

Abstract
Cyclic adenosine monophosphate (cAMP) is an important mediator of hormonal stimulation of cell growth and differentiation through its activation of the extracellular signal-regulated kinase (ERK) cascade. Two small G proteins, Ras and Rap1 have been proposed to mediate this activation. Using HEK293 cells as a model system, we have recently shown that both Ras and Rap1 are required for cAMP signaling to ERKs. However, cAMP-dependent Ras signaling to ERKs is transient and rapidly terminated by PKA phosphorylation of the Raf isoforms C-Raf and B-Raf. In contrast, cAMP-dependent Rap1 signaling to ERKs and Rap1 is potentiated by PKA. We show that this is due to sustained binding of B-Raf to Rap1. One of the targets of PKA is Rap1 itself, directly phosphorylating Rap1a on serine 180 and Rap1b on serine 179. We show that these phosphorylations create potential binding sites for the adaptor protein 14-3-3 that links Rap1 to the scaffold protein KSR. These results suggest that Rap1 activation of ERKs requires PKA phosphorylation and KSR binding. Because KSR and B-Raf exist as heterodimers within the cell, this binding also brings B-Raf to Rap1, allowing Rap1 to couple to ERKs through B-Raf binding to Rap1 independently of its Ras-binding domain.
Keywords: 14-3-3 protein, extracellular-signal-regulated kinase (ERK), protein kinase A (PKA), Raf kinase, Ras-related protein 1 (Rap1)
Introduction
Hormones that are coupled to the second messenger cyclic adenosine monophosphate (cAMP) signal to multiple intracellular pathways. One of these, the MAP kinase, or ERK (extracellular signal-regulated kinase) cascade, mediates many physiological functions of cAMP in development (1), metabolism (2, 3), cognition (4–6), and cell growth (7–12). In all cases, cAMP signaling to ERKs requires the recruitment of the MAP kinase kinase kinase Raf to one of two small G proteins, Ras or Rap1. The ability to signal to both Ras and Rap1 allows cAMP to trigger the sustained activation of ERKs, which is required for many physiological functions, including the transcription/stabilization of many transcription factors (13, 14).
In addition to its ability to activate both Ras and Rap1, cAMP can inhibit signaling to Ras. PKA antagonizes Ras signaling through its phosphorylation of the Raf isoform C-Raf on serine 259 (Ser-259) (15). This phosphorylation and subsequent recruitment of 14-3-3 result in the release of C-Raf from active Ras (15, 16). We have recently shown that PKA phosphorylation of the homologous site in B-Raf (Ser-365) similarly prevents the direct binding of B-Raf to Ras (16). Despite this phosphorylation of B-Raf, B-Raf is still capable of binding to Rap1. This is physiologically relevant, as Rap1/B-Raf links PKA to ERKs (17, 18). How Rap1 is able to couple to B-Raf following PKA phosphorylation of B-Raf is unknown.
We and others have identified Rap1 as a target for phosphorylation by PKA (19–21). Here, we show that Rap1 phosphorylation plays a role in the ability of B-Raf to remain bound to Rap1 following cAMP stimulation. Rap1 phosphorylation creates a novel 14-3-3 binding site that connects Rap1 to the scaffold KSR. Because KSR can exist as a dimer with B-Raf, this provides Rap1 an indirect link to B-Raf and the MAP kinase cascade. This indirect binding of B-Raf to Rap1 is independent of its Ras-binding domain.
Results
cAMP Triggers the Indirect Binding of B-Raf to Rap1
Forskolin, an activator of endogenous adenylate cyclases, mimics the actions of hormones coupled through cAMP (22, 23). Additional elevation of cAMP levels can be achieved through the application of 3-isobutyl-1-methylxanthine (IBMX),2 a general phosphodiesterase. Stimulation of HEK293 cells by this mixture of forskolin and IBMX (herein referred to as “F/I treatment”) activated ERKs (measured by phospho-ERK antibodies) within 10 min and this activation was sustained for at least 30 min (Fig. 1A). B-Raf is the major Raf isoform that is capable of coupling cAMP to ERKs in many cell types (1, 3, 8, 12, 18, 24–28). This is shown in HEK293 cells as well, using shRNA specific for either human B-Raf or C-Raf (Fig. 1A). cAMP signaling to ERKs can utilize either the small G protein Ras or the small G protein Rap1 (1, 13, 29–31). We have recently shown that Ras activation contributes only to the early portion, whereas Rap1 contributes to both the early and later portions of ERK activation by cAMP (13). That study showed that the effects of F/I recapitulated those of isoproterenol, rationalizing the use of F/I in this study.
FIGURE 1.

B-Raf binding to Rap1 following cAMP stimulation is indirect. A, HEK293 cells were transfected with scrambled shRNA, or shRNA against B-Raf or C-Raf, as indicated. A, HEK293 cells were treated with F/I for 0, 10, 20, and 30 min. The endogenous levels of pERK and total ERK2, and the efficacies of the shRNAs are shown. B, cells transfected with wild type GFP-B-Raf (WT) or GFP-B-Raf R188L (R188L) and either FLAG-RasV12 or FLAG-RapE63 were treated with F/I for the indicated times. The presence of GFP-B-Raf following FLAG immunoprecipitation (IP) was detected by Western blotting (upper panel). The levels of FLAG-RasV12/RapE63 and GFP-B-Raf in the TCL are shown in the lower two panels. C, cells transfected with FLAG-Rap1 and wild type GFP-B-Raf (WT) or GFP-B-Raf R188L (R188L) were treated with F/I, as indicated. The presence of GFP-B-Raf following FLAG IP was examined. The levels of transfected proteins in the TCL are shown. D, wild type B-Raf, but not B-Raf R509H, can rescue cAMP activation of ERKs in cells depleted of B-Raf. HEK293 cells that were stably transfected with shRNA against B-Raf were transiently transfected with FLAG-ERK2 and either GFP-tagged vector control, shRNA-resistant (*) wild type B-Raf (WT*), or the R509H mutant (RH*) as indicated or vector (not shown), and treated with F/I as indicated. Phosphorylated FLAG-ERK2 was detected within the FLAG IPs (pFlag-ERK2). The levels of FLAG-ERK2 and endogenous B-Raf within total cell lysates (TCL) are shown. E, cAMP activation of ERKs by Rap1 requires B-Raf dimerization and KSR. HEK293 cells transfected with wild type GFP-B-Raf (WT) or GFP-B-Raf R509H (R509H), and either FLAG-RasV12 or FLAG-RapE63 were treated with F/I, as indicated. The presence of GFP-B-Raf following FLAG IP and the levels of transfected proteins in the TCL are shown.
