

Abstract
Over half a century ago, D. S. Falconer first reported a mouse with a reeling gate. Four decades later, the Reln gene was isolated and identified as the cause of the reeler phenotype. Initial studies found that loss of Reelin, a large, secreted glycoprotein encoded by the Reln gene, results in abnormal neuronal layering throughout several regions of the brain. In the years since, the known functions of Reelin signaling in the brain have expanded to include multiple postdevelopmental neuromodulatory roles, revealing an ever increasing body of evidence to suggest that Reelin signaling is a critical player in the modulation of synaptic function. In writing this review, we intend to highlight the most fundamental aspects of Reelin signaling and integrate how these various neuromodulatory effects shape and protect synapses.
Keywords: amyloid-β (Aβ), apolipoprotein E (ApoE), neurodegeneration, neurotransmitter receptor, receptor, tyrosine-protein kinase (tyrosine kinase), protein-tyrosine kinase, ApoE receptors, Reelin, synaptic function, neurodegenerative diseases
Reelin Signaling during Development
Besides the obvious motor deficits in Reelin-deficient mice (1), the most overt reeler phenotype is the abnormal layering of neurons in the brain (2). Reelin is essential for normal cortical, hippocampal, and cerebellar neuronal lamination (reviewed in Refs. 3 and 4). While the neocortex is developing, Reelin is expressed and secreted by Cajal-Retzius neurons in the outer layers of the developing neocortex, where Reelin guides newly born neurons to their correct positions in the cortex in an inside-out fashion (3, 4). Similarly, in the prenatal cerebellum, Reelin is expressed in the external granule layer, where it mediates Purkinje cell localization (reviewed in Refs. 3 and 4).
The mechanism of Reelin-mediated neuronal guidance was elucidated through the genetic ablation of its downstream signaling partners. Double knock-out of low-density lipoprotein receptor family members, ApoE receptor2 (ApoER2) and very low density lipoprotein receptor (VLDLR),2 or loss of the cytoplasmic adaptor protein Disabled-1 (Dab1) recapitulated the reeler phenotype (reviewed in Ref. 5), suggesting that these molecules are critical for the action of Reelin during neuronal migration. Interestingly, singular knock-out of either Reelin receptor resulted in a milder migration deficit, indicating divergent roles for the ApoER2 and VLDLR during neuronal migration (reviewed in Ref. 5). These studies ultimately clarified the core components of the Reelin signaling pathway whereby Reelin binding to ApoER2 and VLDLR results in a Src family tyrosine kinase (SFK)-mediated tyrosine phosphorylation of Dab1 (reviewed in Ref. 5).
Reelin Signaling after Neuronal Migration
Postnatally, Reelin is repurposed as a neuromodulator. At this point, inhibitory GABAergic interneurons begin to express and secrete Reelin (6) as the Cajal-Retzius cells begin to die out in the cerebral cortex and later in the hippocampus (7). This postnatally secreted Reelin acts to modulate axonal and dendritic outgrowth through multiple independent and interconnected pathways by regulating the stability of the cytoskeleton (Fig. 1).
FIGURE 1.
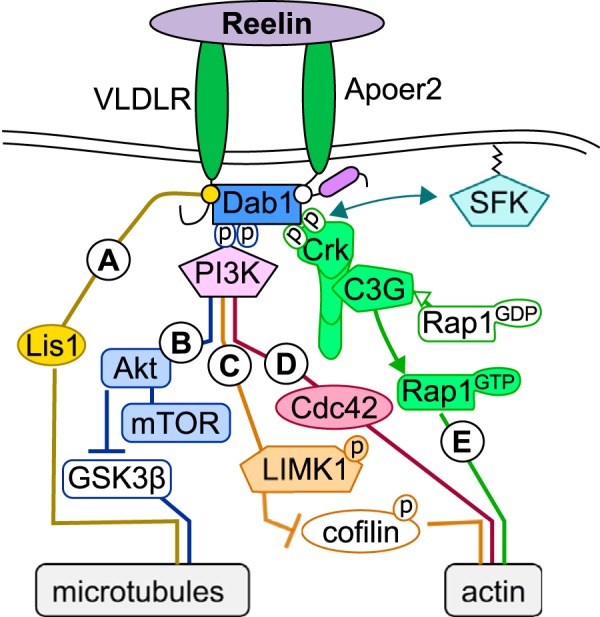
Reelin's role in stabilizing the cytoskeleton. Reelin signaling participates in axonal and dendritic outgrowth and maturation by stabilizing the cytoskeleton. A and B, microtubule stability is promoted by Reelin through VLDLR-dependent activation of Lis1 (reviewed in Refs. 4, 5, and 11) (A) and inhibition of GSK-3β (reviewed in Ref. 31) through activation of the Dab1/PI3K/Akt pathway, which also activates an mTOR-dependent process that promotes the outgrowth and stabilization of dendrites (reviewed in Refs. 5, 10, and 11) (B). C, Reelin promotes actin stability through the ApoER2/Dab1/PI3K pathway by inducing LIMK1-dependent inactivation of cofilin (reviewed in Ref. 3). D, this ApoER2/Dab1/PI3K pathway can also mediate the formation of axons by activating the RhoGTPase Cdc42 (8). E, independent of PI3K, Reelin stabilizes actin by triggering C3G activation of Rap1 through Crk family proteins (Crk), which is essential for normal neocortical lamination and postnatal hippocampal dendritogenesis (12, 13).
The formation of neuronal connections begins with the outcropping of neuritic filopodia, which develop into axon and dendrites. Acute and chronic Reelin application enhances cortical neuritic outgrowth mobility and size (respectively) through an ApoER2/Dab1/PI3K pathway, which activates the RhoGTPase Cdc42 and stimulates the delivery of membrane and extension of the growth cone (8) (Fig. 1D). Neurons from reeler mice exhibit reduced dendritic branching, and these dendrites produce fewer dendritic spines in vivo and in vitro (9). A similar, more subtle effect is observed in heterozygous reeler mice (HRM), which lack only one allele of the Reln gene and express 50% less Reelin. However, neuronal positioning in HRM brains is not affected, indicating that the Reelin deficiency is driving the dendritic abnormalities in the reeler mouse and that they are not due to improper neuronal positioning. Acute application of Reelin can enhance dendritic outgrowth in both wild-type and reeler neurons in a lipoprotein receptor-dependent fashion that requires the presence and phosphorylation of Dab1 by Src kinases (9). Promotion of the outgrowth and stabilization of dendrites by Reelin also requires activation of mTOR through PI3K and AKT (reviewed in Refs. 5, 10, and 11) (Fig. 1B). Crk family proteins are also important downstream components of the Reelin signaling pathway in the regulation of both neocortical lamination and postnatal hippocampal dendritogenesis (12, 13) (Fig. 1E). Microtubule and actin stabilities are also promoted by Reelin through the activation of lissencephaly 1 (Lis1) through VLDLR (reviewed in Refs. 4, 5, and 11) (Fig. 1A) or by the phosphorylation and inactivation of cofilin through LIM kinase 1 (LIMK1) (reviewed in Ref. 3) (Fig. 1C). Each of these actions contributes to the fine-tuning of dendritic outgrowth and helps shape neuronal connectivity.
Spine Formation and Maturation
The neuromodulatory effects of Reelin extend beyond the dendrite to the dendritic spine, where pre- and postsynaptic contacts form. The major components of excitatory postsynaptic compartments contain ionotropic glutamate receptors, both AMPA and NMDA receptors (AMPAR and NMDAR), which bind presynaptically released glutamate. The number of synaptic connections and the molecular composition of these compartments determine the efficacy of neuronal networks, and Reelin signaling is a critical regulator of both the number and the strength of these connections (14). HRM and reeler hippocampal neurons have fewer dendritic spines along their dendrites when compared with wild-type neurons, and the extent of this effect is proportional to the reduction in Reelin protein abundance (14, 15). Exogenous recombinant Reelin recovers this deficit in cultured hippocampal slices from HRM and reeler mice as well as increases dendritic spine density in wild-type control slices (14). The downstream signaling partners, ApoER2/VLDLR and both Dab1 and SFKs, are essential for this Reelin-mediated spinogenesis (14). Intriguingly, overexpression of the Reelin receptor ApoER2 in dissociated hippocampal neuron cultures can dramatically increase dendritic spine numbers in wild-type neuron cultures, suggesting a critical role of the receptor in promoting synaptogenesis (16).
