show that an autoinflammatory syndrome causing mutant H443P of NLRC4 constitutively activates caspase-8 dependent on adapter protein FADD. This mutant shows enhanced interaction with SUG1 and ubiquitinated proteins; in addition it shows increased ubiquitination. Ubiquitination plays an important role in caspase-8 activation and cell death induced by this mutant. NLRC4 requires Ser533 phosphorylation for optimal caspase-8 activation, and H443P mutation overcomes requirement of this phosphorylation. Overall our results provide insight into mechanism of caspase-8 activation by NLRC4 and its H443P mutant. Therefore, individuals with the H443P mutation are likely to show other defects in addition to autoinflammatory disease.
Experimental Procedures
Cell Culture and Transfections
Lung epithelial adenocarcinoma A549 cells and HEK293T cells were maintained in DMEM supplemented with 10% FBS at 37 °C in a CO2 incubator. THP1 cells were maintained in RPMI supplemented with 10% heat-inactivated FBS at 37 °C in a CO2 incubator. For transient expression of proteins, plasmids (purified using Qiagen plasmid mini kit) were transfected using Lipofectamine2000 or Lipofectamine3000 (Invitrogen) as per the manufacturer's protocol. In general, ∼20% transfection efficiency was achieved in A549 cells, whereas HEK cells showed ∼80% expressing cells. Treatment with 25 μm MG132 was for 6 h. Lysates of A549 cells treated with TNF-α (20 ng/ml) and cycloheximide (20 μg/ml) for 8 h were used as positive control for cleaved caspase-8 (Cl.Casp-8) in Western blot experiments.
Antibodies and Chemicals
Mouse monoclonal anti-GFP (catalog no. SC-9996), rabbit polyclonal anti-HA (catalog no. SC-805), anti-FADD (catalog no. SC-5559), anti-ubiquitin, anti-actin (catalog no. SC-47778), anti-caspase-8 (catalog no. SC-7890), anti-caspase-1 (catalog nos. SC-622 and SC515), and anti-Myc (SC-40) antibodies were obtained from Santa Cruz Biotechnology. Antibodies against cleaved caspase-8 (catalog no. 9496S) and NLRC4 (catalog no. 12421) were from Cell Signaling Technology, SUG1 antibody (611066) was from BD Biosciences, and GAPDH antibody (MAB-374) was from Millipore. Agarose-conjugated GFP antibody (GFPTrap; gta-20) and control agarose beads (bab-20) for immunoprecipitation were purchased from ChromoTek. HRP-conjugated rabbit and mouse secondary antibodies were obtained from GE Healthcare. FemtoLUCENTTM PLUS HRP kit (786–10) was procured from GBiosciences. Cy3 (Indocarbocyanine) and Alexa 633-conjugated mouse or rabbit secondary antibodies were purchased from Millipore and Thermo Fisher, respectively.
Expression Vectors
Full-length NLRC4 cDNA, various deletion constructs, and NLRC4-Ub2 cloned in pEGFP-C1 mammalian expression vector (Clontech) are described (15). Phosphodefective S533A mutants of pEGFP-C1-NLRC4, pEGFP-C1-ΔLRR-NLRC4, and pEGFP-C1-NLRC4-Ub2 were generated by TCT-GCT nucleotide substitution (GenBank® accession number NM_001199139.1) using site-directed mutagenesis. Similarly, phosphomimetic S533D mutants were generated by TCT-GAT nucleotide substitution. Disease-associated mutants NLRC4-H443P, NLRC4-T337S, and NLRC4-V341A were generated by CAC-CCC, ACC-TCC, and GTG-GCG nucleotide substitutions respectively. Sequences of all the mutants were confirmed by automated DNA sequencing. Constructs expressing HA-tagged SUG1, catalytically inactive K196M-SUG1, catalytically inactive C360A caspase-8 and C285A caspase-1, HA-tagged ubiquitin, and its K48R and K63R mutants have been described previously (15). Constructs expressing HA-tagged dnFADD were subcloned from expression vector pCDNA3-AU1-dnFADD (23) (a kind gift from Dr. Julian Downward) by EcoRI and XhoI restriction digestion in pCDNA3.1-HA-mammalian expression vector (Clontech). pCruzMycB-NLRC4 and NLRC4-V341A expression vectors were a kind gift from Dr. Barbara Kazmierczak (Yale University). Myc-tagged NLRC4-H443P, NLRC4-T337S, and NLRC4-S533D were generated using site-directed mutagenesis as described above.
Construction of Vectors Expressing shRNA
shRNA SUG1 mRNA was cloned in an U6 promoter-based pDsRed-Monomer-Hyg-C1 vector (Clontech) earlier in our lab (15). The shRNA (RFP-SUG1-shD) targets SUG1 sequence (GenBank® accession number NM_002805) at nucleotides 235–254. A vector expressing shRNA of unrelated sequence (Con-Sh) was used as control.