Ras can couple to ERKs via either of the two major Raf isoforms, C-Raf and B-Raf (13, 32). In contrast, Rap1 can couple to ERKs only through B-Raf (3, 17, 33, 34). The heavy reliance on B-Raf during cAMP signaling to ERKs is consistent with the predominant role for Rap1 in ERK activation at these time points, in these cells (13, 27). To better understand the role of B-Raf in cAMP signaling, we examined its binding to GTP-loaded Ras and Rap1. In the absence of additional stimulation, B-Raf bound to the constitutively GTP-loaded Ras mutant RasV12. This basal binding of Ras to B-Raf required an intact Ras-binding domain (RBD), as it was absent in the RBD mutant B-Raf R188L (Fig. 1B), which, like the analogous C-Raf mutant C-Raf R89L, cannot bind to RasV12 (14, 44). Binding of wild type B-Raf was terminated within 10 min following F/I stimulation (Fig. 1B) (16). This was expected as the rapid phosphorylation of B-Raf on serine 365 by PKA interferes with the binding of B-Raf with GTP-loaded Ras (16).
The binding of B-Raf to the constitutively active Rap1 mutant RapE63 was regulated by cAMP/PKA differently than the binding of B-Raf to RasV12. Binding of B-Raf to Rap1E63 was seen basally, but unlike the binding of B-Raf to RasV12, binding of B-Raf to RapE63 remained at 10 and 20 min following F/I treatment (Fig. 1B). Similar results were seen with wild type Rap1, although Rap1 needed to be activated by F/I prior to binding B-Raf (Fig. 1C). Binding of B-Raf to wild type Rap1 was absent basally, was rapidly induced within 5 min of F/I treatment, and remained at 20 min (Fig. 1C). The need for the initial binding of B-Raf to GTP-loaded Rap1 required the B-Raf RBD, as the binding of the B-Raf R188L was absent at 5 min of F/I stimulation. Surprisingly, binding of the B-Raf R188L mutant was seen at 20 min (Fig. 1C), suggesting that the binding of B-Raf to Rap1-GTP at 20 min of F/I stimulation was no longer dependent on its RBD at this time point.
The absence of direct binding of B-Raf to Rap1 suggested that the binding of B-Raf to Rap1 might be mediated by an additional protein. B-Raf can bind to related proteins through a defined dimerization domain. This allows B-Raf to homodimerize with itself and to heterodimerize with either C-Raf or the related scaffold protein KSR (35). Both homo- and heterodimers of B-Raf can be eliminated using the mutant B-Raf R509H within the dimer interface (35, 36). To test the requirement or dimerization in cAMP activation of ERKs, we utilized cells in which B-Raf expression was reduced by shRNA. As expected, B-Raf was required for ERK activation by cAMP (F/I, 20 min), as ERK activation was lost in these B-Raf-deficient cells (Fig. 1D). The loss of ERK activation following knockdown of B-Raf could be rescued by transfection of an shRNA-resistant wild type B-Raf, but not by an shRNA-resistant B-Raf R509H (Fig. 1D), suggesting that B-Raf dimerization participates in cAMP activation of ERKs.
To ask whether dimerization was required for B-Raf to bind to either Ras or Rap1, we examined the binding of B-Raf R509H. In the absence of F/I, constitutively active Ras (RasV12) and Rap1 (RapE63) bound to both wild type B-Raf and the dimer-deficient mutant B-Raf R509H (Fig. 1E). This is consistent with B-Raf binding directly to GTP-loaded Ras and Rap1. After 20 min of F/I treatment, the binding of RasV12 to both wild type B-Raf and B-Raf R509H was terminated, as predicted by previous studies (16). In contrast, the binding of B-Raf to RapE63 seen after F/I treatment required dimerization, as binding of B-Raf R509H was lost (Fig. 1E). Because C-Raf is not required for cAMP activation of ERKs by Rap1 (30, 37), we examined the possibility that KSR could serve as the obligate dimer partner in this pathway.
cAMP Activation of ERKs Requires KSR Dimers
Signaling through Raf kinases is aided by the scaffolding function of KSR (38, 39), and role for KSR in cAMP signaling to ERKs has been proposed (40). KSR and B-Raf can exist as heterodimers (41, 42). Heterodimerization of B-Raf with KSR was detected in unstimulated cells and the level of dimerization was not affected by cAMP treatment (Fig. 2A). As a control, we showed that the KSR mutant R615H, which is homologous to the B-Raf dimerization mutant B-Raf R509H (35), was unable to dimerize with B-Raf (Fig. 2A). Using shRNA for the KSR isoform KSR1, we show that KSR is required for both the early and late phases of cAMP activation of ERKs (Fig. 2B). The efficacy of this shRNA is demonstrated in Fig. 2C. The requirement of KSR dimerization for cAMP activation of ERKs was seen in mouse embryonic fibroblasts (MEFs) derived from mice deficient in KSR1 (KSR−/− MEF cells) (39) (Fig. 2D). ERK activation could be rescued with a wild type KSR1 but not with the dimerization mutant KSR R615H (Fig. 2D).
FIGURE 2.

KSR is required for ERK activation by cAMP. A, cAMP does not increase the levels of B-Raf/KSR dimers. HEK293 cells were transfected with FLAG-tagged KSR (WT), or the mutant KSR R615H, as indicated, and treated with F/I for 20 min (20) or left untreated (0). The association of B-Raf with FLAG-KSR or FLAG-KSR R615H was determined by examining the level of endogenous (endo) B-Raf within each FLAG immunoprecipitation (IP). The levels of FLAG KSR within each IP are shown as a control. Levels of endogenous B-Raf are shown. B, cells transfected with scrambled short hairpin RNA (scrambled) or shRNA against KSR1 were treated with F/I, as indicated. The levels of endogenous pERK and ERK2 are shown. C, the efficacy of the KSR1 shRNA is shown. HEK293 cells were transfected with scrambled shRNA (scram), or shRNA against human KSR, as indicated. Cells were treated with F/I as indicated. The levels of endogenous (endo) KSR were examined following KSR IP, using an anti-KSR Ab. The endogenous levels of activated phospho-ERK1/2 (pERK), ERK2, B-Raf, and C-Raf were detected by Western blotting. D, KSR−/− MEF cells transfected with GFP-ERK2 and vector (not shown), wild type KSR (WT), or the R615H mutant, were treated with F/I for 20 min (20) or left untreated (0). The levels of phosphorylated GFP-ERK2 (pGFP-ERK2) following GFP IP and the levels of GFP-ERK2 and FLAG-KSR in the TCL are shown.