Reelin signaling also modulates the molecular composition of synapses. During development, the majority of NMDARs at hippocampal synapses are composed of NR2B subunits, which have a higher conductance than NR2A-containing receptors. As synapses mature, the subunit composition of NMDARs shifts from NR2B-containing receptors to the NR2A-containing receptors (reviewed in Ref. 17). This switch is accelerated in hippocampal neuron cultures treated with exogenous Reelin over 24 h, an effect that requires lipoprotein receptors and Src kinase activity (18). Alternately, this switch is prevented by inhibiting Reelin signaling via antisense knockdown of Reelin, perfusion with a Reelin antibody (CR-50), or blocking the GABAergic release of Reelin (18, 19). Chronic Reelin treatment of hippocampal slice cultures (6–8 days) augmented AMPAR currents by increasing GluA1 surface expression while reducing the NMDA-mediated currents by promoting the insertion of NR2A-containing NMDARs and removing NR2B-containing NMDARs (reviewed in Refs. 20–22). This chronic Reelin treatment also facilitates the insertion of AMPARs into synaptic membranes containing only NMDARs (reviewed in Ref. 22); these synapses, containing NMDAR and no AMPAR, are known as “silent synapses” and are unresponsive to glutamate at the resting membrane potential (23). Additionally, Reelin regulates the surface diffusion of NR2B-containing NMDARs without affecting those with NR2A subunits (24). Prolonged treatment with Reelin facilitates the mobility and decreases the synaptic dwell time of NR2B-containing NMDARs, whereas inhibition of Reelin signaling with CR-50 stabilizes these receptors at the synapse (24). Constitutive decreases in Reelin also alter NMDAR composition. The postsynaptic compartments of both homozygous and heterozygous reeler synapses have a drastic and comparable reduction in the NR2A subunit of the NMDAR and PSD-95 in comparison with wild-type synapses (14).
Reelin also acts presynaptically (25, 26). Acute application of Reelin increases the spontaneous fusion of vesicle-associated membrane protein 7 (VAMP7)-containing presynaptic vesicles in hippocampal cultures, and this effect is rapid (within 5 min) and robust as early as 6 days in vitro when spontaneous fusion is minimal (25). This increase in VAMP7-containing mobilization of presynaptic vesicles requires release of Ca2+ from intracellular compartments, both ApoER2 and VLDLR, and PI3K (25). Taken together, Reelin signaling appears to act as a potent neuromodulator of the structure and function of synapses and their connectivity.
Modulation of Synaptic Plasticity
Reelin also enhances long-term potentiation (LTP) of synapses, a proposed cellular correlate to memory (27) (Figs. 2, A and B, and 3, A and B). When Reelin binds postsynaptic lipoprotein receptors, ApoER2 and VLDLR, the receptors cluster and phosphorylate Dab1, leading to an SFK-mediated tyrosine phosphorylation of the NR2 subunits of the NMDAR (reviewed in Ref. 5) (Fig. 2A). The SFK, Fyn, is mainly responsible for the phosphorylation of the NR2 subunits of the NMDAR, which is known to enhance their conductance (reviewed in Ref. 28) (Figs. 2A and 3A). Therefore, acute application of Reelin to cortical neurons enhances the NMDAR-mediated Ca2+ conductance and phosphorylation of cAMP-response element-binding protein (CREB) (29), leading to a potent enhancement in LTP (27) (Fig. 2B). This Reelin-mediated LTP enhancement requires a unique 59-amino acid insert in the ApoER2 cytoplasmic tail, which interacts with PSD-95 (30) (Fig. 2A). Inclusion of this proline-rich domain is regulated by activity through alternative splicing of exon 19 (exon 18 in humans) (30). The consolidation of LTP requires the postsynaptic translation of activity-regulated cytoskeletal (Arc) mRNA, and Reelin increases Arc mRNA translation in an mTOR-dependent manner (reviewed in Refs. 5 and 20).
FIGURE 2.
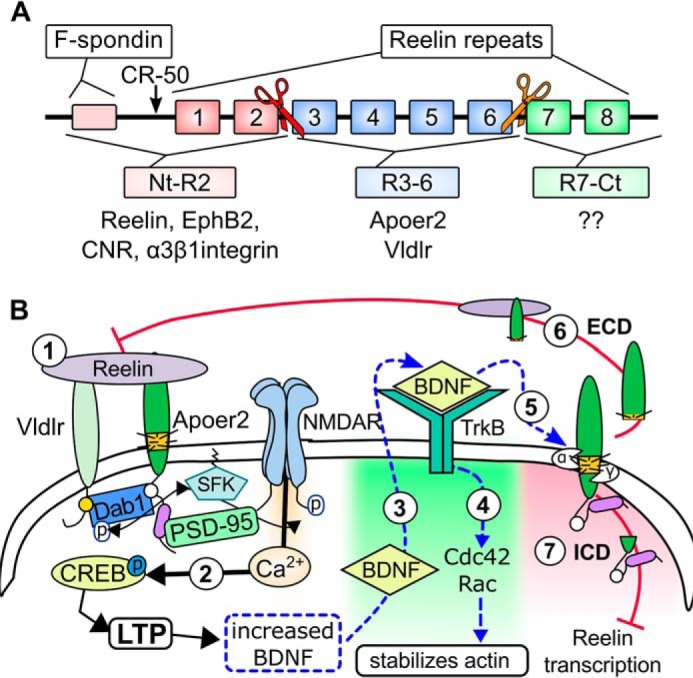
Regulating Reelin signaling. A, the function of Reelin is regulated by proteolytic processing. The diagram depicts the domain structure of Reelin and the N- and C-terminal proteolytic cleavage sites (red and orange scissors, respectively). B, Reelin regulates BDNF signaling (hypothetical). Reelin increases the phosphorylation of CREB by enhancing Ca2+-influx through NMDARs (1) (29). CREB activation (phosphorylation) drives BDNF expression (2) (74). This BDNF can be released at the activated synapse and bind TrkB receptors (3), which promote actin stability through Cdc42 and Rac activity (4) (48). Both Reelin binding and TrkB/BDNF signaling can inhibit Reelin-mediated NMDAR phosphorylation by inducing the proteolytic processing of ApoER2 (5) (36, 47), whereby the ECD acts as a dominant negative receptor (6) (37) and the ICD can inhibit the transcription of Reelin (7) (38).
FIGURE 3.
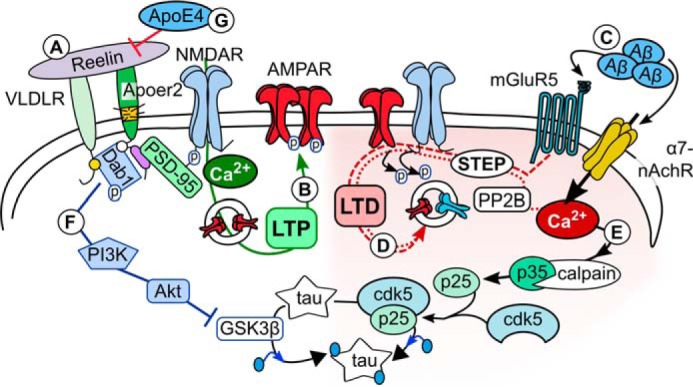
Reelin's protective role against synaptic dysfunction. A and B, Reelin enhances the Ca2+ conductance of NMDARs (reviewed in Ref. 5) (A), leading to enhanced AMPAR insertion and LTP (B). C and D, Aβ can induce synaptic weakening (LTD) through agonizing mGluR5 and α7-nAChRs (C) (reviewed in Refs. 56 and 57), which promote glutamate receptor (NMDAR and AMPAR) endocytosis (D). E, elevated Ca2+ concentrations can activate calpain and drive Cdk5-mediated tau hyperphosphorylation (reviewed in Ref. 62), which destabilizes microtubules. F, Reelin can inhibit tau phosphorylation by inhibiting GSK-3β (reviewed in Ref. 31). G, ApoE4 inhibits the Reelin signal by sequestering ApoER2 in the endosome (85, 87).
