

Abstract
A number of point mutations in the intracellular Ca2+-sensing protein calmodulin (CaM) are arrhythmogenic, yet their underlying mechanisms are not clear. These mutations generally decrease Ca2+ binding to CaM and impair inhibition of CaM-regulated Ca2+ channels like the cardiac Ca2+ release channel (ryanodine receptor, RyR2), and it appears that attenuated CaM Ca2+ binding correlates with impaired CaM-dependent RyR2 inhibition. Here, we investigated the RyR2 inhibitory action of the CaM p.Phe142Leu mutation (F142L; numbered including the start-Met), which markedly reduces CaM Ca2+ binding. Surprisingly, CaM-F142L had little to no aberrant effect on RyR2-mediated store overload-induced Ca2+ release in HEK293 cells compared with CaM-WT. Furthermore, CaM-F142L enhanced CaM-dependent RyR2 inhibition at the single channel level compared with CaM-WT. This is in stark contrast to the actions of arrhythmogenic CaM mutations N54I, D96V, N98S, and D130G, which all diminish CaM-dependent RyR2 inhibition. Thermodynamic analysis showed that apoCaM-F142L converts an endothermal interaction between CaM and the CaM-binding domain (CaMBD) of RyR2 into an exothermal one. Moreover, NMR spectra revealed that the CaM-F142L-CaMBD interaction is structurally different from that of CaM-WT at low Ca2+. These data indicate a distinct interaction between CaM-F142L and the RyR2 CaMBD, which may explain the stronger CaM-dependent RyR2 inhibition by CaM-F142L, despite its reduced Ca2+ binding. Collectively, these results add to our understanding of CaM-dependent regulation of RyR2 as well as the mechanistic effects of arrhythmogenic CaM mutations. The unique properties of the CaM-F142L mutation may provide novel clues on how to suppress excessive RyR2 Ca2+ release by manipulating the CaM-RyR2 interaction.
Keywords: calcium intracellular release, calcium-binding protein, calmodulin (CaM), protein-protein interaction, ryanodine receptor, CaM-F142L, arrhythmia, cardiac ryanodine receptor
Introduction
Point mutations in one of the three extremely conserved calmodulin (CaM)2-encoding genes, CALM1–3, result in life-threatening ventricular arrhythmias likely due to altered CaM-regulation of the ion channels that govern cardiac excitation-contraction (1–8). The CaM-N54I and -N98S mutations (numbering includes start-Met) were identified in individuals with catecholaminergic polymorphic ventricular tachycardia (CPVT). The CaM-D96V, -D130G, and -F142L mutations were found in individuals with long QT syndrome (LQTS) (1, 2). Interestingly, the CaM-N98S mutant also apparently causes LQTS or a mixed phenotype, depending on the genetic background (4). Not only do these various mutations impose different cardiac arrhythmias, but it appears that their disease mechanisms differ at the molecular level for each CaM target, even within the same arrhythmia type (6–11). One such CaM target is the cardiac Ca2+ release channel/ryanodine receptor (RyR2). RyR2 mediates Ca2 release from the sarcoplasmic reticulum (SR) in cardiomyocytes (12, 13). The RyR2 protein forms homotetrameric channels in the SR membrane with a large cytosolic domain that interacts with numerous proteins and ligands, which regulate RyR2 Ca2+ release (12, 13). During cardiac excitation-contraction coupling, RyR2 channels are activated by Ca2+ entry into the cytosol through sarcolemmal voltage-gated Ca2+ channels (CaV1.2). This Ca2+ entry triggers RyR2-mediated SR Ca2+ release via a process called Ca2+-induced Ca2+ release, which results in the rise in cytosolic free Ca2+ ([Ca2+]cyt) and thereby drives contraction (14). RyR2 channels are sensitive to both [Ca2+]cyt and the SR luminal free Ca2+ ([Ca2+]SR) as well as a plethora of regulatory signals (13). One of these regulatory signals is CaM binding to RyR2, which generally inhibits Ca2+ release both at the diastolic and systolic [Ca2+]cyt.
CaM is a ubiquitously expressed sensor of cytosolic Ca2+ signals that has two Ca2+-binding domains (N- and C-domain), each containing two EF-hand motifs. These two domains are separated by a flexible linker, and thus one CaM protein binds up to four Ca2+ (Fig. 1). The two domains of CaM display distinct affinities and kinetics for binding to Ca2+. This allows the CaM domains to have both independent and correlated interactions with different CaM targets (3, 15–17). Moreover, the Ca2+ binding attributes of either domain are affected by Ca2+ binding to the other domain as well as by the binding of CaM to protein targets (8, 17–20). In addition, protein complexes regulated by CaM generally contain more than one region for their interaction with the two CaM domains (3, 21, 22). For example, the binding of the CaM C-domain to the RyR2 CaM-binding domain (CaMBD) (Arg-3581–Pro-3607, human RyR2) is a prerequisite for CaM-dependent RyR2 inhibition. The RyR2 CaMBD also interacts with the CaM N-domain (Fig. 1), although less is known about this interaction (3, 8, 23–26). The pivotal role of this CaMBD in CaM-dependent RyR2 inhibition has been demonstrated unequivocally, but some studies suggest that other putative CaMBD in RyR2 may be involved as well (8, 26–29).
FIGURE 1.
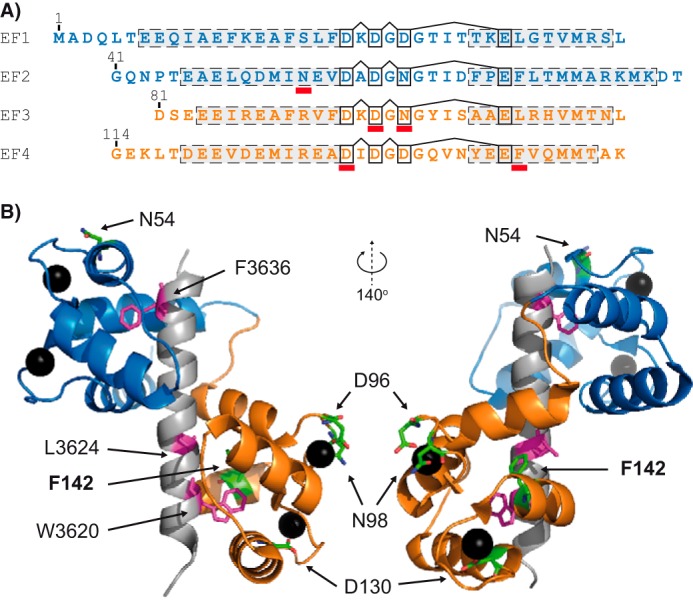
Overview of CaM and selected CaM mutations. A, the primary structure of CaM (N-domain, Met-1–Thr-80; C-domain, Asp-81–Lys-149) with EF-hands 1–4 aligned. Dashed boxes indicate α-helices and connected black boxes the Ca2+ coordinating residues. Red bars highlight individual sites for arrhythmogenic mutations (N54I, D96V, N98S, D130G, and F142L). B, representative tertiary structure of CaM. Ca2+-saturated CaM binding to a 27-residue peptide corresponding to part of the CaMBD in RyR1 (Lys-3614–Pro-3640) (Protein Data Bank code 2BCX) is shown. Protein and peptide secondary structures are represented schematically. The N-domain of CaM is indicated in blue, the C-domain in orange, and the peptide in gray. Sites of arrhythmogenic CaM mutations are highlighted as green stick representations (non-mutated residues) and Ca2+ ions as black spheres. RyR1 CaMBD residues (Trp-3620, Leu-3624, and Phe-3636) corresponding to RyR2 CaMBD residues Trp-3587, Leu-3591, and Phe-3603 are highlighted as magenta stick representations.
Recently, we showed that both the CPVT-causing CaM-N54I and the CPVT- and LQTS-causing CaM-N98S, as well as the LQTS-causing CaM-D96V and -D130G mutations markedly reduce inhibition of RyR2 Ca2+ release during store overload-induced Ca2+ release (SOICR) (8). The CaM-D96V, -N98S, and -D130G mutations directly affect Ca2+-coordinating residues in the C-domain. Thus, the diminished ability of these mutations to regulate RyR2 function is potentially explained by the reduced C-domain Ca2+ binding. Unlike the CaM-D96V, -N98S, and -D130G mutations, CaM-F142L does not affect a Ca2+-coordinating residue but still reduces CaM C-domain Ca2+ binding (2). In the X-ray structure of CaM complexed with the RyR1 CaMBD, the Phe-142 residue directly contributes to the CaM-CaMBD binding interface in contrast to the CaM Asn-54, Asn-98, Asp-96, and Asp-130 residues. We used a combination of functional, biophysical, and structural assays to investigate in detail the action of the LQTS-causing CaM-F142L mutation on RyR2 regulation. Unexpectedly, we found that the F142L mutation caused only a minor reduction in RyR2 inhibition by CaM (compared with CaM-WT), despite the markedly reduced CaM-F142L C-domain Ca2+ binding. Even more surprisingly, the F142L mutation enhanced the inhibitory action of CaM in RyR2 single channel experiments (i.e. displayed a CaM gain-of-function (GoF) effect). These actions are unique to the CaM-F142L mutation as compared with the CaM-N54I, -D96V, -N98S, and -D130G mutations. Discovery of this unique GoF property, conferred by the CaM-F142L mutation, may potentially serve as a molecular guide for how to manipulate CaM-dependent RyR2 inhibition as a therapeutic strategy for treating arrhythmias and/or heart failure.