KSR Binding to Rap1 Requires Phosphorylation of Rap1
KSR does not contain a Ras-binding domain suggesting that KSR can only bind to Ras indirectly. We show here that KSR bound basally to RasV12, and this binding was lost following F/I treatment (Fig. 3A). In contrast, KSR bound to RapE63 both in the presence and absence of F/I (Fig. 3A). The requirement for KSR/B-Raf dimerization for KSR binding to Rap1 was tested using the dimerization-deficient mutant KSR R615H. KSR R615H did not bind to RapE63 in the absence of additional stimulation. However, KSR R615H was able to bind to RapE63 following F/I stimulation (Fig. 3A). This demonstrates that KSR can bind Rap1 independently of any Raf isoform. Because F/I treatment induces the PKA phosphorylation of Rap1, we asked whether PKA phosphorylation of Rap1 could be involved.
FIGURE 3.

KSR/B-Raf dimerization correlates with ERK activation by cAMP. A, HEK293 cells transfected with GFP-RasV12, GFP-RapE63 (E63), or GFP-RapE63AA (E63AA) and either FLAG-tagged WT KSR or KSR R615H (R615H) were treated with F/I, as indicated. The levels of GFP-Ras/Rap following FLAG immunoprecipitation (IP) are shown. The levels of FLAG-KSR (WT/RH) and the levels of GFP-RasV12, GFP-RapE63, and GFP-RapE63AA (GFP-Ras/Rap) in the TCL are shown. B, cells transfected with FLAG-KSR, and GFP-Rap1 (WT) or GFP-RapAA (AA), were treated with F/I, as indicated. The levels of GFP-Rap1 (WT/AA) following FLAG IP, and the levels of GFP-Rap1 (WT/AA) and FLAG-KSR in the TCL are shown.
PKA phosphorylation of Rap1 occurs within the carboxyl terminus of Rap1 itself (19, 21). For Rap1b and its constitutively active mutant RapE63, this phosphorylation occurs on Ser-179 (21). The single mutant Rap1b S179A (but not WT Rap1b) can be phosphorylated on the adjacent serine 180 (21). Therefore, to eliminate phosphorylation of Rap1b, we used the double mutation Rap1 S179A/S180A, either within wild type Rap1 (RapAA) or the constitutively active version (RapE63AA). Both RapE63 and RapE63AA bound to KSR basally, but unlike RapE63, the binding of KSR to RapE63AA was eliminated after 20 min of F/I treatment (Fig. 3A).
The interaction of KSR with wild type Rap1 was also dependent on Rap1 phosphorylation, as 20 min of F/I treatment stimulated the association of KSR with wild type Rap1, but not with RapAA (Fig. 3B). Taken together, these results are consistent with a two-step model of the binding of B-Raf/KSR dimers to Rap1. Immediately following Rap1 activation by F/I, B-Raf/KSR dimers bind to Rap1 through B-Raf. At later time points of F/I stimulation, B-Raf/KSR dimers bind to phosphorylated Rap1 through KSR.
Rap1 Phosphorylation Is Necessary for cAMP Signaling from Rap1 to ERKs
We asked whether the requirement for Rap1 phosphorylation for its sustained binding to KSR would also be true for B-Raf. As shown above, the binding of B-Raf to RapE63 (but not RasV12) was sustained during F/I treatment (Fig. 4A). The sustained binding of B-Raf to Rap1 required Rap1 phosphorylation, as this was lost in the phosphorylation-deficient RapE63AA (Fig. 4A, right three lanes). This pattern was similar to that seen with RasV12 (Fig. 1B, first three lanes). We propose that Rap1 phosphorylation allows B-Raf/KSR dimers to remain bound to Rap1 following F/I treatment.
FIGURE 4.

Rap1 activation of ERKs requires PKA phosphorylation of Rap1. A, HEK293 cells transfected with wild type GFP-B-Raf and FLAG-RasV12, FLAG-RapE63, or FLAG-RapE63AA were treated with F/I for the indicated times. The presence of GFP-B-Raf following FLAG immunoprecipitation (IP) was detected by Western blotting (upper panel). The levels of GFP-B-Raf and FLAG-Ras/Rap in the TCL are shown in the lower two panels. B, cells transfected with FLAG-ERK2, and either vector or GFP-Rap1AA were treated with F/I for the times indicated. The levels of phosphorylated FLAG-ERK2 (pFlag-ERK2) following FLAG IP are shown in the first panel. The levels of FLAG ERK2 and GFP-Rap1AA in the TCL are also shown. C, cells transfected with FLAG-ERK2, scrambled shRNA (scram), or shRNA against Rap1a/b, and GFP-tagged shRNA-resistant (*) wild type Rap1b (GFP-Rap1*), or the corresponding phosphorylation-deficient mutant (GFP-Rap1AA*) were treated with F/I, as indicated. The levels of pFLAG-ERK2 following FLAG IP and the levels of FLAG ERK2, GFP-Rap (WT/AA), and the endogenous Rap1 (endo Rap1) in the TCL are also shown.
Given the requirement of Rap1 phosphorylation in maintaining the binding of B-Raf to Rap1, we asked whether this phosphorylation would help sustain B-Raf-dependent ERK signaling. To examine this requirement for Rap1 phosphorylation in cAMP activation of ERKs, we expressed GFP-Rap1AA and FLAG-ERK2 in HEK293 cells and examined a time course of ERK activation by F/I. In the absence of GFP-Rap1AA, FLAG-ERK activation was rapid and sustained. In the presence of GFP-Rap1AA, the sustained portion of ERK activation was selectively lost, whereas the early portion of ERK activation remained unchanged (Fig. 4B). This suggests that Rap1AA was selectively interfering with the signaling of endogenous Rap1 to ERKs. The lack of effect of Rap1AA at the early time points is consistent with previous findings that the early time points of ERK activation were largely Ras-dependent (13).