Role in Learning and Memory
Reelin takes part in both spatial and fear memory. In adult wild-type mice, injection of Reelin in vivo enhances not only synaptic plasticity but also spatial memory (reviewed in Refs. 5, 10, 21, and 31). Mice deficient in either ApoER2 or VLDLR have reduced ability to acquire hippocampus-dependent contextual fear memory (27). Interestingly, VLDLR KO mice had a significant deficiency in cued fear memory when compared with wild-type littermates, whereas ApoER2 KO mice did not (27). The contextual deficit in fear learning in ApoER2 KOs is recapitulated in mice expressing ApoER2 lacking the alternatively spliced proline-rich domain of ApoER2 (30), indicating that this domain is critical for the downstream action of Reelin in fear memory acquisition. Reelin's role in fear memory is further supported by the observations that HRM mice have reduced short-term contextual fear learning (32) and deficient long-term association of both forms of fear memory (33). These studies demonstrate a critical modulatory role of Reelin in both spatial and fear memory.
Regulating Reelin Signaling
Many of Reelin's effects require ApoER2, so an important regulatory factor in Reelin signaling is the alternative splicing of ApoER2. ApoER2 RNA can undergo numerous splicing events; the two most notable for synapses are the cytoplasmic proline-rich region and a highly glycosylated extracellular domain. The ability of Reelin to modulate synaptic strength depends upon the inclusion of the proline-rich cytoplasmic domain of the Reelin receptor, ApoER2 (30). Differential inclusion of the glycosylated region of ApoER2 has a very different role. This juxtamembranous O-linked sugar (OLS) domain regulates the abundance of the receptor by protecting it from proteolytic processing in a glycosylation-dependent manner (34, 35). Loss of ApoER2 cleavage leads to increased ApoER2 expression and a concomitant enhancement of dendritic spine density (34), which is consistent with the published results of ApoER2 overexpression in wild-type neuron cultures (16).
Alternative splicing of ApoER2 can dramatically modify this extracellular proteolytic processing of ApoER2. ApoER2 is sequentially cleaved by extracellular proteases such as ADAM10, resulting in the release of a soluble extracellular domain (ECD), followed by the subsequent γ-secretase-mediated release of the intracellular domain (ICD) (35). Acute application of Reelin can induce this cleavage (36). Both ApoER2 cleavage fragments are negative feedback regulators of the Reelin signal. The ApoER2-ECD acts as a dominant-negative receptor for ApoER2 ligands (37), and the ApoER2-ICD can translocate to the nucleus and alter transcription of genes including inhibition of Reelin transcription (38, 39). Alternative splicing of ApoER2 can affect this sequential cleavage. In addition to increased abundance of the receptor, exclusion of the OLS domain, which contains the extracellular cleavage site, imparts a strong resistance to the cleavage of ApoER2 and thus prevents the release of both the extracellular and the intracellular fragments of ApoER2 (34).
In addition to proteolytic processing of ApoER2, the function of Reelin is also influenced by proteolytic processing (reviewed in Refs. 11 and 21). After secretion, Reelin is cleaved as it diffuses through the extracellular space (reviewed in Ref. 21). This processing occurs at two specific sites within the protein, which consists of eight repeated Reelin domains flanked by an F-spondin domain on the N-terminal (Nt) side and a C-terminal (Ct) stretch of acidic residues (reviewed in Refs. 20 and 21). Reelin is cleaved within the third repeat and between the sixth and seventh repeats (Fig. 2A), whereby “completely cleaved” Reelin produces three fragments: the Nt-(N-R2), Ct-(R7-C), and the central-(R3–6) fragments (reviewed in Ref. 11).
The central fragment contains the binding site for ApoER2 and VLDLR (Fig. 2A) and is sufficient to rescue the abnormal neuronal migration in reeler slice cultures (reviewed in Refs. 5, 11, and 20–22). However, the homodimerization of Reelin through disulfide linkage of the N-terminal region of the fragment is required for efficient Dab1 phosphorylation (reviewed in Refs. 5, 11, and 22). Not much is known about the role of the Ct cleavage of Reelin between R6 and R7; however, Reelin-mediated Dab1 phosphorylation is reduced when Reelin lacks the Ct region, so this cleavage would reduce the efficacy of Reelin signaling (reviewed in Refs. 11, 21, and 22). Interestingly, neuronal activity can drive this cleavage by activating tissue plasminogen activator (tPA) (reviewed in Ref. 21), thus diminishing the effect of Reelin after synaptic potentiation.
More is known about the role of the Nt fragment, which has been reported to bind α3β1 integrins, EphB receptors, and the cadherin-related neuronal receptors (CNRs) (Fig. 2A) (reviewed in Refs. 5 and 11). The interaction of Reelin with α3β1 integrins is required for the Reelin-mediated surface diffusion of NR2B-containing NMDARs (24). The interaction of Reelin with EphB receptors may contribute to the proper lamination of the CA3 region of the hippocampus (reviewed in Ref. 11). Recently, EphB receptors were shown to be critical for the development of innate fear in mice, whereby loss of EphB receptors disrupted normal axonal innervation in the amygdala (40). Cleavage of Reelin between R2 and R3 would prevent these known interactions. Additionally, this cleavage is known to reduce the distance Reelin diffuses and the potency of downstream signaling by Reelin (reviewed in Refs. 5, 11, and 20–22).
This cleavage at the N-terminal site plays a potential role in temporal lobe epilepsies (TLEs). The most common risk factor for TLEs is an initial fever-induced seizure. Postmortem studies of TLE patients suggest that in most cases, the seizures are accompanied by dispersion of the dentate gyrus granule cells with a correlated decrease of Reelin-expressing neurons in the hilus (reviewed in Ref. 41). Induction of seizures by intrahippocampal perfusion of kainate (KA), a potent glutamate receptor agonist, in mice recapitulated this granule cell dispersion (GCD) and loss of Reelin-positive neurons (reviewed in Ref. 41). Later studies were able to recapitulate the KA-induced GCD in rodent organotypic hippocampal slice cultures, which resulted in a rapid loss of Reelin-expressing hilar neurons. These organotypic hippocampal slice cultures also had elevated expression of the matrix metalloprotease (MMP) inhibitor, tissue inhibitor of MMP-1 (TIMP1), which led to reduced Nt cleavage and diffusion of Reelin (42–44). Inhibition of TIMP1 restored the Nt cleavage of Reelin and reversed the KA-induced GCD (43). Furthermore, perfusion of the central fragment of Reelin could prevent this GCD (44). Although these studies demonstrate that GCD results from both the loss of Reelin-positive neurons and hindered proteolysis of Reelin after epileptic insult, the question still remains whether the recurrence of seizures in TLEs is due to GCD.
Alternative splicing of the Reelin receptor, ApoER2, can differentially modulate the acquisition of fear memory (30, 34). Mice expressing ApoER2 splice variants lacking the proline-rich domain (ApoER2[Δ19]) have reduced contextual fear memory when compared with mice expressing the wild-type ApoER2 or ApoER2 with exon 19 (ApoER2[+19]) (30). Interestingly, these mice behave differently when exon 16 (exon 15 in humans) encoding the OLS domain of ApoER2 is also excluded. Mice lacking exon 16 either with or without exon 19 (ApoER2[Δ16+19] or ApoER2[Δ16Δ19], respectively) acquire contextual fear memory as well as wild-type mice; however, mice lacking both exons appear hypersensitive to cued fear memory, whereas the inclusion of exon 19 completely blocked the acquisition of cued fear memory (34).