Results
The CaM-F142L Mutation Slightly Decreases the Termination Threshold for Store Overload-induced Ca2+ Release
To test whether the CaM-F142L mutation affects the regulation of RyR2 during SOICR, we transfected RyR2-expressing HEK293 cells with CaM-WT or -F142L and then monitored the endoplasmic reticulum (ER) Ca2+ concentration using the D1ER Ca2+ probe (27). Perfusion of the transfected cells with increasing extracellular Ca2+ concentrations induced SOICR in the form of spontaneous ER Ca2+ oscillations (Fig. 2) (27, 30). The oscillating D1ER signal was then used to determine the ER Ca2+ level at which SOICR occurred (activation threshold) and the ER Ca2+ depletion at which SOICR ended (termination threshold; Fig. 2A and “Experimental Procedures”). The difference between the activation and termination thresholds was specified as the fractional ER Ca2+ release. Fig. 2, B and D, shows that expression of the CaM-F142L mutant decreased the termination threshold by 5% compared with the control with only endogenous CaM-WT (F142L 55% versus control 60%, p < 0.001). This in turn increased the fractional ER Ca2+ release during Ca2+ release oscillations by 6% (Fig. 2E, F142L 38% versus control 32%, p < 0.001). Note that the percentages listed here refer to the unit for ER Ca2+ load and not the relative effects of the CaM variants. On the other hand, expression of CaM-WT increased the termination threshold by 4% (WT 64% versus control 60%, p < 0.01) and minutely reduced the fractional ER Ca2+ release (WT 30% versus control 32%, p ≈ 0.1), although the latter was not statistically significant (Fig. 2, A and E). Neither CaM-WT nor CaM-F142L expression affected the activation threshold (Fig. 2, A–C). The control and CaM-WT results here are highly consistent with those reported previously (27). Taken together, CaM-F142L slightly reduced the SOICR termination threshold or, in other words, had a slightly less inhibitory action on RyR2-mediated Ca2+ release compared with CaM-WT. For comparison, we previously reported equivalent experiments showing that CPVT-causing (N54I), CPVT- and LQTS-causing (N98S), and LQTS-causing CaM mutations (D96V and D130G) all dramatically alter the Ca2+ release termination threshold (8). Specifically, the CaM-N54I, -D96V, -N98S, and -D130G mutations reduced the termination threshold by 12, 17, 18, and 16%, respectively. These variants also minimally, but significantly, reduced the activation threshold (∼5%) (8). Thus, the action of the CaM-F142L mutant on RyR2 function appeared very different from that of CaM-N54I, -D96V, -N98S, and -D130G.
FIGURE 2.
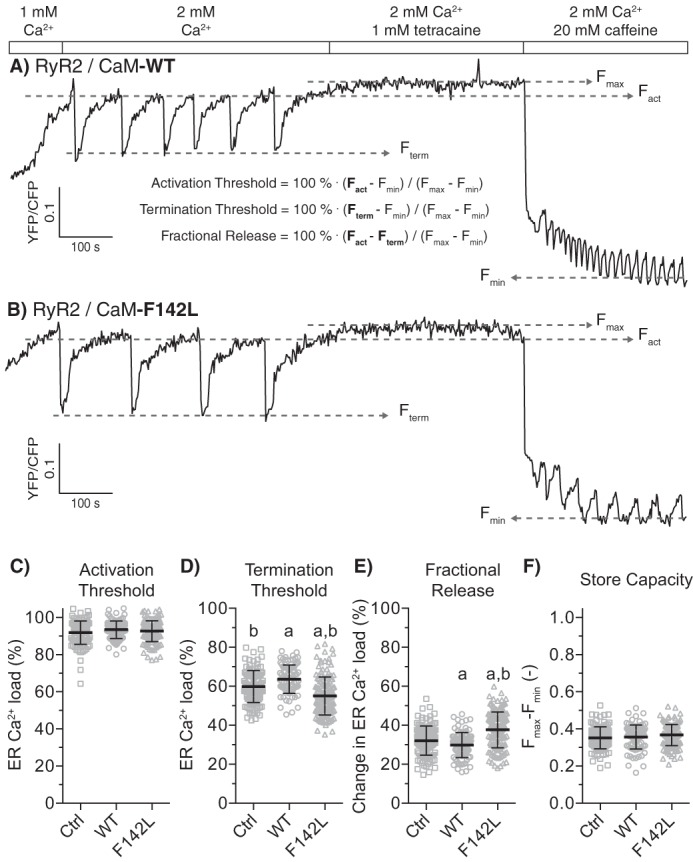
CaM F142L minutely affects regulation of RyR2 Ca2+ release during SOICR. A and B, example traces for the D1ER signal during SOICR from RyR2 expressing HEK293 cells co-transfected with CaM-WT (A) or CaM-F142L (B). The FRET signal from the ER luminal D1ER [Ca2+]free indicator oscillated as Ca2+ was released through RyR2. The rectangular bars are labeled to indicate the concentrations of Ca2+, tetracaine (RyR2 channel inhibitor), and caffeine (activator) in the perfusion solution. The increase in the ER Ca2+ concentration elicited RyR2 SOICR oscillations; the tetracaine blocked Ca2+ release, filling ER to the maximum [Ca2+]free; and finally caffeine depleted ER Ca2+ to the minimum [Ca2+]free (Fmax and Fmin). C–F, the activation (C) and termination (D) thresholds and the fractional Ca2+ release (E) relative to Fmax and Fmin; their difference was the fractional ER Ca2+ release (F). The expression of CaM-WT or -F142L affected the measured termination thresholds compared with endogenous CaM (Ctrl). Error bars show S.D., and values were calculated from 90–140 single cell traces. The a and b indicate values significantly different from those for the Ctrl or CaM-WT (one-way ANOVA, p < 0.05), respectively.
The CaM-F142L Mutation Enhances Inhibition of Single RyR2 Channels
Next, we tested the action of the CaM mutations on single RyR2 channels incorporated into lipid bilayers. Luminal [Ca2+]free was kept at 1 mm, and the cytosolic [Ca2+]free was set at 10 μm. The cytosolic solution also contained 1 mm [Mg2+]free and 5 mm ATP to approximate the levels present in cells. The 10 μm cytosolic [Ca2+]free approximates the level that RyR2 encounter during systole (see “Experimental Procedures”). Single RyR2 channel function was measured before and after the addition of 1 μm CaM-WT, -F142L, -N54I, -D96V, -N98S, or -D130G to the cytosolic solution (Fig. 3). Control (Ctrl) recordings in the absence of CaM were also included. The addition of CaM-WT significantly lowered the RyR2 open probability (PO) from 0.47 to 0.35 compared with control, consistent with CaM-WT inhibition of RyR2-mediated Ca2+ release (Fig. 3D). CaM-WT reduced the mean open time (MOT) and increased the mean closed time (MCT), although these changes individually were not statistically significant (p = 0.18 and 0.05 against Ctrl) (Fig. 3, E and F). In contrast, the C-domain mutations, CaM-D96V, -N98S, and -D130G, did not lower RyR2 PO compared with the control (PO 0.54, 0.46, and 0.52, respectively). The CaM-D96V, -N98S, and -D130G mutations all significantly decreased the RyR2 MCT compared with both CaM-WT and the control. The CaM-N98S mutation significantly decreased MOT compared with the control. These results suggest that: 1) these CaM mutations do interact with RyR2 as also shown in previous studies (7, 8, 10); and 2) CaM mutations D96V, N98S, and D130G, in contrast to CaM-WT, do not appear to inhibit RyR2 function (but this is due to almost equal decreases in both MOT and MCT compared with no CaM present). In contrast, the CaM N-domain N54I mutation did not significantly affect RyR2 function (PO, MCT or MOT) under these experimental conditions, suggesting that aberrant RyR2 regulation by CaM-N54I is mechanistically distinct from that caused by the CaM C-domain mutations (8, 17).
FIGURE 3.

Effect of CaM-WT and mutants on single RyR2 channel properties. A–C, example of single RyR2 channel current traces without CaM (left traces) and after the addition of 1 μm CaM. D–F, RyR2 channel PO, MOT, and MCT after the addition of CaM mutants (Ctrl with no added CaM). Measurements were done at +40 mV with 1 mm free Ca2+ at the luminal face and 10 μm free Ca2+, 1 mm free Mg2+, and 1 mm ATP at the cytosolic face. The a indicates values significantly different from CaM-WT, and b indicates values significantly different from Ctrl (t test, p < 0.05). Error bars show S.E. from 6–8 channels for each CaM variant and 32 channels before or without CaM addition (Ctrl).