Indirect Immunofluorescence and Microscopy
Immunostaining of cells fixed with formaldehyde was carried out as described previously (37, 38). Immunostained cells were observed, and images were taken with an automated AxioImager Z.1 (Zeiss) fluorescence microscope using Axiovision software under ×40/0.75 NA dry objective.
Quantitation of Cell Death
The cells were grown on coverslips for 24 h before transfection with the desired plasmids. Transfected cells expressing indicated plasmids were fixed 20 h post-transfection (or later as indicated) with 4% formaldehyde and stained with required antibodies as described previously (37, 38). The cells were mounted in PBS with 90% glycerol, 1 mg/ml anti-fade (para-phenylenediamine), and 0.5 mg/ml DAPI. Expressing and non-expressing cells were monitored for apoptotic features like cytoplasmic shrinkage, nuclear constriction, loss of refractility, membrane blebbing, and formation of apoptotic bodies under fluorescence microscope (-40/0.75NA dry objective; Nikon). Cell death was quantitated as the percentage of cells showing apoptotic morphology in expressing cells and non-expressing cells from the same coverslips. The data were obtained from three independent experiments carried out on duplicate coverslips (or indicated otherwise) upon observation of at least 200 expressing cells and 800 non-expressing cells from each coverslip and represented as means ± S.D.
Cells transiently expressing desired proteins were assayed for apoptosis using GFP certified apoptosis assay kit, (Enzo Life Sciences, catalog no. ENZ-51002-100) as per manufacturer's protocol. The cells were washed with PBS and incubated with annexin V and 7-aminoactinomycin D (7-AAD) for 15 min at room temperature, washed with PBS, and fixed in 2% formaldehyde, mounted in DAPI-containing medium, and observed under a fluorescence microscope. Apoptotic cells show annexin V binding to externalized phosphatidylserine and/or nuclear staining of 7-AAD. Cells positive for either or both of annexin V and 7-AAD were scored as apoptotic, and the data were gathered as means ± S.D. of the percentage of annexin V/7-AAD positivity after monitoring at least 200 expressing as well as non-expressing cells from at least two independent experiments performed on duplicate coverslips.
Co-immunoprecipitation and Western Blotting
A549 or HEK293T cells were transfected with the desired plasmids for 16 h (or 24 h as indicated) followed by lysis at 4 °C for 10 min in buffer containing 20 mm Tris-Cl (pH 7.4), 1% TritonX-100, 150 mm NaCl, 5 mm EDTA, 1 mm PMSF, 0.1% BSA, 10 mm N-ethylmaleimide (a deubiquitinase inhibitor; Calbiochem) and protease inhibitor mixture (GBiosciences). Lysate was centrifuged at 10,000 × g for 10 min at 4 °C to remove cellular debris. Supernatant was incubated with 2 μg of HA antibody or control IgG on a Roto-torque for 8 h at 4 °C followed by 2 h of incubation with protein A/G plus agarose beads (Santa Cruz) to immunoprecipitate HA-tagged proteins. The bound fraction was washed, and immunoprecipitates were lysed in SDS containing Laemmli sample buffer. The samples were then subjected to Western blotting analysis as described earlier (37). For immunoprecipitation of GFP-tagged proteins, GFPTrap antibody (ChromoTek) was used as per the manufacturer's protocol. The cells expressing the desired plasmids were lysed in buffer containing 10 mm Tris-Cl (pH 7.5), 0.5% Nonidet P-40 detergent, 1 mm PMSF, 150 mm NaCl, 5 mm EDTA, 10 mm N-ethylmaleimide, and protease inhibitor mixture at 4 °C for 30 min. The cell lysates were further incubated with agarose bead-conjugated GFPTrap antibody or control antibody for 2 h on a Roto-torque at 4 °C. The blots were quantitated using ImageJ software.
Statistical Analysis
Bar diagrams represent means ± S.D. values. Two-tailed Student's t test was used to calculate significance of differences between two means, and one-tailed t test was used to determine significance of relative difference of a test sample compared with control as 1.
Author Contributions
A. K. R., A. S., P. P., G. G., and Y. K. performed the experiments. G. S., V. R., and A. K. R. planned the experiments, analyzed the data, and wrote the paper. G. S. and V. R. conceived and obtained funding for this study. All authors approved final version of the manuscript.
Acknowledgments
We are grateful to Dr. Julian Downward for providing pCDNA3-AU1-FADD and pCDNA3-AU1-dnFADD plasmid vectors and Dr. Barbara Kazmierczak (Yale University) for providing pCruzMycB-NLRC4 and NLRC4-v341A expression vectors.