Another potential feedback loop involves the induction of brain-derived neurotrophic factor (BDNF). Reelin-mediated enhancement induces the activation of CREB (29) (Fig. 2B, 1), which increases the expression of BDNF (45) (Fig. 2B, 2). This neurotrophin can increase the expression of extracellular metalloproteinases (46) and induces ApoER2 cleavage through binding TrkB receptors (47) (Fig. 2B, 5). Interestingly, experiments utilizing the focal uncaging of glutamate have revealed that Ca2+ influx from NMDAR can increase BDNF rapidly and locally. This BDNF is released into the synaptic cleft of the same synapse, where it activates TrkB receptors and induces the actin-stabilizing GTPases Cdc42 and Rac (48) (Fig. 2B, 3 and 4), much like Reelin during axogenesis (8) (Fig. 1D). This would provide a focal, synapse-specific homeostatic feedback regulation whereby local synaptic production of BDNF can both strengthen the dendritic spine and prevent subsequent excessive Reelin-mediated potentiation through cleavage of ApoER2 (Fig. 2B, 6 and 7).
Reelin's Protective Role against Synaptic Dysfunction
A role for Reelin in several neuropsychiatric and neurodegenerative diseases, such as autism, schizophrenia, bipolar disorder, major depression, and Alzheimer's disease, has been reported (reviewed in Refs. 20, 22, and 49). Each disorder destroys the quality of life in very different ways; however, reduced Reelin expression seems to be a common factor among them (reviewed in Refs. 5 and 21).
Alzheimer's Disease
Alzheimer's disease (AD) is a progressive neurodegenerative disorder, which first presents as memory loss brought on by synaptic dysfunction. Two pathological hallmarks of AD have been identified: extracellular amyloid β (Aβ) plaques and intracellular neurofibrillary tau tangles. Aβ, the central component in the trademark plaques that build up in the brains of people with AD, is a product of the amyloid precursor protein, APP (reviewed in Refs. 31 and 50). Tau neurofibrillary tangles, the other pathological hallmark of AD, form when the microtubule-associated tau protein is hyperphosphorylated, promoting aggregation and intraneuronal tangle formation (reviewed in Refs. 22 and 31).
Aβ and Tau
Both Aβ and tau are released with normal network activity, so each protein most likely serves a physiological purpose (51, 52). In AD, oligomeric Aβ particles are detectable before tau aggregation and are known to impart toxic effects at the synapse (reviewed in Ref. 53), inducing synaptic dysfunction and likely causing the early manifestation of memory deficits in AD. In a recent study, modification of APP cleavage could prevent Aβ formation and prevent cognitive decline, further supporting this amyloid-induced hypothesis of AD (54). Oligomeric Aβ particles suppress LTP and enhance long-term depression (LTD) through interactions with synaptic proteins (and reviewed in Refs. 31 and 55), such as metabotropic glutamate receptors (mGluRs) as well as potentially the Ca2+-permeable α7-nicotinic acetylcholine receptor (α7-nAChR) (reviewed in Refs. 56 and 57). (Fig. 3C). Activation of mGluRs and α7-nAChRs can activate striatal-enriched tyrosine phosphatase 61 (STEP61) (58, 59), leading to NMDAR and AMPAR endocytosis by inhibiting Fyn-mediated phosphorylation of the receptors (59, 60) (Fig. 3D).
The phosphorylation of tau inhibits microtubule stability, which is essential for the formation and maintenance of dendritic spines (reviewed in Ref. 61). Cyclin-dependent kinase5 (cdk5) can phosphorylate tau, which is activated by co-factors p35 or p39. Elevated Ca2+ concentrations activate calpain, which cleaves p35 to p25 (reviewed in Ref. 62). Cdk5 binds p25 longer than p35, which prolongs Cdk5 activity, leading to enhanced tau phosphorylation (Fig. 3E). Both p25 and calpain are elevated in AD brains, and Aβ can induce tau phosphorylation through deregulating Cdk5 activation (reviewed in Ref. 62). At the same time, Reelin signaling through the Dab1/PI3K/Akt pathway reduces this phosphorylation by inhibiting glycogen synthase kinase-3β (GSK-3β) (reviewed in Ref. 31) (Figs. 1B and 3F), which would stabilize microtubules and help maintain synaptic stability. VLDLR and ApoER2 KO mice show increased tau phosphorylation in the brain (63), supporting the role of Reelin signaling in the regulation of tau protein hyperphosphorylation and a potential role in AD pathogenesis.
Regulation of Aβ Formation by the Reelin Receptor ApoER2
The production of oligomeric Aβ depends on how APP is processed. The amyloidogenic fate of APP is determined by the initial secretase cleavage step. Non-amyloidogenic cleavage of APP is mediated by extracellular α-secretases, whereas amyloidogenic processing starts primarily in endosomal compartments by β-secretase cleavage (reviewed in Refs. 31 and 50). Thus, subcellular localization plays an important role in whether APP cleavage produces oligomer-prone Aβ.
Multiple intracellular and extracellular adaptor proteins can influence APP localization and processing. Interestingly, the majority of these proteins also interact with ApoER2, which adds an additional layer of regulation to APP localization and processing. Co-expression of ApoER2 with APP promotes APP surface expression and the lipid raft association of APP, both of which reduce Aβ formation (64). The extracellular ligand, F-spondin, binds both ApoER2 and APP, enhances their surface expression, and reduces Aβ formation (65, 66). Intracellularly, APP and ApoER2 bind the adaptor proteins X11α/β (67–70), Fe65 (69, 71, 72), Snx17 (73, 74), Dab1 (75–79), and Dab2 (73, 80), and these interactions regulate their surface expression and processing (Fig. 4).
FIGURE 4.
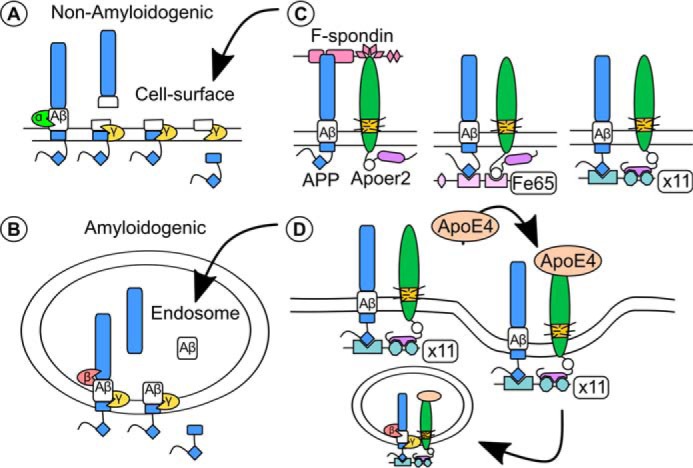
ApoER2, ApoE, and APP all converge to regulate Aβ formation. A and B, the production of oligomeric Aβ depends on the proteolytic processing of APP, which is sequentially cleaved initially by a α- or β-secretase followed by γ-secretase cleavage. A, Non-amyloidogenic processing of APP. The α-secretases are present in the extracellular space and cleave APP within the Aβ region of the protein, preventing the release of Aβ upon γ-secretase cleavage. B, amyloidogenic processing of APP. β-Secretases are found mostly in the endosome and cleave APP to yield the Aβ peptide (reviewed in Ref. 50). C and D, APP and ApoER2 share multiple adaptor proteins that affect their localization and Aβ formation (C). C, the surface localization of APP and ApoER2 and non-amyloidogenic processing of APP are altered by the extracellular binding of F-spondin (left panel) (66) and intracellular interactions with Fe65 (middle panel) (72) and X11α/β (right panel) (67–70). ApoER2 binds the thrombospondin repeats 1–4 (pink triangles) of F-spondin, whereas APP binds the Reelin and Spondin domain of F-spondin (65, 66). The NPXY domain of APP (blue diamond) binds to the phosphotyrosine binding domains (PTB) of both Fe65 and X11α/β (69), whereas ApoER2 binds a second PTB domain of Fe65 (via the NPXY domain, white circle) (72) or the PDZ domains of X11α/β via the alternatively spliced proline-rich insert (purple oval) (68). D, the addition of ApoE4 induced an X11α/β-dependent co-endocytosis of APP and ApoER2, leading to increased amyloidogenic processing of APP (67).