Strikingly, the C-domain CaM-F142L mutation had an opposite action on single RyR2 channel function. CaM-F142L significantly lowered the RyR2 PO, even more so than CaM-WT (PO 0.18 versus 0.35) (Fig. 3D). The CaM-F142L mutation caused a significant MOT decrease compared with both CaM-WT and the control, yet appeared to have approximately the same effect on MCT as CaM-WT (22 versus 24 ms). Hence, the CaM-F142L mutation resulted in a stronger RyR2 inhibitory action (compared with that of CaM-WT). In other words, the CaM F142L mutation generates a GoF defect with regard to RyR2 inhibition, which is in stark contrast to CaM-D96V, -N98S, and -D130G, none of which inhibited RyR2 function. Hence, CaM-WT, CaM-F142L, and the other C-domain mutations (D96V, N98S, and D130G), as well as the CaM N-domain N54I mutation tested here, have distinctive actions on RyR2 modulation.
The CaM-F142L Mutation Reduces Ca2+ Binding Affinity in the Presence of the RyR2 CaMBD Peptide
The ability of CaM to inhibit RyR2 Ca2+ release depends both on Ca2+ binding to CaM and CaM binding to the RyR2 CaMBD (2, 29, 31). The CaM-F142L mutation clearly impairs Ca2+ binding to the CaM C-domain in the absence of a CaM binding target (2, 29, 31). Given the limited action of the CaM-F142L mutant on SOICR in HEK293 cells and its GoF action at the single RyR2 channel level, we assessed whether the CaM-F142L mutation differentially affects CaM Ca2+ affinity when the CaM is bound to the RyR2 CaMBD. The binding of Ca2+ to CaM in the presence of the CaMBD (i.e. the RyR2(R3581-L3611) peptide) was determined using a Ca2+ titration while monitoring the intrinsic protein fluorescence specific for each CaM domain (Phe for the N-domain and Tyr for the C-domain) (Fig. 4). Fitting the apparent dissociation constant (appKD) for either CaM domain using a two-site Adair model revealed that the CaM-F142L mutation lowered the affinity of the C-domain for binding Ca2+ in the presence of the CaMBD more than 10-fold compared with CaM-WT (appKD 0.32 versus 0.03 μm). The CaM-F142L mutation did not significantly affect N-domain Ca2+ binding, albeit there was a tendency toward increased affinity (appKD 0.61 versus 0.78 μm). Moreover, the CaM-F142L C-domain Ca2+ binding affinity in the presence of the RyR2 CaMBD appeared ∼2-fold greater than that observed for the CaM-N98S and -D96V mutations (appKD 0.15 and 0.14 μm, respectively). Note that in the absence of a CaM target, the CaM-F142L mutant C-domain Ca2+ binding has an affinity intermediate of these other mutations (appKD 10, 15, and 31 μm for CaM-N98S, -F142L, and -D96V, respectively) (2, 8). Thus, the CaM-F142L mutation causes a loss of function (LoF) in terms of Ca2+ binding to the CaM C-domain when complexed with its RyR2 target, and this LoF is at least as severe as for CaM-D96V or -N98S.
FIGURE 4.
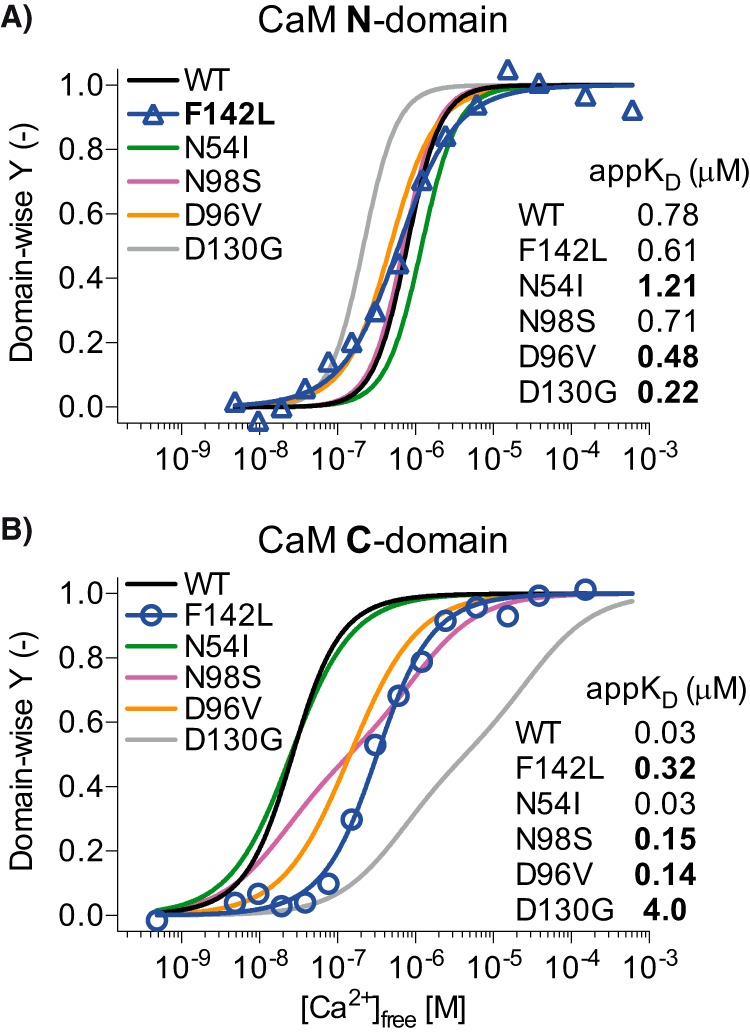
Domain-specific titration curves for Ca2+ binding to CaM in the presence of RyR2(R3581-L3611) peptide. CaM-F142L/RyR2(R3581-L3611) Ca2+ titration points for CaM N-domain (A, triangles) and C-domain (B, circles) were fitted with a two-site Adair model (solid blue lines), and normalized Y (see “Experimental Procedures”) was plotted as a function of [Ca2+]free. The solid lines in either panel show the fit to the equivalent titrations of CaM-WT (black), -N54I (green), -N98S (magenta), -D96V (tangerine), and -D130G (gray) with data from Søndergaard et al. (8). Bold, highlighted appKD values were significantly different from those for CaM-WT/RyR2(R3581-L3611) (one-way ANOVA p < 0.05).
Thermodynamically Distinct Interactions of CaM-F142L and CaM-WT with the RyR2 CaMBD
To further probe the interaction between the CaM-F142L mutant and the RyR2 CaMBD, we followed the titration of CaMBD (i.e. the RyR2(R3581-P3607) peptide) with CaM variants under Ca2+-free (apoCaM) and saturating Ca2+ conditions (CaCaM) using ITC (Fig. 5). This measurement compares the differences in the CaMBD interaction aside from those caused by differences in CaM Ca2+ binding affinity. The binding of apo- or CaCaM-WT to the CaMBD peptide represent two thermodynamically distinct reactions. The apoCaM-WT/CaMBD interaction is comparatively low affinity (μm KD) and is entropy-driven (ΔH° > 0, −T·ΔS° < 0). The CaCaM-WT/CaMBD interaction is high affinity (nm KD) and is enthalpy-driven (ΔH° ≪ 0, −T·ΔS° > 0) (Figs. 5–7) (26).
FIGURE 5.

ITC example data for the titration of the RyR2(R3581-P3607) peptide with CaM. Example ITC thermograms for apoCaM-WT and -F142L (A and B) and for CaCaM conditions (D and E). C and F, averaged integrated change in enthalpy per injection (ΔH°) comparing CaM-WT, -D96V, -N98S, and -F142L under apo (C) or CaCaM conditions (F). Without Ca2+ present, the binding of apoCaM-WT to the peptide is an endothermic reaction (positive ΔH°), whereas for the apoCaM-F142L the interaction is exothermic (A and B). With saturating Ca2+ present, both CaCaM-WT and -F142L display exothermic binding reactions (D and E). Thermograms for CaM-D96V and -N98S were not visibly different from those for the CaM-WT and therefore are not shown. DP, change in ITC instrument heat effect.
FIGURE 6.

Fitted thermodynamic parameters for the binding of CaM variants to the RyR2(R3581-P3607) peptide as calculated from ITC. Panels in the upper row summarize values for the apoCaM condition, and those in the lower row summarize values for CaCaM. A, the affinity of CaM for binding to the peptide expressed as the dissociation constant (KD). B and C, enthalpy (ΔH°) and entropy contribution (−T·ΔS°), respectively, for the binding interaction between CaM and the peptide. D, the change in Gibb's free energy (ΔG°) as conferred by the CaM mutations. Error bars show S.D. The a and b indicate values significantly different from those for CaM-WT or CaM-F142L, respectively (one-way ANOVA, p < 0.05).