It is possible that Rap1AA might be acting to interfere with endogenous Rap1. To examine the actions of Rap1AA in the absence of endogenous Rap1, we reduced endogenous Rap1 using siRNA. The knockdown of Rap1a/b eliminated the activation of ERKs following 20 min of F/I stimulation (Fig. 4C, left four lanes). This activation was restored by expressing an shRNA-resistant version of Rap1b (Fig. 4C, center four lanes). To examine the action of Rap1AA, we used an shRNA-resistant version of Rap1AA. In contrast to shRNA-resistant wild type Rap1b, shRNA-resistant Rap1AA was unable to rescue the loss of cAMP-dependent ERK activation (Fig. 4C, right four lanes). These data demonstrate that Rap1 phosphorylation is essential for cAMP signaling from endogenous Rap1 to ERKs.
14-3-3 Binding to Rap1 Requires KSR
It has been proposed that PKA phosphorylation of Rap1 creates a consensus novel binding motif for the scaffold protein 14-3-3 (43). This has been demonstrated for Rap1a (43). Like Rap1a, Rap1b has a single PKA consensus site at its carboxyl terminus (21, 44). The phosphorylation of Rap1b also creates a potential 14-3-3 binding site (43). Using Myc-tagged 14-3-3γ isoform, we demonstrate that 14-3-3γ binding to Rap1b does occur in cells (Fig. 5A). This binding was phosphorylation-dependent, as it was absent in the phosphorylation-deficient RapAA mutants (Fig. 5A). Similar results were seen when examining binding of Rap1b to endogenous 14-3-3 isoforms (Fig. 5B).
FIGURE 5.

Both Rap1a and Rap1b contain putative 14-3-3 binding sites. A, HEK293 cells transfected with Myc-14-3-3 and either GFP-Rap1b (WT) or the phosphorylation mutant Rap1bAA (AA), constitutively active mutant Rap1bE63 (E63), or Rap1bE63AA (E63AA), were treated with F/I, as indicated. The levels of Myc-14-3-3 and GFP-Rap1 following GFP immunoprecipitation (IP) are shown. The levels of Myc-14-3-3 in the TCL are shown in the bottom panel. B, cells transfected with the constitutively active mutants Rap1bE63 or Rap1bE63AA were treated with F/I, as indicated. The levels of endogenous 14-3-3 and FLAG-Rap1 following FLAG IP are shown. The levels of endogenous 14-3-3 in the TCL are shown in the bottom panel. C, KSR−/− MEF cells transfected with FLAG-Rap1b or vector and GFP-KSR were treated with F/I, as indicated. Endogenous 14-3-3 and FLAG-Rap1b following FLAG IP are shown. The levels of endogenous 14-3-3 and FLAG-RapE63, and GFP-KSR in the TCL are shown. D, HEK293 cells transfected with FLAG-RapE63 and vector and HA-14-3-3γ, HA-14-3-3β, and HA-14-3-3σ, and were treated with F/I, as indicated. HA-14-3-3 and GFP-Rap1b following FLAG IP are shown. The levels of HA-14-3-3 and GFP-RapE63 in the TCL are shown.
Because the association of both KSR and 14-3-3 with Rap1 require the same phosphorylated site within Rap1, it is possible that KSR enables the direct binding of 14-3-3 to Rap1. To test whether KSR participates in 14-3-3 binding to Rap1b, we examined binding in KSR−/− MEF cells. In KSR−/− MEF cells, no binding of endogenous 14-3-3 to Rap1b was detected. However, upon expression of KSR, the binding of endogenous 14-3-3 to Rap1b was observed, and this binding required F/I stimulation (Fig. 5C). It has been shown that KSR binds selectively to 14-3-3γ, but not 14-3-3β or 14-3-3σ (45). Only 14-3-3γ, but neither 14-3-3β nor 14-3-3σ, could bind RapE63 (Fig. 5D), consistent with the participation of KSR in 14-3-3 binding to RapE63.
KSR Ser-297 Links 14-3-3 to Rap1
14-3-3 exists as a dimer and can either link two 14-3-3 binding sites with the same protein or bring together two distinct proteins that each contain a 14-3-3 binding site. 14-3-3 proteins can bind to KSR through either mechanism. For example, two 14-3-3 binding sites are created by the phosphorylations of Ser-297 and Ser-392, and one dimer can bind simultaneously to both phosphorylation sites (46). This binding helps maintain KSR within the cytoplasm, as 14-3-3 binding masks a membrane association domain within KSR (47, 48). Both Ser-297 and Ser-392 are basally phosphorylated and constitutively bound to 14-3-3 (49).
Phospho-Ser-297 remains phosphorylated following growth factor stimulation (49). In contrast, phospho-Ser-392 is rapidly dephosphorylated following growth factor stimulation. This releases 14-3-3 to allow KSR to associate with the plasma membrane, a requirement for growth factors to stimulate ERK (49). Unlike stimulation by growth factor, stimulation by cAMP does not induce the dephosphorylation of phospho-Ser-392 (Fig. 6A). Therefore, the phosphorylated Ser-297 and Ser-392 sites are potential candidates for linking 14-3-3 dimers to phosphorylated Rap1 during cAMP stimulation.
FIGURE 6.

KSR 14-3-3 sites differentially interact with Rap1. A, HEK293 cells transfected with FLAG-KSR were treated with F/I as indicated. Lysates were subjected to FLAG immunoprecipitation (IP) and probed with a phospho-specific antibody recognizing phosphorylated Ser-392 of KSR. The levels of Ser(P)-392 are shown in the upper panel. The levels of FLAG-KSR within the IP are shown in the lower panel. B, HEK293 cells transfected with GFP-Rap1bE63 (RapE63) and FLAG-KSR (WT), FLAG-KSR S297A (S297A), FLAG-KSR S392A (S392A), FLAG-KSR S297A/S392A (S297A/S392A), or FLAG-KSR S838A (S838A). Cells were treated with F/I for 20 min (20) or left untreated (0). The levels of GFP-RapE63 following FLAG IP are shown. The levels of GFP-RapE63 and FLAG-KSR WT/mutants within the TCL are shown. C, KSR S838A cannot dimerize with B-Raf. HEK293 cells were transfected with FLAG-tagged KSR (WT), or mutants KSR S392A, KSR S838A, or KSR R615H, as indicated, and treated with F/I for 20 min (20) or left untreated (0). The association of B-Raf with KSR and/or KSR mutants was determined by examining the level of endogenous B-Raf within each FLAG IP. The levels of FLAG KSR within each IP are shown as a control. Levels of endogenous B-Raf in the TCL are shown. D, KSR S297A can dimerize with B-Raf. HEK293 cells were transfected with FLAG-tagged KSR (WT) or KSR S297A, and treated with F/I for 20 min (20) or left untreated (0). The association of B-Raf with KSR and/or KSR mutants was determined by examining the level of endogenous B-Raf within each FLAG IP. The levels of FLAG KSR within each IP are shown as a control. Levels of endogenous B-Raf in the TCL are shown.