In addition to protecting against Aβ- and tau-related AD pathology, Reelin signaling is implicated in protecting against pathological effects of the most common genetic risk factor for late-onset AD, the ϵ4 allele of apolipoprotein E (ApoE4) (reviewed in Refs. 31 and 81). Human carriers of ApoE4 are at increased risk for AD when compared with those carrying the more common ϵ3 allele (ApoE3), or the protective ϵ2 allele (ApoE2). ApoE transports cholesterol to cells via lipoprotein receptors through the endosome, where they recycle back to the membrane or shuttle to the lysosome for degradation. Initial studies in cell lines found that endocytosed particles of lipidated ApoE4 remained in endosomes longer (reviewed in Ref. 31). The lipidation of ApoE4 is also less efficient than ApoE3. Of note, enhancing ApoE4 lipidation by stimulating the activity of the astrocytic ABCA1 could actually rescue known behavior deficit in ApoE4 knock-in mice (82–84).
In central and peripheral synapses, ApoE is known to bind the low-density lipoprotein receptor-related protein 1 and 4 (Lrp1 and Lrp4) as well as ApoER2 and VLDLR. In a study from our laboratory, we demonstrated that exogenous ApoE4 sequesters neuronal ApoER2 and glutamate receptors in the recycling endosome (85) and endogenous ApoE4 reduced the protective effect of Reelin against Aβ-induced synaptic suppression (86, 87) (Fig. 3G). This ApoE4-mediated ApoER2 sequestration was recapitulated in cells transfected with ApoER2 and APP (67), resulting in enhanced Aβ formation. This increased amyloidogenic processing of APP by ApoE4 required the cytosolic adaptor X11α/β, which binds to the NPXY motif of APP and the alternatively spliced proline-rich insert of ApoER2, leading to enhanced co-endocytosis of the receptors (67). Without X11α/β, ApoER2 actually reduces amyloidogenic processing of APP. Reelin can interrupt this interaction between X11α/β and ApoER2 (68), indicating another protective role of Reelin against Aβ toxicity.
Studies exploring the pathology of AD in both human patients and mouse models have unveiled multiple paths that could repress Reelin signaling. The numbers of Reelin-expressing cells are reduced in the entorhinal cortex, where the earliest signs of AD pathogenesis emerge, in both an AD mouse model as well as postmortem human AD brains (88). Altered cleavage of Reelin was also found in AD mice as well as postmortem brain tissue and the cerebrospinal fluid of human AD subjects (89). This altered cleavage may enhance the adsorption of Reelin into Aβ plaques (90), which would further exacerbate the deficit in Reelin signaling. Furthermore, alternative splicing deficits observed in both AD mice and human brains leads to reduced inclusion of the proline-rich cytoplasmic domain of ApoER2 (91), which would reduce the ability of Reelin to modulate synaptic strength.
The likelihood that Reelin signaling is in fact repressed in AD is further solidified by evidence of suppressed activity of pathways downstream of Reelin. mTOR activity, which is increased by Reelin signaling (92), is lower in AD mice (93). Reduced Reelin expression in HRM mice exacerbates the accumulation of both Aβ and phosphorylated tau (94), whose production is inhibited by Reelin signaling (Ref. 76 and reviewed in Ref. 31). Similarly, the learning deficits in an AD mouse model are exacerbated by loss of Reelin expression after development (49), further solidifying the protective role of Reelin in AD. Enhancing Reelin signaling by either exogenous supplementation of Reelin (95) or reversing the ApoER2 splicing deficit with antisense oligonucleotides (91) can improve the cognitive performance of AD mice in a murine model of AD.
Schizophrenia and Autism
Genetic linkage analyses implicate Reelin polymorphisms in the pathophysiology of two complex neurodevelopmental diseases, schizophrenia and autism spectrum disorders (ASDs) (reviewed in Ref. 10). Postmortem studies reveal a decrease in the expression of Reelin in schizophrenia and ASD brains as well as the cerebrospinal fluid of autistic subjects (reviewed in Refs. 10, 22, and 96). Spine density of schizophrenic individuals is reduced, similar to the reduction observed with Reelin deficiency (reviewed in Refs. 10 and 22). Interestingly, expression of downstream signaling partners is also altered in postmortem ASD brains, with reduced Dab1 expression and increased VLDLR (96). The time and place at which Reelin signaling is reduced may prove to be the deciding factor for the extent of cognitive impact that defective Reelin signaling may have (97). Many parallels have been drawn between the pathology of synaptic dysfunction and behavior in the HRM and schizophrenia and ASD phenotypes (reviewed extensively in Ref. 20).
Concluding Remarks
The implication of Reelin in a variety of neurological conditions is intriguing, with the most obvious common thread being reduced Reelin signaling during development and at the synapse. Considering the multitude of modulatory mechanisms by which Reelin alters brain function during development and in the adult CNS, the diversity of pathologies linked to faulty Reelin signaling is not surprising. Because of the vast role of Reelin throughout the brain at all ages, future investigation of suspected deficits in Reelin signaling linked to a disease should pay special attention to the spatial and temporal appearance of these deficits. Clinical application of targeted therapies like the antisense oligonucleotide-mediated reversal of ApoER2 splicing deficits will require understanding not only how disease-associated abnormalities in Reelin or associated downstream effectors contribute to disease but also when and where. A convincing example of the benefit of this approach is a study that focused on Reelin expression in the entorhinal cortex and hippocampus. This focus revealed that Reelin-expressing neurons in the entorhinal cortex are especially vulnerable to Aβ toxicity and die off before obvious signs of Aβ accumulation. As the site of the earliest neuronal loss in AD pathogenesis, loss of Reelin-expressing cells at this time and place in both an AD mouse model as well as postmortem human AD brains clarifies how Aβ might instigate AD pathogenesis by reducing Reelin levels and increasing the vulnerability of synapses to the toxic effects of Aβ (88). As both schizophrenia and autism are believed to arise from early neurodevelopmental deficits, special attention should be given to the effect of genetically associated Reln mutations during development: a time at which Reelin is critical for neuronal migration and the formation of axons and dendrites as well as the maturation of the synapses formed between them.
Intriguingly, recent studies have thrown a spotlight on novel functions of Reelin expressed in the periphery (98) that are independent of its roles in the CNS, which we have reviewed in depth here. These recent findings have demonstrated that Reelin controls inflammatory responses in the vascular wall or colon, respectively (99, 100). To date, there is no known inflammatory role of Reelin in the CNS, and these newly uncovered peripheral inflammatory actions raise the possibility that Reelin may also play a role in inflammatory syndromes of the CNS, such as multiple sclerosis.
Acknowledgments
We are indebted to Theresa Pohlkamp for critical reading of the manuscript and to all past and present members of the Herz lab for their many contributions, which are in part reviewed here. Due to citation limitations, we were unable to cite many of the primary publications.