FIGURE 7.
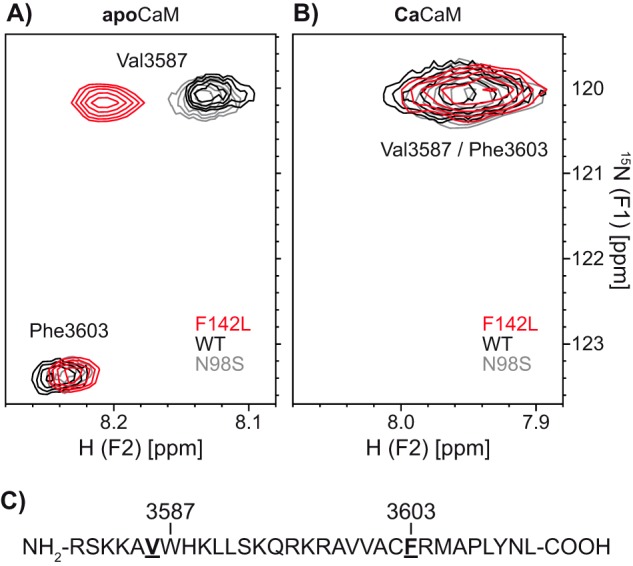
2D [15N,1H]HSQC NMR spectra of CaM in complex with the RyR2(R3581-L3611) peptide. 15N-hydrogen HSQC NMR spectra of the RyR2(R3581-L3611) peptide with 15N-labeled Val-3586 and Phe-3603 in complex with CaM-WT, -N98S, or -F142L. The labeled residues correspond to Phe-3636 and the Val adjacent to Trp-3620 highlighted in Fig. 1. Spectra were recorded without any Ca2+ (A) and with saturating Ca2+ (B). The peaks are color-coded according to the CaM variant in the protein-peptide complex. C, amino acid sequence of the RyR2(R3581-L3611) peptide with 15N-labeled residues highlighted in bold and underlined. The CaM C-domain binds around Trp-3587 and the N-domain around Phe-3603.
The ITC measurements showed a fundamental difference in the thermodynamics of apoCaM-F142L binding to the CaMBD compared with the apoCaM-WT (Fig. 5, A–C). The binding reaction between apoCaM-F142L and the CaMBD peptide was exothermic (ΔH° < 0) in marked contrast to the endothermic (ΔH° > 0) interaction between apoCaM-WT and the CaMBD. Moreover, titration curve analysis showed that apoCaM-F142L binding affinity for the CaMBD was ∼3-fold greater compared with that of apoCaM-WT (KD 9 versus 28 μm) (Fig. 6A). ApoCaM-F142L binding also displayed a small negative ΔH° (−1.8 kJ/mol)) compared with the larger, positive ΔH° (11.8 kJ/mol) for apoCaM-WT binding (Fig. 6B). A comparison of the fitted ΔH° values to the −T·ΔS° values (−38 and −27 kJ/mol for apoCaM-WT and -F142L) indicated that the binding of both apoCaM-WT and -F142L to the CaMBD peptide remained governed by entropy (−T·ΔS°) relative to enthalpy (ΔH°) (Fig. 6C). The change in ΔH° conferred by the CaM-F142L mutation translated into a significant 11% decrease (−29 versus −26 kJ/mol) in ΔG° for the apoCaM-F142L interaction with the CaMDB peptide (Fig. 6D). Generally, an enthalpy-driven reaction is indicative of specific molecular bonding, whereas an entropy-driven reaction indicates hydrophobic interactions and solvent effects (32). Thus, these ITC results indicated that the CaM-F142L mutation transformed the apoCaM interaction from mainly entropy-driven to one more dominated by molecular bonds. Under saturating Ca2+ conditions, the interaction between CaCaM-F142L and the CaMBD peptide was indistinguishable from that for the CaCaM-WT (Figs. 5, C and D, and 6). Interestingly, the increase in affinity comparing Ca2+-free to saturating Ca2+ conditions was still 3 orders of magnitude (KD 8 μm versus 14 nm) for CaM-F142L, attributable to an increased ΔH° contribution under the CaCaM condition. Thus, the thermodynamic difference between apo and CaCaM binding to the CaMBD peptide remained similar for CaM-WT and -F142L.
The interactions of CaM-D96V and -N98S with the RyR2 CaMBD peptide were also investigated using ITC, and for both Ca2+ conditions neither of the thermograms (not shown) was visibly different from those for the CaM-WT. However, detailed titration curve analysis indicated a slightly decreased affinity of CaM-D96V for CaMBD binding with and without Ca2+ present (Figs. 5, C and F, and 6A). Small changes to ΔH° were also detected for apoCaM-D96V and -N98S and for CaCaM-D96V and -N98S (Fig. 6B). Interestingly, the minute effects observed for CaM-D96V and -N98S were all significantly distinct from those observed for CaM-F142L under both Ca2+ conditions. Thus, CaM-F142L generally showed CaMBD binding properties different from not only the CaM-WT but also from the other arrhythmogenic CaM mutations, D96V and N98S.
ApoCaM-WT and -F142L Binding to RyR2 CaMBD Peptide Are Structurally Distinct Interactions
The apoCaM C-domain binds to RyR2 CaMBD around Trp-3587 and Leu-3591. Upon CaM C-domain Ca2+ binding, this interaction shifts toward Trp-3587 (1, 8, 23, 25, 26, 33). The CaCaM N-domain binds to the RyR2 CaMBD peptide around Phe-3603, but it is unclear whether this interaction occurs for the apoCaM N-domain or is dependent on N-domain Ca2+ binding (3, 8, 27).
Motivated by the results from the ITC experiment, we used 2D NMR (15N-HSQC) to measure the effect of Ca2+ on the chemical shifts from a CaMBD peptide with 15N-labeled Val-3586 and Phe-3603 in a complex with CaM-WT, -N98S, or -F142L. The chemical shifts of these labeled residues depend on their immediate structural surroundings (i.e. the binding of the CaM C- and N-domain, respectively). The peaks corresponding to Val-3586 and Phe-3603 were easily discernable under apoCaM conditions (Fig. 7A). Unfortunately, their chemical shifts overlapped under saturating Ca2+ conditions (Fig. 7B). Nonetheless, a clear difference under apoCaM conditions was observed for CaM-CaMBD complexes containing CaM-F142L compared with CaM-WT or -N98S. ApoCaM-F142L displayed a higher chemical shift for the Val-3586 HN but showed no differences for the Phe-3603 HN or N. The latter observation may reflect that the N-domains of apoCaM-F142L and -WT do not bind to the CaMBD peptide. Under saturating Ca2+ conditions, no differences in the spectra were observed, albeit the addition of Ca2+ clearly affected the structural surroundings of the labeled residues (i.e. chemical shifts for both Val-3586 and Phe-3603 changed markedly). Opposite CaM-F142L, the spectra recorded using CaM-N98S were identical to those for CaM-WT, with and without Ca2+ (Fig. 7, A and B). Taken together, these results support the notion that the apoCaM-F142L C-domain binds to the RyR2 CaMBD peptide close to the Val-3586 residue (next to Trp-3587) in a unique conformation structurally distinct from that of the apoCaM-WT and -N98S. Further, Ca2+ binding to CaM-F142L changes this conformation into one that is indistinguishable from that of the CaCaM-WT and -N98S.
Discussion
CaM is a constitutive Ca2+ sensor of the RyR2 macromolecular complex, where it inhibits RyR2 Ca2+ release in a [Ca2+]cyt dependent, allosteric manner (3, 13, 34). This inhibition is critical for maintaining a low RyR2 activity at diastole (i.e. at low [Ca2+]cyt) and also for a sufficient termination of the Ca2+ release during cardiac excitation (i.e. as [Ca2+]cyt increases) (8, 23, 27–29, 33, 35, 36). The details of how the two Ca2+-sensing domains of CaM interact with Ca2+ and RyR2 to facilitate this complex inhibition are not well understood (3, 13, 26, 34). However, the interactions between the RyR2 CaMBD, the CaM C-domain and Ca2+ are critical for the physiological regulation of RyR2 Ca2+ release (8, 23, 24, 27–29, 36). Binding of the apoCaM C-domain to the CaMBD inhibits RyR2 Ca2+ release, whereas binding of Ca2+ to the CaM C-domain further increases this CaM-dependent inhibition (8, 23, 27–29, 35, 36). Based on the biophysical results for the tripartite interaction (CaM, RyR2 CaMBD, and Ca2+) and the effects of arrhythmogenic CaM mutations, we proposed previously that the CaM C-domain at diastolic [Ca2+]cyt binds to RyR2 CaMBD in a near Ca2+-saturated state (1, 8, 27). In this scheme, the reduced affinity for Ca2+ binding of the arrhythmogenic CaM C-domain mutations (D96V, N98S, and D130G) causes the CaM C-domain to be less Ca2+-saturated and thereby reduces the CaM-dependent inhibition of RyR2 (8).