This work was supported by Department of Biotechnology, Government of India Grant BT/PR14917/BRB/10/888/2010 (to G. S. and V. R.), a J. C. Bose National fellowship (to G. S.), and a fellowship from the Council for Scientific and Industrial Research of India (to A. K. R.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
- NLRC4
- Nod-like receptor family card containing 4
- Cl.Casp-8
- cleaved caspase-8
- dn
- dominant negative
- FCAS
- familial cold autoinflammatory syndrome
- CARD
- caspase activation and recruitment domain
- NBD
- nucleotide binding domain
- LRR
- leucine-rich repeat
- WHD
- winged helix domain
- 7-AAD
- 7-aminoactinomycin D.
References
- 1. Dowling J. K., and O'Neill L. A. (2012) Biochemical regulation of the inflammasome. Crit. Rev. Biochem. Mol. Biol. 47, 424–443 [DOI] [PubMed] [Google Scholar]
- 2. Kofoed E. M., and Vance R. E. (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tenthorey J. L., Kofoed E. M., Daugherty M. D., Malik H. S., and Vance R. E. (2014) Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol. Cell 54, 17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao Y., Yang J., Shi J., Gong Y. N., Lu Q., Xu H., Liu L., and Shao F. (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 [DOI] [PubMed] [Google Scholar]
- 5. Hu Z., Yan C., Liu P., Huang Z., Ma R., Zhang C., Wang R., Zhang Y., Martinon F., Miao D., Deng H., Wang J., Chang J., and Chai J. (2013) Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175 [DOI] [PubMed] [Google Scholar]
- 6. Diebolder C. A., Halff E. F., Koster A. J., Huizinga E. G., and Koning R. I. (2015) Cryoelectron tomography of the NAIP5/NLRC4 inflammasome: implications for NLR activation. Structure 23, 2349–2357 [DOI] [PubMed] [Google Scholar]
- 7. Hu Z., Zhou Q., Zhang C., Fan S., Cheng W., Zhao Y., Shao F., Wang H. W., Sui S. F., and Chai J. (2015) Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404 [DOI] [PubMed] [Google Scholar]
- 8. Zhang L., Chen S., Ruan J., Wu J., Tong A. B., Yin Q., Li Y., David L., Lu A., Wang W. L., Marks C., Ouyang Q., Zhang X., Mao Y., and Wu H. (2015) Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lightfield K. L., Persson J., Trinidad N. J., Brubaker S. W., Kofoed E. M., Sauer J. D., Dunipace E. A., Warren S. E., Miao E. A., and Vance R. E. (2011) Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect. Immun. 79, 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qu Y., Misaghi S., Izrael-Tomasevic A., Newton K., Gilmour L. L., Lamkanfi M., Louie S., Kayagaki N., Liu J., Kömüves L., Cupp J. E., Arnott D., Monack D., and Dixit V. M. (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 [DOI] [PubMed] [Google Scholar]
- 11. Matusiak M., Van Opdenbosch N., Vande Walle L., Sirard J. C., Kanneganti T. D., and Lamkanfi M. (2015) Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc. Natl. Acad. Sci. U.S.A. 112, 1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nordlander S., Pott J., and Maloy K. J. (2014) NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 7, 775–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sellin M. E., Müller A. A., Felmy B., Dolowschiak T., Diard M., Tardivel A., Maslowski K. M., and Hardt W. D. (2014) Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 16, 237–248 [DOI] [PubMed] [Google Scholar]
- 14. Kong H., Wang Y., Zeng X., Wang Z., Wang H., and Xie W. (2015) Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumour Biol. 36, 7501–7513 [DOI] [PubMed] [Google Scholar]
- 15. Kumar Y., Radha V., and Swarup G. (2010) Interaction with Sug1 enables Ipaf ubiquitination leading to caspase 8 activation and cell death. Biochem. J. 427, 91–104 [DOI] [PubMed] [Google Scholar]
- 16. Canna S. W., de Jesus A. A., Gouni S., Brooks S. R., Marrero B., Liu Y., DiMattia M. A., Zaal K. J., Sanchez G. A., Kim H., Chapelle D., Plass N., Huang Y., Villarino A. V., Biancotto A., et al. (2014) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 46, 1140–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Romberg N., Al Moussawi K., Nelson-Williams C., Stiegler A. L., Loring E., Choi M., Overton J., Meffre E., Khokha M. K., Huttner A. J., West B., Podoltsev N. A., Boggon T. J., Kazmierczak B. I., and Lifton R. P. (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 46, 1135–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kitamura A., Sasaki Y., Abe T., Kano H., and Yasutomo K. (2014) An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 211, 2385–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makino Y., Yamano K., Kanemaki M., Morikawa K., Kishimoto T., Shimbara N., Tanaka K., and Tamura T. (1997) SUG1, a component of the 26 S proteasome, is an ATPase stimulated by specific RNAs. J. Biol. Chem. 272, 23201–23205 [DOI] [PubMed] [Google Scholar]