The possible roles of KSR Ser-297 and Ser-392 in linking KSR to Rap1 were examined using KSR mutants lacking each phosphorylation site. We reasoned that mutation of the relevant phosphorylation site(s) would disrupt binding of KSR to phosphorylated RapE63 only following F/I treatment, but not in the absence of F/I treatment. This pattern was seen for the mutant KSR S297A. KSR S297A bound to RapE63 in the absence of F/I treatment, but this binding was lost upon F/I treatment (Fig. 6B). These data suggested that Ser-297 was a candidate 14-3-3 binding site linking KSR to phosphorylated Rap1. In contrast, KSR S392A and the double mutant KSR S297A/S392A were both unable to bind to RapE63 in either the presence or absence of F/I (Fig. 6B). Therefore, because KSR S392A did not bind RapE63 in the absence of F/I, we could not rule out a role for phospho-Ser-392 in linking KSR to phospho-Rap1.
A third potential 14-3-3 binding site, KSR Ser-838, has been proposed as a site of PKA phosphorylation (40), and shown to be required for cAMP signaling to ERKs (40). Phosphorylated Ser-838 may bind 14-3-3, possibly to promote B-Raf/KSR dimerization (35). Consistent with this notion, we show that of all the KSR mutants examined, only KSR S838A was unable to form dimers with B-Raf (Fig. 6, C and D). Like WT KSR, KSR S838A bound to RapE63 modestly in untreated cells, but bound to RapE63 robustly after F/I treatment (Fig. 6B), demonstrating that phosphorylation of Ser-838 was not required to allow KSR to bind RapE63. Taken together, these data are consistent with a role for Ser-297, and possibly Ser-392, but not Ser-838, as the 14-3-3 binding site linking KSR to phosphorylated Rap1.
Activation of ERKs by Rap1 Requires KSR and PKA
These data suggest strongly that a novel protein complex containing Rap1, 14-3-3, and KSR is formed upon F/I stimulation, and this is dependent on PKA phosphorylation of Rap1. We have identified two distinct mechanisms by which PKA regulates the binding of B-Raf/KSR dimers to Ras and Rap1. Ras recruitment of B-Raf/KSR is terminated by PKA phosphorylation of B-Raf on Ser-365 (16). Rap1 recruitment of B-Raf/KSR is maintained despite B-Raf phosphorylation, through the PKA phosphorylation of Rap1, which permits the indirect link to B-Raf via KSR binding to phosphorylated Rap1. Given the well known scaffold function of KSR (38, 39, 50, 51), this complex may also facilitate the coupling of B-Raf to MEK and ERK. This is depicted in Fig. 7.
FIGURE 7.
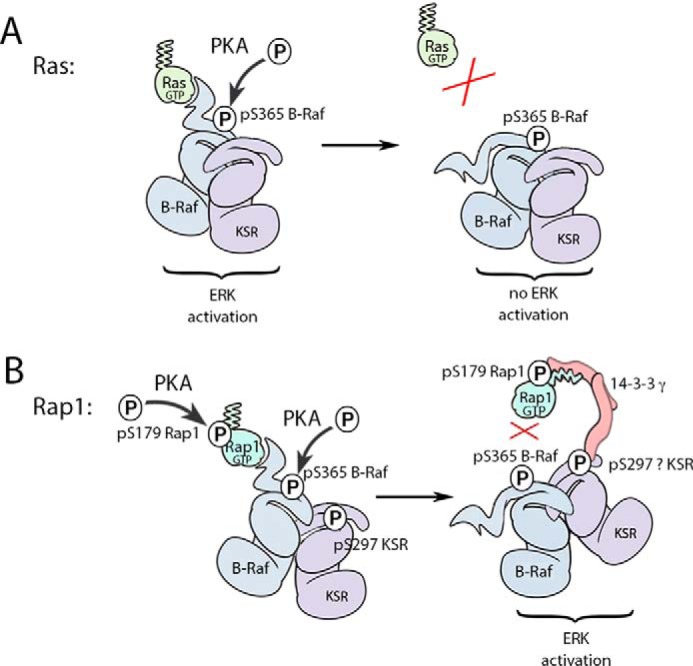
Model of cAMP activation of ERK. A, Ras activation recruits B-Raf/KSR to trigger ERK activation. This occurs through the direct binding of Ras-GTP to the B-Raf RBD. The binding of B-Raf to Ras is regulated by PKA phosphorylation of B-Raf on Ser-365 (pS365). This phosphorylated site interrupts B-Raf binding to Ras-GTP, terminating signals to ERK. This occurs through the binding of 14-3-3 (not shown). The release of B-Raf also releases KSR, as part of the B-Raf/KSR dimer. Ras-GTP also recruits C-Raf and a similar mechanism involving PKA phosphorylation of C-Raf S295 releases C-Raf from Ras (not shown). B, Rap1 recruitment of B-Raf/KSR is regulated by two phosphorylations induced by cAMP/PKA, one on B-Raf and one on Rap1. Activated Rap1 binds to a B-Raf/KSR dimer through the B-Raf RBD. This direct binding is inhibited by the PKA-dependent phosphorylation on B-Raf (pS365). At the same time, the indirect binding of B-Raf/KSR to Rap1 is enhanced by the PKA-dependent phosphorylations of Rap1. (Here, Rap1b and pS179 are shown.) The inhibitory PKA phosphorylation of B-Raf on Ser-365 functions to release the B-Raf/KSR dimer from Rap1-GTP in a similar fashion to that shown above for Ras. Although we have not definitively proved that 14-3-3 is required for KSR to bind Rap1, the association of both KSR and 14-3-3 with Rap1 requires the same phosphorylated site within Rap1. This PKA phosphorylation of Rap1 on Ser-179 creates a novel 14-3-3 binding site at the carboxyl terminus of Rap1. 14-3-3 binding accommodates this phosphorylation in conjunction with the adjacent the C-terminal geranylgeranyl lipid. We propose that the binding of 14-3-3 to phosphorylated Rap1 occurs as part of a 14-3-3γ dimer that also binds KSR. Therefore, we have drawn 14-3-3 as a bridge between Rap1 and KSR in this model. Rap1 binds to KSR via established 14-3-3 binding sites. Mutational analysis of potential 14-3-3 sites in KSR suggested Ser-297 as a candidate site, although Ser-392 could not be ruled out. We propose that whereas the B-Raf/KSR dimer is released from direct binding to Rap1, the dimer remains associated with phosphorylated Rap1 through a phospho-specific 14-3-3 binding site that links KSR and its dimer partner B-Raf to Rap1. This model explains how KSR and B-Raf can bind to phosphorylated Rap1 even after direct binding of Rap1 to B-Raf is blocked.