The LQTS-causing CaM-F142L mutation clearly reduces the C-domain Ca2+ binding affinity (appKD 15 μm), compared with CaM-WT (appKD 2.5 μm). This is similar to what was observed for the CaM-N98S and -D96V mutants (appKD 10 and 31 μm), albeit less than for CaM-D130G (appKD 84 μm) (2, 17). Here, we found that this reduced affinity for the CaM-F142L C-domain Ca2+ binding was retained in the presence of the RyR2 CaMBD peptide, even to the extent that CaM-F142L displayed a lower affinity than CaM-D96V (appKD 0.32 versus 0.14 μm) (Fig. 4). Thus, we expected that in HEK293 cells where [Ca2+]cyt oscillates between ∼0.1 and 2 μm, CaM-F142L would abnormally regulate RyR2 function (compared with CaM-WT) to a similar extent as CaM-N98S and -D96V (Fig. 2) (37, 38). Curiously, the action of CaM-F142L on RyR2 function in the HEK293 SOICR assay was relatively benign and distinctly different from the actions of the CaM-N54I, -D96V, -N98S, and -D130G mutants (8). Moreover, these other CaM mutants were expressed using low expressing plasmids, resulting in a CaM ratio of ∼1.4 relative to endogenous CaM (8). The expression plasmid used in this study, and also in Tian et al. (27), results in a CaM ratio of ∼4 relative to endogenous CaM as judged from Western blotting analysis (Fig. 8). This further supports that the decrease in RyR2 inhibition caused by CaM-F142L was strikingly less than the decreases caused by CaM-N54I, -D96V, -N98S, and -D130G. Lastly, we observed no difference between CaM-F142L and -WT when the low expressing plasmid was used (data not shown). Consistent with these results, Hwang et al. (7) report similar spontaneous Ca2+ wave frequencies in permeabilized cardiomyocytes ([Ca2+]free at 0.12 μm) with CaM-WT or CaM-F142L present. Also, Vassilakopoulou et al. (10) report that CaM-F142L inhibits binding of [3H]ryanodine to porcine cardiac SR vesicles to the same extent as the CaM-WT. Taken together, these results indicate that the F142L mutation causes little or no loss of CaM-dependent RyR2 inhibition, despite a pronounced LoF in terms of CaM C-domain Ca2+ binding. Even more strikingly though, we showed that at 10 μm cytosolic [Ca2+]free, CaM-F142L is a more potent inhibitor of single RyR2 channels than CaM-WT. Specifically, CaM-F142L promoted a much faster RyR2 closing (Fig. 3). In other words, CaM-F142L displayed a GoF action in that it increased the CaM-dependent RyR2 inhibition.
FIGURE 8.
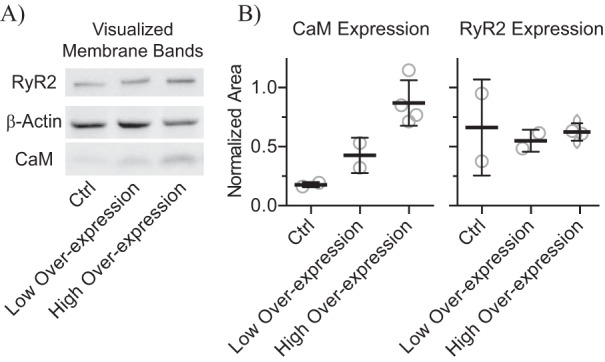
Estimation of CaM and RyR2 expression levels in HEK293 cells. A, example of Western-blotted protein bands visualized using chemiluminescence imaging. HEK293 cells were cultured with CaM-WT overexpression from a low or high expression plasmid and also without overexpression (Ctrl). B, the expression levels of CaM and RyR2 were normalized to that of β-actin. Averaged CaM expression levels were 0.18, 0.43, and 0.87 for endogenous CaM expression (Ctrl), and high and low plasmid expression, respectively. Opposite from CaM, the stable and inducible RyR2 expression levels appeared constant across all samples.
How can the F142L mutation confer both a LoF in terms of Ca2+ binding with seemingly little consequence to the RyR2 inhibitory action in cells and a robust RyR2 inhibitory action at the single channel level? Our ITC experiments hint at a potential explanation. ApoCaM-F142L showed increased affinity for binding the RyR2 CaMBD peptide and bound in a manner thermodynamically in between that of the apoCaM-WT and CaCaM-WT interactions (Figs. 5 and 6). The NMR HSQC spectra also support this view demonstrating that the apoCaM-F142L C-domain bound around Val-3586 in the CaMBD in a conformation that was structurally distinct from that of the apoCaM-WT. Based on our functional, biophysical, and structural results, we propose that the F142L mutation has two opposing actions on RyR2 regulation.
First, the F142L mutation enhances the CaM C-domain interactions with the RyR2 CaMBD, thus increasing RyR2 inhibition (GoF). Second, the CaM-F142L C-domain has impaired Ca2+ binding (LoF), thus decreasing RyR2 inhibition. Under the comparatively low [Ca2+]cyt conditions in HEK293 cells or the permeabilized cardiomyocytes (7), the effect of reduced CaM-F142L C-domain Ca2+ binding would be partially offset by the enhanced CaM binding to the RyR2 CaMBD, explaining why little aberrant RyR2 regulation was observed. However, increasing Ca2+ saturates the CaM-F142L C-domain (appKD: 0.32 μm), in effect ablating any LoF action from the reduced C-domain Ca2+ affinity. Accordingly, at high Ca2+ concentrations only the GoF action would remain and may explain why CaM-F142L was a more potent inhibitor than the CaM-WT in our single RyR2 channel studies. The molecular basis for this GoF effect was not clear from these experiments, as no differences between CaCaM-WT and CaCaM-F142L binding to the RyR2 CaMBD peptide were detected. This implies perhaps that interactions between CaM and RyR2 not recapitulated in our biophysical experiments, e.g. RyR2 regions outside the CaMBD studied here, are responsible for the GoF action. Structurally delineating this GoF action may provide a molecular guide for how to manipulate the CaM-dependent RyR2 inhibition. Also, increasing this inhibition reduces SR Ca2+ release and/or leak and thus represents a therapeutic approach for treating heart failure and arrhythmia (28, 31, 39–41).
Aside from the novel insights into the effects of CaM-F142L on RyR2 regulation, our single RyR2 channel experiments support our previous finding that both CPVT- and LQTS-causing CaM mutations (N98S, D96V, and D130G) result in aberrant RyR2 regulation (8). Using different experimental conditions (sheep cardiomyocytes, 0.1 mm luminal [Ca2+]free, 2 mm ATP cytosolic, and no Mg2+), Hwang et al. (7) report that CaM-N54I and -N98S increased RyR2 PO at 0.1 and 1 μm cytosolic [Ca2+]free, whereas CaM-D96V did not. The apparent CaM-D96V discrepancy between our study and the Hwang et al. study (7) may be explained by differences in the experimental conditions. Our study shows that CaM mutations N54I, D96V, N98S, D130G, and F142L alter the RyR2 CaM regulation via distinct molecular mechanisms. The CaM-D96V, -N98S, and -D130G mutations diminish C-domain Ca2+ binding and thereby the inhibitory interaction of CaM with the RyR2 CaMBD. The N-domain CaM-N54I mutation likely affects CaM-RyR2 interactions that are outside the canonical CaMBD and/or increases Ca2+ binding kinetics (17). The CaM-F142L mutation diminishes C-domain Ca2+ binding but also enhances CaM interaction with the RyR2 CaMBD and possibly other regions (7, 8, 10, 17). Because LQTS-causing CaM-D96V, -N98S, and -D130G mutations diminish CaM-dependent RyR2 inhibition, increased RyR2 Ca2+ release may contribute to the LQTS phenotypes in individuals with these CaM mutations. Generally, it is thought that spontaneous diastolic RyR2 Ca2+ release causes CPVT, and augmented Ca2+ influx through CaV1.2 channels causes LQTS (see Refs. 42 and 43 for details). Studies using recombinant CaV1.2 and patch clamping show that Ca2+-dependent inactivation of CaV1.2 is reduced when CaM-D96V, -N98S, -D130G and -F142L are present (compared with CaM-WT) (6, 9).