This work was supported by National Institutes of Health Grants HL063762, NS093382, and AG053391 (to J. H.), Consortium for Frontotemporal Dementia Research Grant A108400, and Brightfocus Foundation Grant A2016396S. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- VLDLR
- very low density lipoprotein receptor
- SFK
- Src family tyrosine kinase
- HRM
- heterozygous reeler mice
- mTOR
- mammalian target of rapamycin
- AMPAR
- AMPA receptor
- NMDAR
- NMDA receptor
- LTP
- long-term potentiation
- LTD
- long-term depression
- BDNF
- brain-derived neurotrophic factor
- CREB
- cAMP-response element-binding protein
- OLS
- O-linked sugar
- ECD
- extracellular domain
- ICD
- intracellular domain
- TLE
- temporal lobe epilepsy
- KA
- kainate
- GCD
- granule cell dispersion
- TrkB
- tropomyosin receptor kinase B
- AD
- Alzheimer's disease
- Aβ
- amyloid β
- APP
- amyloid precursor protein
- mGluR
- metabotropic glutamate receptor
- α7-nAChR
- α7-nicotinic acetylcholine receptor
- GSK-3β
- glycogen synthase kinase-3β
- ASD
- autism spectrum disorder
- Nt
- N-terminal
- Ct
- C-terminal.
References
- 1. Falconer D. S. (1951) Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 50, 192–201 [DOI] [PubMed] [Google Scholar]
- 2. Curran T., and D'Arcangelo G. (1998) Role of reelin in the control of brain development. Brain Res. Brain Res. Rev. 26, 285–294 [DOI] [PubMed] [Google Scholar]
- 3. Frotscher M. (2010) Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 33, 407–414 [DOI] [PubMed] [Google Scholar]
- 4. D'Arcangelo G. (2014) Reelin in the years: controlling neuronal migration and maturation in the mammalian brain. Adv. Neurosci. 2014, 1–19 [Google Scholar]
- 5. Bock H. H., and May P. (2016) Canonical and non-canonical Reelin signaling. Front. Cell. Neurosci. 10, 166, 10.1155/2014/597395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pesold C., Impagnatiello F., Pisu M. G., Uzunov D. P., Costa E., Guidotti A., and Caruncho H. J. (1998) Reelin is preferentially expressed in neurons synthesizing γ-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. U.S.A. 95, 3221–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Del Río J. A., Heimrich B., Supèr H., Borrell V., Frotscher M., and Soriano E. (1996) Differential survival of Cajal-Retzius cells in organotypic cultures of hippocampus and neocortex. J. Neurosci. 16, 6896–6907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leemhuis J., Bouche E., Frotscher M., Henle F., Hein L., Herz J., Meyer D. K., Pichler M., Roth G., Schwan C., and Bock H. H. (2010) Reelin signals through apolipoprotein E receptor 2 and Cdc42 to increase growth cone motility and filopodia formation. J. Neurosci. 30, 14759–14772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niu S., Renfro A., Quattrocchi C. C., Sheldon M., and D'Arcangelo G. (2004) Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 41, 71–84 [DOI] [PubMed] [Google Scholar]
- 10. Ishii K., Kubo K. I., and Nakajima K. (2016) Reelin and neuropsychiatric disorders. Front. Cell. Neurosci. 10, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee G. H., and D'Arcangelo G. (2016) New insights into Reelin-mediated signaling pathways. Front. Cell. Neurosci. 10, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsuki T., Pramatarova A., and Howell B. W. (2008) Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J. Cell Sci. 121, 1869–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jossin Y., and Cooper J. A. (2011) Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 14, 697–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Niu S., Yabut O., and D'Arcangelo G. (2008) The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 28, 10339–10348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glantz L. A., and Lewis D. A. (2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73 [DOI] [PubMed] [Google Scholar]
- 16. Dumanis S. B., Cha H. J., Song J. M., Trotter J. H., Spitzer M., Lee J. Y., Weeber E. J., Turner R. S., Pak D. T., Rebeck G. W., and Hoe H. S. (2011) ApoE receptor 2 regulates synapse and dendritic spine formation. PLoS ONE 6, e17203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cull-Candy S., Brickley S., and Farrant M. (2001) NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335 [DOI] [PubMed] [Google Scholar]
- 18. Sinagra M., Verrier D., Frankova D., Korwek K. M., Blahos J., Weeber E. J., Manzoni O. J., and Chavis P. (2005) Reelin, very-low-density lipoprotein receptor, and apolipoprotein E receptor 2 control somatic NMDA receptor composition during hippocampal maturation in vitro. J. Neurosci. 25, 6127–6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Campo C. G., Sinagra M., Verrier D., Manzoni O. J., and Chavis P. (2009) Reelin secreted by GABAergic neurons regulates glutamate receptor homeostasis. PLoS ONE 4, e5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folsom T. D., and Fatemi S. H. (2013) The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 68, 122–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lussier A. L., Weeber E. J., and Rebeck G. W. (2016) Reelin proteolysis affects signaling related to normal synapse function and neurodegeneration. Front. Cell. Neurosci. 10, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knuesel I. (2010) Reelin-mediated signaling in neuropsychiatric and neurodegenerative diseases. Prog. Neurobiol. 91, 257–274 [DOI] [PubMed] [Google Scholar]
- 23. Malenka R. C., and Nicoll R. A. (1997) Silent synapses speak up. Neuron 19, 473–476 [DOI] [PubMed] [Google Scholar]
- 24. Groc L., Choquet D., Stephenson F. A., Verrier D., Manzoni O. J., and Chavis P. (2007) NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J. Neurosci. 27, 10165–10175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bal M., Leitz J., Reese A. L., Ramirez D. M., Durakoglugil M., Herz J., Monteggia L. M., and Kavalali E. T. (2013) Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 80, 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hellwig S., Hack I., Kowalski J., Brunne B., Jarowyj J., Unger A., Bock H. H., Junghans D., and Frotscher M. (2011) Role for Reelin in neurotransmitter release. J. Neurosci. 31, 2352–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weeber E. J., Beffert U., Jones C., Christian J. M., Forster E., Sweatt J. D., and Herz J. (2002) Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 277, 39944–39952 [DOI] [PubMed] [Google Scholar]
- 28. Waxman E. A., and Lynch D. R. (2005) N-Methyl-d-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist 11, 37–49 [DOI] [PubMed] [Google Scholar]
- 29. Chen Y., Beffert U., Ertunc M., Tang T. S., Kavalali E. T., Bezprozvanny I., and Herz J. (2005) Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 25, 8209–8216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beffert U., Weeber E. J., Durudas A., Qiu S., Masiulis I., Sweatt J. D., Li W. P., Adelmann G., Frotscher M., Hammer R. E., and Herz J. (2005) Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 47, 567–579 [DOI] [PubMed] [Google Scholar]
- 31. Lane-Donovan C., Philips G. T., and Herz J. (2014) More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 83, 771–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qiu S., Korwek K. M., Pratt-Davis A. R., Peters M., Bergman M. Y., and Weeber E. J. (2006) Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 85, 228–242 [DOI] [PubMed] [Google Scholar]
- 33. Iafrati J., Orejarena M. J., Lassalle O., Bouamrane L., Gonzalez-Campo C., and Chavis P. (2014) Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 19, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wasser C. R., Masiulis I., Durakoglugil M. S., Lane-Donovan C., Xian X., Beffert U., Agarwala A., Hammer R. E., and Herz J. (2014) Differential splicing and glycosylation of Apoer2 alters synaptic plasticity and fear learning. Sci. Signal. 7, ra113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. May P., Bock H. H., Nimpf J., and Herz J. (2003) Differential glycosylation regulates processing of lipoprotein receptors by γ-secretase. J. Biol. Chem. 278, 37386–37392 [DOI] [PubMed] [Google Scholar]