Discussion
PKA Phosphorylation of Rap1 Allows RBD-independent Binding of B-Raf to Rap1
The classical view of Ras activation is that GTP loading helps coordinate the binding of effectors to the Ras switch region. These interactions between Ras and its effectors are mediated by RBDs and related Ras-association domains present in all effectors (52–54). This has been well studied for activation of the Raf family of effectors, where GTP-dependent binding to Ras participates in Raf activation through membrane recruitment and release of autoinhibition (55). Similar RBDs are thought to define the binding of Raf effectors to the small G protein Rap1, as predicted from early structural studies (56). RBDs within both B-Raf and C-Raf are themselves targets for phosphorylation that inhibit binding to Ras and terminate signaling from Ras to ERKs (15, 16, 57).
PKA phosphorylation of the B-Raf RBD disrupts its interactions with both Ras and Rap1 (16). However, despite interference of PKA with the direct binding of B-Raf to Rap1, B-Raf can still bind Rap1 indirectly after PKA stimulation (16). PKA reverses its block of direct binding with a second phosphorylation, on Rap1 Ser-179, which permits the indirect binding of B-Raf to Rap1 through KSR. Importantly, the effects of Rap1 phosphorylation were dominant over the effects of B-Raf phosphorylation, and when Rap1 phosphorylation was eliminated by RapAA mutation, the PKA-dependent blockade of B-Raf binding to Rap1 was revealed. The indirect binding was confirmed using a mutant of B-Raf (B-Raf R188L) that could not bind directly to Rap1 in the absence of F/I, but could still bind PKA phosphorylated Rap1 after F/I stimulation. This is the first example of Raf signaling that is independent of its RBD.
KSR Is Required for B-Raf to Bind Rap1 and Signal to ERKs
The indirect binding of B-Raf to Rap1 required dimerization, as the dimerization mutant B-Raf R509H could not bind to phosphorylated Rap1 after F/I treatment. We propose that KSR is an obligate dimer partner for B-Raf in this action, as both KSR and B-Raf dimerization are required for cAMP signaling to ERKs.
Many functions have been suggested for KSR. KSR is a scaffold that brings the MAP kinase kinase MEK in proximity to B-Raf (38, 39, 50, 51). KSR may also provide allosteric activation of B-Raf within the B-Raf/KSR dimer (36). We propose another role KSR, to link B-Raf to phosphorylated Rap1. KSR does not contain an RBD or Ras-association domain and therefore, cannot be recruited directly to Ras or Rap1. Here, we propose that the binding of KSR to Ras is indirect via the binding of B-Raf through its RBD. This occurs rapidly following F/I stimulation as Ras becomes transiently GTP-loaded (13) and is terminated by the PKA phosphorylation of B-Raf itself (16).
The indirect binding of KSR via B-Raf can also characterize its binding to GTP-loaded Rap1. In the absence of F/I stimulation, KSR binding to RapE63 required KSR dimerization, as the B-Raf RBD provided the direct interaction with Rap1-GTP. However, after F/I stimulation and upon Rap1 phosphorylation by PKA, KSR binding to RapE623 no longer required B-Raf. We propose that the binding of the B-Raf/KSR dimer to Rap1 following F/I stimulation has two stages. Initially, B-Raf/KSR binding to Rap1 is similar to Ras, in that it required the direct binding of the B-Raf RBD to Rap1. F/I induces the GTP loading of Rap1. This transiently maintains KSR/B-Raf binding via the B-Raf RBD. Upon further F/I stimulation, PKA phosphorylation of B-Raf disrupts the binding of the RBD to Rap1. Concurrent PKA phosphorylation of Rap1 promotes the binding of KSR to Rap1 itself. Because B-Raf and KSR maintain their dimerization during cAMP stimulation, this allows B-Raf to remain associated with Rap1, albeit indirectly after the direct binding of B-Raf to Rap1 is terminated.
14-3-3 Bridges Rap1 and KSR during cAMP Signaling to ERKs
PKA phosphorylation of Rap1 is analogous to phosphorylations within the C terminus of the Rho family G protein Rnd3 (43) and other prenylated small G proteins that can form bona fide 14-3-3 binding sites (called Mode III/IV sites), whose consensus is (R/K)(R/K)XpSC-geranylgeranyl) (43). This consensus motif lacks the essential proline of Mode I/II, allowing the entire peptide and a portion of the lipid motif to be accommodated within the hydrophobic groove of 14-3-3 (58). Rap1a contains a perfect Mode III/IV consensus sequence (RRApSC-geranylgeranyl) and Rap1b contains a related sequence (RRpSSC-geranylgeranyl) (43). All 14-3-3 isoforms show similar affinities for the hybrid prenyl/phosphorylation motif in Rnd3 described by Riou et al. (43). However, only 14-3-3γ, but neither 14-3-3β nor 14-3-3σ, could bind RapE63. Similarly, only 14-3-3γ, but neither 14-3-3β nor 14-3-3σ, can bind KSR (45). This is consistent with the contribution of KSR to 14-3-3 binding to phosphorylated Rap1.