Thus, some mechanistic overlap between CPVT and LQTS caused by CaM mutations likely exist, and how arrhythmogenic CaM mutations manifest probably depends on their relative effects on RyR2, CaV1.2, and other CaM-regulated targets. The best example of this is CaM-N98S, which affects both RyR2-mediated Ca2+ release and CaV1.2 Ca2+-dependent inactivation, likely contributing to both CPVT and LQTS. Another scenario is represented by the strictly CPVT-causing CaM-N54I mutation, which does not affect CaV1.2 Ca2+-dependent inactivation (1, 4, 6, 44). In addition, some CaM C-domain mutations (N98S, D132E, and Q136P) are reported to cause a LQTS phenotype with some features of CPVT overlapping (4, 44). β-Blockers are the common drug treatment for both CPVT and LQTS (42, 43). Given the differential influence of CaM mutants on CaV1.2 and RyR2, drugs that preferentially alter RyR2 or CaV1.2 function might provide better treatments for CaM-mediated arrhythmias (42, 43, 45). Novel drugs affecting RyR2 and CaV1.2 are being sought (45–47), and as antiarrhythmic therapies advance, defining their differential action on CaV1.2 and RyR2 function will almost certainly become increasingly relevant.
Experimental Procedures
Plasmid Constructs
Plasmid constructs for the recombinant expression and purification of CaM (pMAL, New England Biolabs) or for overexpression of CaM in HEK293 cells (pcDNA3.1, Invitrogen) were prepared as described previously (8). Sanger sequencing verified the CaM-encoding inserts in all plasmids.
Endoplasmic Reticulum Luminal Ca2+ Imaging of HEK293 Cells Expressing RyR2
Stable expression of murine RyR2, or a RyR2 variant with the CaMBD deleted (murine ΔK3583-F3603), in HEK293 cells co-transfected with plasmids encoding CaM and the D1ER Ca2+ probe was done as described previously (27). Briefly, D1ER FRET signals reflecting ER luminal [Ca2+]free in individual cells were monitored by using an epifluorescent microscope (27, 48). Each FRET signal trace was used to measure the Ca2+ release properties of the RyR2 channels relative to the ER Ca2+ store capacity: the activation and termination thresholds and their difference taken as the fractional Ca2+ release. The ER Ca2+ store capacities were calculated from the difference between maximum and minimum FRET signal (Fmax − Fmin) (Fig. 2). The measured RyR2 Ca2+ release properties were compared using one-way ANOVA with Tukey's multiple comparisons test for all possible combinations, with adjusted p < 0.05 taken as significant. The experiments included a control without plasmid expression of CaM.
Estimation of CaM-WT and RyR2 Expression Levels in HEK293 Cells
HEK293 cells were cultured as described above with CaM-WT overexpression from a low (8) or high expressing plasmid (this study and Ref. 27) and without overexpression (Ctrl). Overexpression plasmids differed in their Kozak sequences upstream of the CALM1 cDNA inserts. 40 μg of total protein from cell lysates (protein assay, Bio-Rad) was subjected to SDS-PAGE (1 h at 20 A) in a gradient gel (Bio-Rad, Mini-Protean TGX 4–20%) alongside a molecular weight marker (Bio-Rad, catalog No. 161-0309), and the electrophoretic separated proteins were blotted (0.5 h at 100 V, ∼0.33 A) to a nitrocellulose membrane in Tris-glycine buffer (Bio-Rad) with 10‰ SDS. The membrane was transiently stained with Ponceau S and cut into three regions: RyR2 (∼500 kDa), β-actin (∼42 kDa), and CaM (∼16 kDa). The membrane pieces were blocked in PBS with 1% casein (Bio-Rad), washed in PBS, and then incubated overnight with different primary antibodies (Ab) against CaM (05-173, EMD Milipore), β-actin (A5316, Sigma), or RyR2 (MA3-925, Pierce) in PBS with 1‰ Ab, 20% fetal bovine serum (Sigma), and 17 mm NaN3. After another wash, the pieces were incubated for 0.5 h with a secondary Ab (in-house anti-mouse IgG conjugated to horseradish peroxidase) and then washed again. The amount of bound secondary Ab was detected using luminol reagent (detection reagent 1–2, Thermo Scientific) with the resulting chemiluminescence imaged (ImageQuant LAS 4000, GE Healthcare Life Sciences). As a measure of protein expression levels, the protein band area intensities were quantified in ImageJ, and the expression levels of RyR2 and CaM in the individual samples were normalized to that of β-actin (49) (Fig. 8, normalized area). Western blotting analysis was done in at least duplicate.
Bilayer Recordings of Single RyR2 Channels
Native SR microsomes isolated from rat ventricular muscle were incorporated into bilayers using a modification of the method described by Chamberlain et al. (47, 50, 51). Briefly, planar lipid bilayers (50 mg/ml in a 5:4:1 mixture of bovine brain phosphatidylethanolamine, -serine, and -choline in n-decane) were formed across a 100-μm-diameter hole in a Teflon partition separating two compartments with cytosolic (114 mm Tris-HEPES, 5 mm ATP, 1 mm free Mg2+, 1 mm EGTA, and 10 μm free Ca2+ at pH 7.4) and luminal (cytosolic solution plus 200 mm Cs-HEPES and 1 mm free Ca2+) recording solutions. Single RyR2 activity was measured before and 20 min after the addition of CaM variants (1 μm) to the cytosolic solution. Recordings were made at room temperature (20–22 °C) with currents sampled at 50 μs/point and filtered at 0.75 kHz (4-pole Bessel). Analysis was done using pCLAMP9 software (Molecular Devices, Sunnyvale, CA). Recapitulating the cytosolic and intra-SR cellular milieu in vitro during planar lipid bilayer studies is impossible. Consequently, experimental compromises were necessary, and here the solutions approximated those in cardiomyocytes during systole. Low cytosolic Ca2+ (0.1–1 μm) reduces RyR2 activity to a level unsuitable for reliable measurements. Some researchers have overcome this issue by omitting Mg2+, but this causes a very non-physiological RyR2 Ca2+ dependence, as in cells Mg2+ normally competes with Ca2+ for occupancy of RyR2 cytosolic Ca2+ activation and inactivation sites (51). Differences in single channel parameters (PO, MOT, and MCT) extracted from time traces were compared using two-tailed t tests against the values following the addition of CaM-WT with p < 0.05 considered significant. Also, comparisons with control (no addition of CaM) were done with the same p value criteria.
Protein Expression and Purification
CaM was expressed from the pMAL vectors and purified as described previously (8). The identity, purity, and integrity of each protein preparation was confirmed by SDS-PAGE and MALDI-TOF mass spectrometry of trypsin-digested proteins.
CaM Ca2+ Titrations in the Presence of the RyR2(R3581-L3611) Peptide
A peptide corresponding to the RyR2 CaMBD (human RyR2 3581RSKKAVWHKLLSKQRKRAVVACFRMAPLYNL3611) was purchased from Peptide 2.0 Inc. (Chantilly, VA) at > 98% purity. Titration experiments were done as described previously (8). Briefly; pH- and Ca 2+-buffered solutions (50 mm HEPES, 100 mm KCl, 0.5 mm EGTA, and 2 mm NTA at pH 7.2 (25 °C)) with or without 7 mm CaCl2 were mixed to obtain different [Ca 2+]free levels (52). CaM (15 μm), RyR2(R3581-L3611) peptide (16.5 μm), 16.5 μm TCEP, and Fura-2 (Invitrogen) Ca2+ probe (0.8 μm) were added to double distilled water for dilution of 1.5× concentrated buffers. A 15% error for the [Ca2+]free was included in data fitting procedures based on measuring [Ca2+]free using Fura-2 and Ca2+ binding to CaM-WT. The intrinsic protein fluorescence from each CaM domain was monitored during CaM/RyR2 CaMBD complex Ca2+ titrations. The titration curves were fitted to a two-site Adair function as described previously (8, 19, 53, 54). Briefly, fluorescence intensities (FI) from the N- and C-domains of CaM were measured as partial Phe and Tyr emission spectra, and the fractional saturations (Y) for each domain were fitted to the raw FI according to Equation 1,
| (Eq. 1) |
where b and a are the initial FI and the span in FI, respectively. Y is the fractional saturation of the monitored CaM domain binding two Ca2+ as described by the two-site Adair model,
| (Eq. 2) |
where K1 is the sum of the microscopic equilibrium constants, and K2 is the equilibrium constant for the domain binding to two Ca2+. The apparent dissociation constant (appKD) for either domain was then calculated as the reciprocal square root of K2. The fitted K2 values were compared using one-way ANOVA with Dunnett's multiple comparison test against the value for CaM-WT/RyR2(R3581-L3611) titrations.