- 36. Duit S., Mayer H., Blake S. M., Schneider W. J., and Nimpf J. (2010) Differential functions of ApoER2 and very low density lipoprotein receptor in Reelin signaling depend on differential sorting of the receptors. J. Biol. Chem. 285, 4896–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koch S., Strasser V., Hauser C., Fasching D., Brandes C., Bajari T. M., Schneider W. J., and Nimpf J. (2002) A secreted soluble form of ApoE receptor 2 acts as a dominant-negative receptor and inhibits Reelin signaling. EMBO J. 21, 5996–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balmaceda V., Cuchillo-Ibáñez I., Pujadas L., García-Ayllón M. S., Saura C. A., Nimpf J., Soriano E., and Sáez-Valero J. (2014) ApoER2 processing by presenilin-1 modulates reelin expression. FASEB J. 28, 1543–1554 [DOI] [PubMed] [Google Scholar]
- 39. Telese F., Ma Q., Perez P. M., Notani D., Oh S., Li W., Comoletti D., Ohgi K. A., Taylor H., and Rosenfeld M. G. (2015) LRP8-Reelin-regulated neuronal enhancer signature underlying learning and memory formation. Neuron 86, 696–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu X. N., Liu X. D., Zhuang H., Henkemeyer M., Yang J. Y., and Xu N. J. (2016) Amygdala EphB2 signaling regulates glutamatergic neuron maturation and innate fear. J. Neurosci. 36, 10151–10162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haas C. A., and Frotscher M. (2010) Reelin deficiency causes granule cell dispersion in epilepsy. Exp Brain Res. 200, 141–149 [DOI] [PubMed] [Google Scholar]
- 42. Tinnes S., Schäfer M. K., Flubacher A., Münzner G., Frotscher M., and Haas C. A. (2011) Epileptiform activity interferes with proteolytic processing of Reelin required for dentate granule cell positioning. FASEB J. 25, 1002–1013 [DOI] [PubMed] [Google Scholar]
- 43. Tinnes S., Ringwald J., and Haas C. A. (2013) TIMP-1 inhibits the proteolytic processing of Reelin in experimental epilepsy. FASEB J. 27, 2542–2552 [DOI] [PubMed] [Google Scholar]
- 44. Orcinha C., Münzner G., Gerlach J., Kilias A., Follo M., Egert U., and Haas C. A. (2016) Seizure-induced motility of differentiated dentate granule cells is prevented by the central Reelin fragment. Front. Cell. Neurosci. 10, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tao X., Finkbeiner S., Arnold D. B., Shaywitz A. J., and Greenberg M. E. (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726 [DOI] [PubMed] [Google Scholar]
- 46. Kuzniewska B., Rejmak E., Malik A. R., Jaworski J., Kaczmarek L., and Kalita K. (2013) Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol. Cell. Biol. 33, 2149–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Larios J. A., Jausoro I., Benitez M. L., Bronfman F. C., and Marzolo M. P. (2014) Neurotrophins regulate ApoER2 proteolysis through activation of the Trk signaling pathway. BMC Neurosci. 15, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harward S. C., Hedrick N. G., Hall C. E., Parra-Bueno P., Milner T. A., Pan E., Laviv T., Hempstead B. L., Yasuda R., and McNamara J. O. (2016) Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538, 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lane-Donovan C., Philips G. T., Wasser C. R., Durakoglugil M. S., Masiulis I., Upadhaya A., Pohlkamp T., Coskun C., Kotti T., Steller L., Hammer R. E., Frotscher M., Bock H. H., and Herz J. (2015) Reelin protects against amyloid β toxicity in vivo. Sci. Signal. 8, ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marzolo M. P., and Bu G. (2009) Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer's disease. Semin. Cell Dev. Biol. 20, 191–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wei W., Nguyen L. N., Kessels H. W., Hagiwara H., Sisodia S., and Malinow R. (2010) Amyloid β from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu J. W., Hussaini S. A., Bastille I. M., Rodriguez G. A., Mrejeru A., Rilett K., Sanders D. W., Cook C., Fu H., Boonen R. A., Herman M., Nahmani E., Emrani S., Figueroa Y. H., Diamond M. I., et al. (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paula-Lima A. C., Brito-Moreira J., and Ferreira S. T. (2013) Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer's disease. J. Neurochem. 126, 191–202 [DOI] [PubMed] [Google Scholar]
- 54. Kounnas M. Z., Lane-Donovan C., Nowakowski D. W., Herz J., and Comer W. T. (2016) NGP 555, a γ-secretase modulator, lowers the amyloid biomarker, Aβ42, in cerebrospinal fluid while preventing Alzheimer's disease cognitive decline in rodents. Alzheimers Dement. Transl Res. Clin Interv. 10.1016/j.trci.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guntupalli S., Widagdo J., and Anggono V. (2016) Amyloid-β-induced dysregulation of AMPA receptor trafficking. Neural Plast. 2016, 3204519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lilja A. M., Porras O., Storelli E., Nordberg A., and Marutle A. (2011) Functional interactions of fibrillar and oligomeric amyloid-β with α7 nicotinic receptors in Alzheimer's disease. J. Alzheimers Dis. 23, 335–347 [DOI] [PubMed] [Google Scholar]
- 57. Ferreira S. T., Lourenco M. V., Oliveira M. M., and De Felice F. G. (2015) Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front. Cell. Neurosci. 9, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen X., Lin R., Chang L., Xu S., Wei X., Zhang J., Wang C., Anwyl R., and Wang Q. (2013) Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience 253, 435–443 [DOI] [PubMed] [Google Scholar]
- 59. Zhang L., Xie J. W., Yang J., and Cao Y. P. (2013) Tyrosine phosphatase STEP61 negatively regulates amyloid β-mediated ERK/CREB signaling pathways via α7 nicotinic acetylcholine receptors. J. Neurosci. Res. 91, 1581–1590 [DOI] [PubMed] [Google Scholar]
- 60. Moult P. R., Gladding C. M., Sanderson T. M., Fitzjohn S. M., Bashir Z. I., Molnar E., and Collingridge G. L. (2006) Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J. Neurosci. 26, 2544–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sabbagh J. J., and Dickey C. A. (2016) The metamorphic nature of the Tau protein: dynamic flexibility comes at a cost. Front. Neurosci. 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shukla V., Skuntz S., and Pant H. C. (2012) Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Arch. Med. Res. 43, 655–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hiesberger T., Trommsdorff M., Howell B. W., Goffinet A., Mumby M. C., Cooper J. A., and Herz J. (1999) Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489 [DOI] [PubMed] [Google Scholar]
- 64. Fuentealba R. A., Barría M. I., Lee J., Cam J., Araya C., Escudero C. A., Inestrosa N. C., Bronfman F. C., Bu G., and Marzolo M. P. (2007) ApoER2 expression increases Aβ production while decreasing Amyloid Precursor Protein (APP) endocytosis: possible role in the partitioning of APP into lipid rafts and in the regulation of γ-secretase activity. Mol. Neurodegener. 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ho A., and Südhof T. C. (2004) Binding of F-spondin to amyloid-β precursor protein: a candidate amyloid-β precursor protein ligand that modulates amyloid-β precursor protein cleavage. Proc. Natl. Acad. Sci. U.S.A. 101, 2548–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hoe H. S., Wessner D., Beffert U., Becker A. G., Matsuoka Y., and Rebeck G. W. (2005) F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol. Cell Biol. 25, 9259–9268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. He X., Cooley K., Chung C. H., Dashti N., and Tang J. (2007) Apolipoprotein receptor 2 and X11 α/β mediate apolipoprotein E-induced endocytosis of amyloid-β precursor protein and β-secretase, leading to amyloid-β production. J. Neurosci. 27, 4052–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Minami S. S., Sung Y. M., Dumanis S. B., Chi S. H., Burns M. P., Ann E. J., Suzuki T., Turner R. S., Park H. S., Pak D. T., Rebeck G. W., and Hoe H. S. (2010) The cytoplasmic adaptor protein X11α and extracellular matrix protein Reelin regulate ApoE receptor 2 trafficking and cell movement. FASEB J. 24, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Borg J. P., Ooi J., Levy E., and Margolis B. (1996) The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell Biol. 16, 6229–6241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Borg J. P., Yang Y., De Taddéo-Borg M., Margolis B., and Turner R. S. (1998) The X11α protein slows cellular amyloid precursor protein processing and reduces Aβ40 and Aβ42 secretion. J. Biol. Chem. 273, 14761–14766 [DOI] [PubMed] [Google Scholar]