- 20. Bhat K. P., Turner J. D., Myers S. E., Cape A. D., Ting J. P., and Greer S. F. (2008) The 19S proteasome ATPase Sug1 plays a critical role in regulating MHC class II transcription. Mol. Immunol. 45, 2214–2224 [DOI] [PubMed] [Google Scholar]
- 21. Durairaj G., and Kaiser P. (2014) The 26S proteasome and initiation of gene transcription. Biomolecules 4, 827–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geetha T., Kenchappa R. S., Wooten M. W., and Carter B. D. (2005) TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 24, 3859–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chinnaiyan A. M., Tepper C. G., Seldin M. F., O'Rourke K., Kischkel F. C., Hellbardt S., Krammer P. H., Peter M. E., and Dixit V. M. (1996) FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J. Biol. Chem. 271, 4961–4965 [DOI] [PubMed] [Google Scholar]
- 24. Man S. M., Tourlomousis P., Hopkins L., Monie T. P., Fitzgerald K. A., and Bryant C. E. (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J. Immunol. 191, 5239–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gurung P., Anand P. K., Malireddi R. K., Vande Walle L., Van Opdenbosch N., Dillon C. P., Weinlich R., Green D. R., Lamkanfi M., and Kanneganti T. D. (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Philip N. H., Dillon C. P., Snyder A. G., Fitzgerald P., Wynosky-Dolfi M. A., Zwack E. E., Hu B., Fitzgerald L., Mauldin E. A., Copenhaver A. M., Shin S., Wei L., Parker M., Zhang J., Oberst A., et al. (2014) Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7385–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chi W., Li F., Chen H., Wang Y., Zhu Y., Yang X., Zhu J., Wu F., Ouyang H., Ge J., Weinreb R. N., Zhang K., and Zhuo Y. (2014) Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc. Natl. Acad. Sci. U.S.A. 111, 11181–11186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lavrik I., Krueger A., Schmitz I., Baumann S., Weyd H., Krammer P. H., and Kirchhoff S. (2003) The active caspase-8 heterotetramer is formed at the CD95 DISC. Cell Death Differ. 10, 144–145 [DOI] [PubMed] [Google Scholar]
- 29. Sadasivam S., Gupta S., Radha V., Batta K., Kundu T. K., and Swarup G. (2005) Caspase-1 activator Ipaf is a p53-inducible gene involved in apoptosis. Oncogene 24, 627–636 [DOI] [PubMed] [Google Scholar]
- 30. Thalappilly S., Sadasivam S., Radha V., and Swarup G. (2006) Involvement of caspase 1 and its activator Ipaf upstream of mitochondrial events in apoptosis. FEBS J. 273, 2766–2778 [DOI] [PubMed] [Google Scholar]
- 31. Denes A., Coutts G., Lénárt N., Cruickshank S. M., Pelegrin P., Skinner J., Rothwell N., Allan S. M., and Brough D. (2015) AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. U.S.A. 112, 4050–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poyet J. L., Srinivasula S. M., Tnani M., Razmara M., Fernandes-Alnemri T., and Alnemri E. S. (2001) Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313 [DOI] [PubMed] [Google Scholar]
- 33. Mariathasan S., Newton K., Monack D. M., Vucic D., French D. M., Lee W. P., Roose-Girma M., Erickson S., and Dixit V. M. (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 [DOI] [PubMed] [Google Scholar]
- 34. Carrington P. E., Sandu C., Wei Y., Hill J. M., Morisawa G., Huang T., Gavathiotis E., Wei Y., and Werner M. H. (2006) The structure of FADD and its mode of interaction with procaspase-8. Mol. Cell 22, 599–610 [DOI] [PubMed] [Google Scholar]
- 35. Tourneur L., and Chiocchia G. (2010) FADD: a regulator of life and death. Trends Immunol. 31, 260–269 [DOI] [PubMed] [Google Scholar]
- 36. Békés M., and Salvesen G. S. (2009) The CULt of caspase-8 ubiquitination. Cell 137, 604–606 [DOI] [PubMed] [Google Scholar]
- 37. Shivakrupa R., Radha V., Sudhakar C., and Swarup G. (2003) Physical and functional interaction between Hck tyrosine kinase and guanine nucleotide exchange factor C3G results in apoptosis, which is independent of C3G catalytic domain. J. Biol. Chem. 278, 52188–52194 [DOI] [PubMed] [Google Scholar]
- 38. Radha V., Sudhakar C., Ray P., and Swarup G. (2002) Induction of cytochrome c release and apoptosis by Hck-SH3 domain-mediated signalling requires caspase-3. Apoptosis 7, 195–207 [DOI] [PubMed] [Google Scholar]