Isothermal Titration Calorimetry of RyR2(R3581-P3607) Peptide with CaM
A peptide corresponding to the CaMBD (human RyR2 3581RSKKAVWHKLLSKQRKRAVVACFRMAP3607) was purchased from GenScript (Piscataway, NJ) at >95% purity. Titration of the RyR2(R3581-L3607) peptide with CaM was investigated under both Ca2+-free (apo) and Ca2+-saturating conditions and followed the use of ITC. Purified CaM variants were dialyzed (10 K Slide-a-lyzerTM, Thermo Scientific) against the titration buffer (10 mm HEPES, 150 mm KCl, pH 7.2) with either 10 mm EDTA or 10 mm CaCl2 added for 40 h at 4 °C (with buffer changed at 24 h). The spent dialysis buffer was filtered (0.22 μm) and used for diluting peptide and CaM preparations. 5 mm final TCEP was added to all solutions. Protein and peptide concentrations were estimated using absorption at 280 nm (NanoDrop ND-1000). Using an Auto-ITC200 (Malvern Instruments Ltd.) isothermal titration calorimeter, the peptide (apo 0.1 mm, Ca2+ 10 μm) in the sample cell was titrated 20 times at 25 °C with CaM (apo 1 mm, Ca2+ 100 μm) in 2-μl increments at 2-min intervals and with 750 rpm stirring (c values: apo 2–10 and Ca2+ 550–850). Two to three titrations were done for each combination of CaM variant and Ca2+ condition. ITC curves (change in instrument heat effect as a function of time, DP) were analyzed using MicroCal PEAQ-ITC software, and the extracted curves of the cumulative reaction heat as a function of total CaM concentration were fitted to a two-component binding equation (32). The reaction stoichiometry (n), standard enthalpy change (ΔH°), and dissociation constant (KD) were estimated from this fitting, and the parameters were compared using one-way ANOVA and Tukey's post hoc test for all possible comparisons. The binding reaction change in standard Gibb's free energy (ΔG°) and entropy contribution (−T·ΔS°) were calculated from Equation 3.
| (Eq. 3) |
NMR Spectra of 15N-Labeled RyR2(R3581-L3611) Peptide in Complex with CaM
A version of the RyR2 CaMBD peptide, 15N-labeled at Val-3586 and Phe-3603 (15N-RyR2(R3581-L3611)), was purchased from ProteoGenix (Schiltigheim, France) at >98% purity. The complex of this peptide with CaM-WT, -F142L, or -N98S was investigated under both Ca2+-free (apo) and Ca2+-saturating conditions using NMR. All four samples contained 20 mm HEPES, 100 mm KCl, 2 mm TCEP, 1 mm PMSF, 5% D2O, 40 μm TSP-d4, 100 μm 15N-RyR2(R3581-L3611), and 200 μm CaM-WT or -F142L along with either 10 mm CaCl2 or 10 mm EDTA. Protein and peptide concentrations were estimated using absorption at 280 nm (NanoDrop ND-1000) and quantitative NMR. The pH of the samples was adjusted to 6.50 ± 0.03 with either 1 m KOH or 1 m HCl, and the final sample volume was 550 μl. 15N-HSQC spectra were recorded on a 600-MHz Bruker AVIII at 298.1 K. TopSpin 3.2. was used for the acquisition and processing of the spectra. To assign the peaks in the 15N-HSQC, a 2D 15N-TOCSY-HSQC with a DIPSI2 mixing of 60 ms and a γB1/2π of 9.6 kHz was performed.
Author Contributions
M. T. S., R. W., F. V. P., M. F., S. R. W. C., and M. T. O. designed the research. M. T. S., Y. L., K. T. L., A. N., X. T., C. H., and R. W. performed the experiments. M. T. S., R. W., K. T. L., C. H., M. F., and M. T. O. analyzed the data. M. T. S., M. F., S. R. W. C., and M. T. O. wrote the paper.
This work was supported by research grants from the Danish Council for Independent Research (DFF-4181-00447), the Obelske Family Foundation, the Novo Nordic Foundation (NNF15OC001776299), and the Lundbeck Foundation (2013-14432) (to M. T. O.) and by a postdoctoral fellowship from the Danish Council for Independent Research (DFF-4093-00242 to M. T. S.). This work was also supported by research grants from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, the Canada Foundation for Innovation, and the Heart and Stroke Foundation/Libin Professorship in Cardiovascular Research (to S. R. W. C.). Support for this research was also provided by National Institutes of Health Grants AR054098 and HL057832 (to M. F.) and HL057832 (to S. R. W. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CaM
- calmodulin
- CPVT
- catecholaminergic polymorphic ventricular tachycardia
- LQTS
- long QT syndrome
- RyR2
- cardiac ryanodine receptor
- SR
- sarcoplasmic reticulum
- CaMBD
- CaM-binding domain
- SOICR
- store overload-induced Ca2+ release
- GoF
- gain of function
- LoF
- loss of function
- ER
- endoplasmic reticulum
- Ctrl
- control
- MOT
- mean open time
- MCT
- mean closed time
- ITC
- isothermal titration calorimetry
- ANOVA
- analysis of variance
- TCEP
- tris(2-carboxyethyl)phosphine
- FI
- fluorescence intensity
- PO
- open probability.
References
- 1. Nyegaard M., Overgaard M. T., Søndergaard M. T., Vranas M., Behr E. R., Hildebrandt L. L., Lund J., Hedley P. L., Camm A. J., Wettrell G., Fosdal I., Christiansen M., and Børglum A. D. (2012) Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 91, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crotti L., Johnson C. N., Graf E., De Ferrari G. M., Cuneo B. F., Ovadia M., Papagiannis J., Feldkamp M. D., Rathi S. G., Kunic J. D., Pedrazzini M., Wieland T., Lichtner P., Beckmann B. M., Clark T., et al. (2013) Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sorensen A. B., Søndergaard M. T., and Overgaard M. T. (2013) Calmodulin in a heartbeat. FEBS J. 280, 5511–5532 [DOI] [PubMed] [Google Scholar]
- 4. Makita N., Yagihara N., Crotti L., Johnson C. N., Beckmann B. M., Roh M. S., Shigemizu D., Lichtner P., Ishikawa T., Aiba T., Homfray T., Behr E. R., Klug D., Denjoy I., Mastantuono E., et al. (2014) Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 7, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marsman R. F., Barc J., Beekman L., Alders M., Dooijes D., van den Wijngaard A., Ratbi I., Sefiani A., Bhuiyan Z. A., Wilde A. A., and Bezzina C. R. (2014) A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J. Am. Coll. Cardiol. 63, 259–266 [DOI] [PubMed] [Google Scholar]
- 6. Limpitikul W. B., Dick I. E., Joshi-Mukherjee R., Overgaard M. T., George A. L. Jr., and Yue D. T. (2014) Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell. Cardiol. 74, 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hwang H. S., Nitu F. R., Yang Y., Walweel K., Pereira L., Johnson C. N., Faggioni M., Chazin W. J., Laver D., George A. L. Jr., Cornea R. L., Bers D. M., and Knollmann B. C. (2014) Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ. Res. 114, 1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Søndergaard M. T., Tian X., Liu Y., Wang R., Chazin W. J., Chen S. R., and Overgaard M. T. (2015) Arrhythmogenic calmodulin mutations affect the activation and termination of cardiac ryanodine receptor-mediated Ca2+ release. J. Biol. Chem. 290, 26151–26162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin G., Hassan F., Haroun A. R., Murphy L. L., Crotti L., Schwartz P. J., George A. L., and Satin J. (2014) Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J. Am. Heart Assoc. 3, e000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vassilakopoulou V., Calver B. L., Thanassoulas A., Beck K., Hu H., Buntwal L., Smith A., Theodoridou M., Kashir J., Blayney L., Livaniou E., Nounesis G., Lai F. A., and Nomikos M. (2015) Distinctive malfunctions of calmodulin mutations associated with heart RyR2-mediated arrhythmic disease. Biochim. Biophys. Acta 1850, 2168–2176 [DOI] [PubMed] [Google Scholar]
- 11. Boczek N. J., Gomez-Hurtado N., Ye D., Calvert M. L., Tester D. J., Kryshtal D. O., Hwang H. S., Johnson C. N., Chazin W. J., Loporcaro C. G., Shah M., Papez A. L., Lau Y. R., Kanter R., Knollmann B. C., and Ackerman M. J. (2016) Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin (CaM) variants in long QT syndrome (LQTS) and functional characterization of a novel LQTS-associated CaM missense variant, E141G. Circ. Cardiovasc. Genet. 9, 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fearnley C. J., Roderick H. L., and Bootman M. D. (2011) Calcium signaling in cardiac myocytes. Cold Spring Harb. Perspect. Biol. 3, a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Petegem F. (2015) Ryanodine receptors: allosteric ion channel giants. J. Mol. Biol. 427, 31–53 [DOI] [PubMed] [Google Scholar]
- 14. Berridge M. J. (2014) Cell signalling biology: Module 7-cellular processes. Biochem. J. 10.1042/csb0001007 [DOI] [Google Scholar]
- 15. Linse S., Helmersson A., and Forsén S. (1991) Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 266, 8050–8054 [PubMed] [Google Scholar]