- 71. Fiore F., Zambrano N., Minopoli G., Donini V., Duilio A., and Russo T. (1995) The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J. Biol. Chem. 270, 30853–30856 [DOI] [PubMed] [Google Scholar]
- 72. Hoe H. S., Magill L. A., Guenette S., Fu Z., Vicini S., and Rebeck G. W. (2006) FE65 interaction with the ApoE receptor ApoEr2. J. Biol. Chem. 281, 24521–24530 [DOI] [PubMed] [Google Scholar]
- 73. Lee J., Retamal C., Cuitiño L., Caruano-Yzermans A., Shin J. E., van Kerkhof P., Marzolo M. P., and Bu G. (2008) Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 283, 11501–11508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sotelo P., Farfán P., Benitez M. L., Bu G., and Marzolo M. P. (2014) Sorting nexin 17 regulates ApoER2 recycling and reelin signaling. PLoS ONE 9, e93672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ando K., Iijima K. I., Elliott J. I., Kirino Y., and Suzuki T. (2001) Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of β-amyloid. J. Biol. Chem. 276, 40353–40361 [DOI] [PubMed] [Google Scholar]
- 76. Hoe H. S., Tran T. S., Matsuoka Y., Howell B. W., and Rebeck G. W. (2006) DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J. Biol. Chem. 281, 35176–35185 [DOI] [PubMed] [Google Scholar]
- 77. Homayouni R., Rice D. S., Sheldon M., and Curran T. (1999) Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J. Neurosci. 19, 7507–7515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Howell B. W., Lanier L. M., Frank R., Gertler F. B., and Cooper J. A. (1999) The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol. Cell. Biol. 19, 5179–5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morimura T., and Ogawa M. (2009) Relative importance of the tyrosine phosphorylation sites of Disabled-1 to the transmission of Reelin signaling. Brain Res. 1304, 26–37 [DOI] [PubMed] [Google Scholar]
- 80. Cuitino L., Matute R., Retamal C., Bu G., Inestrosa N. C., and Marzolo M. P. (2005) ApoER2 is endocytosed by a clathrin-mediated process involving the adaptor protein Dab2 independent of its Rafts' association. Traffic 6, 820–838 [DOI] [PubMed] [Google Scholar]
- 81. Strittmatter W. J., and Roses A. D. (1996) Apolipoprotein E and Alzheimer's disease. Annu. Rev. Neurosci. 19, 53–77 [DOI] [PubMed] [Google Scholar]
- 82. Boehm-Cagan A., and Michaelson D. M. (2014) Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J. Neurosci. 34, 7293–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Salomon-Zimri S., Boehm-Cagan A., Liraz O., and Michaelson D. M. (2014) Hippocampus-related cognitive impairments in young apoE4 targeted replacement mice. Neurodegener. Dis. 13, 86–92 [DOI] [PubMed] [Google Scholar]
- 84. Boehm-Cagan A., Bar R., Liraz O., Bielicki J. K., Johansson J. O., and Michaelson D. M. (2016) ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J. Alzheimers Dis. 54, 1219–1233 [DOI] [PubMed] [Google Scholar]
- 85. Chen Y., Durakoglugil M. S., Xian X., and Herz J. (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. U.S.A. 107, 12011–12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chen Q. S., Kagan B. L., Hirakura Y., and Xie C. W. (2000) Impairment of hippocampal long-term potentiation by Alzheimer amyloid β-peptides. J. Neurosci. Res. 60, 65–72 [DOI] [PubMed] [Google Scholar]
- 87. Durakoglugil M. S., Chen Y., White C. L., Kavalali E. T., and Herz J. (2009) Reelin signaling antagonizes β-amyloid at the synapse. Proc. Natl. Acad. Sci. U.S.A. 106, 15938–15943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chin J., Massaro C. M., Palop J. J., Thwin M. T., Yu G. Q., Bien-Ly N., Bender A., and Mucke L. (2007) Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer's disease. J. Neurosci. 27, 2727–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Herring A., Donath A., Steiner K. M., Widera M. P., Hamzehian S., Kanakis D., Kölble K., ElAli A., Hermann D. M., Paulus W., and Keyvani K. (2012) Reelin depletion is an early phenomenon of Alzheimer's pathology. J. Alzheimers Dis. 30, 963–979 [DOI] [PubMed] [Google Scholar]
- 90. Knuesel I., Nyffeler M., Mormède C., Muhia M., Meyer U., Pietropaolo S., Yee B. K., Pryce C. R., LaFerla F. M., Marighetto A., and Feldon J. (2009) Age-related accumulation of Reelin in amyloid-like deposits. Neurobiol. Aging 30, 697–716 [DOI] [PubMed] [Google Scholar]
- 91. Hinrich A. J., Jodelka F. M., Chang J. L., Brutman D., Bruno A. M., Briggs C. A., James B. D., Stutzmann G. E., Bennett D. A., Miller S. A., Rigo F., Marr R. A., and Hastings M. L. (2016) Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol. Med. 8, 328–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jossin Y., and Goffinet A. M. (2007) Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell. Biol. 27, 7113–7124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ma T., Hoeffer C. A., Capetillo-Zarate E., Yu F., Wong H., Lin M. T., Tampellini D., Klann E., Blitzer R. D., and Gouras G. K. (2010) Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS ONE 5, e12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kocherhans S., Madhusudan A., Doehner J., Breu K. S., Nitsch R. M., Fritschy J. M., and Knuesel I. (2010) Reduced Reelin expression accelerates amyloid-β plaque formation and tau pathology in transgenic Alzheimer's disease mice. J. Neurosci. 30, 9228–9240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pujadas L., Rossi D., Andrés R., Teixeira C. M., Serra-Vidal B., Parcerisas A., Maldonado R., Giralt E., Carulla N., and Soriano E. (2014) Reelin delays amyloid-β fibril formation and rescues cognitive deficits in a model of Alzheimer's disease. Nat. Commun. 5, 3443. [DOI] [PubMed] [Google Scholar]
- 96. Fatemi S. H., Snow A. V., Stary J. M., Araghi-Niknam M., Reutiman T. J., Lee S., Brooks A. I., and Pearce D. A. (2005) Reelin signaling is impaired in autism. Biol. Psychiatry 57, 777–787 [DOI] [PubMed] [Google Scholar]
- 97. Brosda J., Dietz F., and Koch M. (2011) Impairment of cognitive performance after reelin knockdown in the medial prefrontal cortex of pubertal or adult rats. Neurobiol. Dis. 44, 239–247 [DOI] [PubMed] [Google Scholar]
- 98. Smalheiser N. R., Costa E., Guidotti A., Impagnatiello F., Auta J., Lacor P., Kriho V., and Pappas G. D. (2000) Expression of reelin in adult mammalian blood, liver, pituitary pars intermedia, and adrenal chromaffin cells. Proc. Natl. Acad. Sci. U.S.A. 97, 1281–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Carvajal A. E., Vázquez-Carretero M. D., García-Miranda P., Peral M. J., Calonge M. L., and Ilundain A. A. (2016) Reelin expression is up-regulated in mice colon in response to acute colitis and provides resistance against colitis. Biochim. Biophys. Acta 1863, 462–473 [DOI] [PubMed] [Google Scholar]
- 100. Ding Y., Huang L., Xian X., Yuhanna I. S., Wasser C. R., Frotscher M., Mineo C., Shaul P. W., and Herz J. (2016) Loss of Reelin protects against atherosclerosis by reducing leukocyte-endothelial cell adhesion and lesion macrophage accumulation. Sci. Signal. 9, ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]