We propose that the binding of 14-3-3 to KSR potentiates signals from Rap1 to ERKs. This is fundamentally different from the function of specific 14-3-3 sites on Raf (B-Raf Ser-365 and C-Raf Ser-259) that inhibit Ras signaling to ERKs (15, 49). This model may help explain the long-standing mystery of why high 14-3-3 levels within cells correlates with the ability of cAMP to activate, rather than inhibit, ERKs (26).
Possible 14-3-3 sites in KSR include phospho-Ser-838, phospho-Ser-392, and phospho-Ser-297. Ser-838 phosphorylation is unlikely to link KSR to Rap1 because the S838A mutant can still bind Rap1 after F/I stimulation. In contrast, KSR S297A binds only to unphosphorylated RapE63, but not to RapE63 that is phosphorylated after F/I treatment. These data suggest that Ser-297 is a candidate site to mediate the PKA-dependent binding of KSR to phosphorylated Rap1.
KSR Ser-392 remains phosphorylated during cAMP stimulation of ERKs, making it a potential candidate to remain bound to 14-3-3 during the course of cAMP stimulation. The mutant KSR S392A cannot bind Rap1, possibly due to its constitutive membrane localization (47, 49), making it difficult to conclude that the lack of Rap1 binding of KSR S392A reflects the lack of 14-3-3 coupling to Rap1 in this mutant.
Model for B-Raf/KSR Function during cAMP Signaling to ERKs
We propose a two-step model for cAMP signaling to ERKs. Ras-dependent ERK activation requires the recruitment of B-Raf/KSR dimers to Ras. This is mediated by the direct binding of B-Raf to the switch region of Ras. B-Raf phosphorylation by PKA at Ser-365 releases B-Raf from Ras to terminate Ras signaling to ERKs (16). This is depicted in Fig. 7A.
In contrast to Ras, Rap1 activation of B-Raf/KSR signaling to ERKs is not terminated following PKA phosphorylation of B-Raf. The simplest model is that preformed B-Raf/KSR dimers are initially recruited to Rap1-GTP through direct binding of B-Raf to the switch region of Rap1. We propose that this PKA phosphorylation of B-Raf may also interrupt RBD interactions with Rap1. However, the phosphorylation of Rap1 opposes the inhibitory actions of B-Raf phosphorylation on Rap1 binding, by providing a novel 14-3-3 binding site for KSR that allows an indirect binding of B-Raf to Rap1. This is depicted in Fig. 7B.
It is possible that upon Rap1 activation, a molecule of B-Raf initially binds directly to Rap1-GTP and then subsequently binds indirectly to phosphorylated Rap1 through KSR. However, the ability of the B-Raf R188L mutant to bind Rap1 after F/I treatment suggests that the direct binding of B-Raf to unphosphorylated Rap1 is not a prerequisite for its subsequent binding to phosphorylated Rap1. In either case, these studies demonstrate that B-Raf can signal independently of direct interactions with small G proteins, and utilizes KSR to help maintain a signaling complex with Rap1 during cAMP activation.
One aspect of Rap1 signaling that has remained unexplained is why the constitutive mutants RapV12 and RapE63 are often poor activators of ERKs, even in cells where a requirement for Rap1 in cAMP signaling to ERKs has been demonstrated (59). We propose that efficient signaling of Rap1 to ERKs requires multiple events in addition to Rap1 activation, including phosphorylation by PKA and recruitment of KSR. This model may explain why constitutively active Rap1 mutants are poor activators of ERKs in the absence of PKA phosphorylation and may explain why constitutively active Rap1 mutants are rarely seen in human cancers (60). In this regard, it is interesting that the mitogenic actions of constitutively active Rap1 in a model of thyroid follicular cell growth required Rap1 phosphorylation (61). In addition, endogenous levels of KSR may be limiting during overexpression of constitutively active Rap1 mutants. KSR levels vary greatly in different cell types (62). It will be interesting to see if the ability of constitutively active Rap1 mutants to activate ERKs correlates with KSR expression.
Experimental Procedures
Chemicals
Forskolin was purchased from Tocris Bioscience (Minneapolis, MN). IBMX was purchased from Calbiochem (Riverside, CA).
Plasmids
Human B-Raf, mouse KSR1, and tagged-14-3-3 isoforms were provided by Andréy Shaw (Washington University, St. Louis, MO) and Peter Hurlin (Oregon Health & Sciences University). Human H-RasG12V (H-RasV12) was purchased from the Missouri S&T cDNA Resource Center (Rolla, MO) and was cloned into a pEGFP-C1 vector. References to the Rap1 cDNA refer to the bovine Rap1b sequence. The cDNA of rat Rap1a was cloned into a pEGFP-C1 vector. The construction of vectors containing FLAG-Rap1b, FLAG-Rap1bS179A/S180A (Rap1AA), FLAG-Rap1bE63, FLAG-Rap1bE63S179A/S180A (RapE63AA), GFP-Rap1b, GFP-Rap1AA, GFP-Rap1bE63, GFP-RapE63AA, HA-B-Raf, HA-B-Raf R509H, GFP-B-Raf, and GFP-B-Raf R188L have been described previously (16, 17, 19). The FLAG-KSR S392A, FLAG-KSR S297A, FLAG-KSR S297A/S392A, FLAG-KSR S838A, and FLAG-KSR R615H mutants were generated by site-directed mutagenesis of the corresponding plasmids using the QuikChangeTM kit (Stratagene, La Jolla, CA). GFP-B-Raf R509H, GFP-Rap1aE63, and GFP-Rap1aE63 S180A mutants were subcloned from the corresponding FLAG-tagged plasmids. shRNA-resistant mutants of HA-B-Raf (WT or R509H) were generated by mutagenesis of ACAGAGACCTCAAGAGTAA to ACAGAGATCTGAAGAGCAA. The mutations are underlined. For an shRNA-resistant version of Rap1b, bovine Rap1b cDNA was used.
RNA Interference
Complementary pairs of shRNA oligonucleotides were synthesized by Integrated DNA Technologies Inc. (Coralville, IA), annealed, and cloned into the pSUPER-neo+GFP vector (OligoEngine, Seattle, WA). The shRNA for Rap1a, Rap1b, B-Raf, and C-Raf were described previously (13, 16, 63–66). The sense KSR1 shRNA oligonucleotide is GTGCCAGAAGAGCATGATA (human sequence, corresponding to the mouse KSR1 siRNA) (67).