- 16. Zhang M., Abrams C., Wang L., Gizzi A., He L., Lin R., Chen Y., Loll P. J., Pascal J. M., and Zhang J. F. (2012) Structural basis for calmodulin as a dynamic calcium sensor. Structure 20, 911–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Søndergaard M. T., Sorensen A. B., Skov L. L., Kjaer-Sorensen K., Bauer M. C., Nyegaard M., Linse S., Oxvig C., and Overgaard M. T. (2015) Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 282, 803–816 [DOI] [PubMed] [Google Scholar]
- 18. Pedigo S., and Shea M. A. (1995) Discontinuous equilibrium titrations of cooperative calcium binding to calmodulin monitored by 1-D 1H-nuclear magnetic resonance spectroscopy. Biochemistry 34, 10676–10689 [DOI] [PubMed] [Google Scholar]
- 19. VanScyoc W. S., Sorensen B. R., Rusinova E., Laws W. R., Ross J. B., and Shea M. A. (2002) Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys. J. 83, 2767–2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang J., Zhou Y., Zou J., Chen Y., Patel P., Yang J. J., and Balog E. M. (2010) Site-specific modification of calmodulin Ca2(+) affinity tunes the skeletal muscle ryanodine receptor activation profile. Biochem. J. 432, 89–99 [DOI] [PubMed] [Google Scholar]
- 21. Saimi Y., and Kung C. (2002) Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 64, 289–311 [DOI] [PubMed] [Google Scholar]
- 22. Ben Johny M., Yang P. S., Bazzazi H., and Yue D. T. (2013) Dynamic switching of calmodulin interactions underlies Ca(2+) regulation of CaV1.3 channels. Nat. Commun. 4, 1717–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi N., Xu L., Pasek D. A., Evans K. E., and Meissner G. (2003) Molecular basis of calmodulin binding to cardiac muscle Ca(2+) release channel (ryanodine receptor). J. Biol. Chem. 278, 23480–23486 [DOI] [PubMed] [Google Scholar]
- 24. Fruen B. R., Black D. J., Bloomquist R. A., Bardy J. M., Johnson J. D., Louis C. F., and Balog E. M. (2003) Regulation of the RYR1 and RYR2 Ca2+ release channel isoforms by Ca2+-insensitive mutants of calmodulin. Biochemistry 42, 2740–2747 [DOI] [PubMed] [Google Scholar]
- 25. Maximciuc A. A., Putkey J. A., Shamoo Y., and Mackenzie K. R. (2006) Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode. Structure 14, 1547–1556 [DOI] [PubMed] [Google Scholar]
- 26. Lau K., Chan M. M., and Van Petegem F. (2014) Lobe-specific calmodulin binding to different ryanodine receptor isoforms. Biochemistry 53, 932–946 [DOI] [PubMed] [Google Scholar]
- 27. Tian X., Tang Y., Liu Y., Wang R., and Chen S. R. (2013) Calmodulin modulates the termination threshold for cardiac ryanodine receptor-mediated Ca2+ release. Biochem. J. 455, 367–375 [DOI] [PubMed] [Google Scholar]
- 28. Yang Y., Guo T., Oda T., Chakraborty A., Chen L., Uchinoumi H., Knowlton A. A., Fruen B. R., Cornea R. L., Meissner G., and Bers D. M. (2014) Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 114, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arnáiz-Cot J. J., Damon B. J., Zhang X. H., Cleemann L., Yamaguchi N., Meissner G., and Morad M. (2013) Cardiac calcium signalling pathologies associated with defective calmodulin regulation of type 2 ryanodine receptor. J. Physiol. (Lond.) 591, 4287–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones P. P., Jiang D., Bolstad J., Hunt D. J., Zhang L., Demaurex N., and Chen S. R. (2008) Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem. J. 412, 171–178 [DOI] [PubMed] [Google Scholar]
- 31. Ono M., Yano M., Hino A., Suetomi T., Xu X., Susa T., Uchinoumi H., Tateishi H., Oda T., Okuda S., Doi M., Kobayashi S., Yamamoto T., Koseki N., Kyushiki H., et al. (2010) Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca(2+) release in heart failure. Cardiovasc. Res. 87, 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holdgate G. A. (2001) Making cool drugs hot: isothermal titration calorimetry as a tool to study binding energetics. BioTechniques 31, 164–184 [PubMed] [Google Scholar]
- 33. Yamaguchi N., Chakraborty A., Huang T. Q., Xu L., Gomez A. C., Pasek D. A., and Meissner G. (2013) Cardiac hypertrophy associated with impaired regulation of cardiac ryanodine receptor by calmodulin and S100A1. Am. J. Physiol. Heart Circ. Physiol. 305, H86–H94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walweel K., Oo Y. W., and Laver D. R. (2016) The emerging role of calmodulin regulation of RyR2 in controlling heart rhythm, the progression of heart failure and the antiarrhythmic action of dantrolene. Clin. Exp. Pharmacol. Physiol. 10.1111/1440-1681.12669 [DOI] [PubMed] [Google Scholar]
- 35. Xu L., and Meissner G. (2004) Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys. J. 86, 797–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamaguchi N., Takahashi N., Xu L., Smithies O., and Meissner G. (2007) Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J. Clin. Invest. 117, 1344–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Breitwieser G. E., and Gama L. (2001) Calcium-sensing receptor activation induces intracellular calcium oscillations. Am. J. Physiol. Cell Physiol. 280, C1412–C1421 [DOI] [PubMed] [Google Scholar]
- 38. Jiang D., Xiao B., Yang D., Wang R., Choi P., Zhang L., Cheng H., and Chen S. R. (2004) RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. U.S.A. 101, 13062–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oo Y. W., Gomez-Hurtado N., Walweel K., van Helden D. F, Imtiaz M. S., Knollmann B. C., and Laver D. R. (2015) Essential role of calmodulin in RyR inhibition by dantrolene. Mol. Pharmacol. 88, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oda T., Yang Y., Uchinoumi H., Thomas D. D., Chen-Izu Y., Kato T., Yamamoto T., Yano M., Cornea R. L., and Bers D. M. (2015) Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell. Cardiol. 85, 240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uchinoumi H., Yang Y., Oda T., Puglisi J. L., Chen-Izu Y., Cornea R. L., Wehrens X. H. T., and Bers D. M. (2016) CaMKII and heart failure promote a pathological ryanodine receptor conformation that reduces calmodulin binding and enhances SR Ca2+ leak. Biophys. J. 110, 599a [Google Scholar]
- 42. Venetucci L., Denegri M., Napolitano C., and Priori S. G. (2012) Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 9, 561–575 [DOI] [PubMed] [Google Scholar]
- 43. Mizusawa Y., Horie M., and Wilde A. A. (2014) Genetic and clinical advances in congenital long QT syndrome. Circ. J. 78, 2827–2833 [DOI] [PubMed] [Google Scholar]
- 44. Jiménez-Jáimez J., Palomino Doza J., Ortega Á., Macías-Ruiz R., Perin F., Rodríguez-Vázquez del Rey M. M., Ortiz-Genga M., Monserrat L., Barriales-Villa R., Blanca E., Álvarez M., and Tercedor L. (2016) Calmodulin 2 mutation N98S is associated with unexplained cardiac arrest in infants due to low clinical penetrance electrical disorders. PLoS ONE 11, e0153851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Driessen H. E., Bourgonje V. J., van Veen T. A., and Vos M. A. (2014) New antiarrhythmic targets to control intracellular calcium handling. Neth. Heart J. 22, 198–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou Q., Xiao J., Jiang D., Wang R., Vembaiyan K., Wang A., Smith C. D., Xie C., Chen W., Zhang J., Tian X., Jones P. P., Zhong X., Guo A., Chen H., et al. (2011) Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. 17, Nat. Med. 1003–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tan Z., Xiao Z., Wei J., Zhang J., Zhou Q., Smith C. D., Nani A., Wu G., Song L. S., Back T. G., Fill M., and Chen S. R. (2016) Nebivolol suppresses cardiac ryanodine receptor-mediated spontaneous Ca2+ release and catecholaminergic polymorphic ventricular tachycardia. Biochem. J. 473, 4159–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang D., Wang R., Xiao B., Kong H., Hunt D. J., Choi P., Zhang L., and Chen S. R. (2005) Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 97, 1173–1181 [DOI] [PubMed] [Google Scholar]
- 49. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chamberlain B. K., Volpe P., and Fleischer S. (1984) Calcium-induced calcium release from purified cardiac sarcoplasmic reticulum vesicles: general characteristics. J. Biol. Chem. 259, 7540–7546 [PubMed] [Google Scholar]
- 51. Qin J., Valle G., Nani A., Chen H., Ramos-Franco J., Nori A., Volpe P., and Fill M. (2009) Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys. J. 97, 1961–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dweck D., Reyes-Alfonso A. Jr., and Potter J. D. (2005) Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 [DOI] [PubMed] [Google Scholar]
- 53. Adair G. (1925) The hemoglobin system: VI. The oxygen dissociation curve of hemoglobin. J. Biol. Chem. 63, 529–545 [Google Scholar]
- 54. Shea M. A., Verhoeven A. S., and Pedigo S. (1996) Calcium-induced interactions of calmodulin domains revealed by quantitative thrombin footprinting of Arg37 and Arg106. Biochemistry 35, 2943–2957 [DOI] [PubMed] [Google Scholar]