Antibodies
Anti-ERK2 (D2), anti-B-Raf (H145), anti-GFP (SC-8334), and anti-Rap1a/b (SC-65) antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Agarose-coupled anti-FLAG (M2) and anti-FLAG (M2) were from Sigma. Anti-Myc (MA1–21316) was from Thermo Fisher Scientific Inc. (Rockford, IL). Anti-KSR1 (611577) was from BD Transduction Laboratories. Anti-14-3-3 (AB9748) was from EMD Millipore (Billerica, MA). Anti-phospho-KSR1 (pS392) (antibody number 4951) and anti-phospho-ERK (Thr-201 and Tyr-204) (Ab number 9101) were purchased from Cell Signaling Technology (Beverly, MA). Anti-mouse and anti-rabbit secondary antibodies for Western blots were from GE Healthcare.
Cell Culture Conditions and Transfections
KSR−/− MEF cells were kindly provided by Andréy Shaw (Washington University, St. Louis, MO). All cells (HEK293 and KSR−/− MEF cells) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and l-glutamine at 37 °C in 5% CO2. Transient transfections were performed using Lipofectamine 2000 reagent (Invitrogen Corp.) according to the manufacturer's instructions, and serum starved prior to treatment. The control vector, pcDNA3, was included in each set of transfections to assure that each plate received the same amount of transfected DNA. Cells were treated with 10 μm forskolin and 100 μm IBMX for 20 min unless otherwise indicated. This treatment is designated “F/I” in the text and figures. To generate stable cell populations, HEK293 cells were transfected with pSUPER-neo+GFP encoding shRNA for B-Raf or a scrambled construct and selected using 0.5 mg/ml of G418 for 4 to 6 weeks. shRNA experiments were conducted as previously described (13).
Western Blotting
Immunoprecipitations and Western blotting were performed as previously described (31, 68). Thermo Scientific PageRuler Prestained Protein Ladder was used as protein size indicators in all gels. Active Rap1 was assayed using GST-Ral-GDS in an in vitro pulldown assay as previously described (69). Active Ras was assayed using a Ras activation assay kit (Cytoskeleton Inc., Denver, CO) and performed according to the manufacturer's instructions. In all cases, representative Western blots of at least three independent experiments are shown.
Author Contributions
P. J. S. S. conceived and coordinated the experiments within this study, designed Fig. 7, and wrote the paper. M. T. designed, performed, and analyzed most of the experiments shown in Figs. 1, 2, 3, 5, and 6. T. J. D. designed and performed most of the experiments shown in Fig. 4, as well as some of the experiments shown in Figs. 1 and 6, and analyzed the experiments shown in Fig. 1. Y. L. performed and analyzed some of the experiments shown in Figs. 1, 2, and 4. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Andréy Shaw (Genentech) and Lawrence Quilliam (University of Indiana, Indianapolis, IN) for selected plasmid constructs.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK090309 and R21 CA191392-01. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IBMX
- 3-isobutyl-1-methylxanthine
- F/I
- forskolin and IBMX
- Ras
- rat sarcoma GTPase
- KSR
- kinase suppressor of Ras
- Rap1
- Ras-related protein 1
- TCL
- total cell lysate
- RBD
- Ras-binding domain
- MEF
- mouse embryonic fibroblast.
References
- 1. Fujita T., Meguro T., Fukuyama R., Nakamuta H., and Koida M. (2002) New signaling pathway for parathyroid hormone and cyclic AMP action on extracellular-regulated kinase and cell proliferation in bone cells: checkpoint of modulation by cyclic AMP. J. Biol. Chem. 277, 22191–22200 [DOI] [PubMed] [Google Scholar]
- 2. Gomez E., Pritchard C., and Herbert T. P. (2002) cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediate Raf-independent activation of extracellular regulated kinase in response to glucagon-like peptide-1 in pancreatic beta-cells. J. Biol. Chem. 277, 48146–48151 [DOI] [PubMed] [Google Scholar]
- 3. Ehses J. A., Pelech S. L., Pederson R. A., and McIntosh C. H. (2002) Glucose-dependent insulinotropic polypeptide activates the Raf-Mek1/2-ERK1/2 module via a cyclic AMP/cAMP-dependent protein kinase/Rap1-mediated pathway. J. Biol. Chem. 277, 37088–37097 [DOI] [PubMed] [Google Scholar]
- 4. Morozov A., Muzzio I. A., Bourtchouladze R., Van-Strien N., Lapidus K., Yin D., Winder D. G., Adams J. P., Sweatt J. D., and Kandel E. R. (2003) Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 39, 309–325 [DOI] [PubMed] [Google Scholar]
- 5. Monaghan T. K., Mackenzie C. J., Plevin R., and Lutz E. M. (2008) PACAP-38 induces neuronal differentiation of human SH-SY5Y neuroblastoma cells via cAMP-mediated activation of ERK and p38 MAP kinases. J. Neurochem. 104, 74–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grewal S. S., Horgan A. M., York R. D., Withers G. S., Banker G. A., and Stork P. J. (2000) Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J. Biol. Chem. 275, 3722–3728 [DOI] [PubMed] [Google Scholar]
- 7. Iacovelli L., Capobianco L., Salvatore L., Sallese M., D'Ancona G. M., and De Blasi A. (2001) Thyrotropin activates mitogen-activated protein kinase pathway in FRTL-5 by a cAMP-dependent protein kinase A-independent mechanism. Mol. Pharmacol. 60, 924–933 [DOI] [PubMed] [Google Scholar]
- 8. Vuchak L. A., Tsygankova O. M., Prendergast G. V., and Meinkoth J. L. (2009) Protein kinase A and B-Raf mediate extracellular signal-regulated kinase activation by thyrotropin. Mol. Pharmacol. 76, 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Kolen K., Dautzenberg F. M., Verstraeten K., Royaux I., De Hoogt R., Gutknecht E., and Peeters P. J. (2010) Corticotropin releasing factor-induced ERK phosphorylation in AtT20 cells occurs via a cAMP-dependent mechanism requiring EPAC2. Neuropharmacology 58, 135–144 [DOI] [PubMed] [Google Scholar]
- 10. Le Péchon-Vallée C., Magalon K., Rasolonjanahary R., Enjalbert A., and Gérard C. (2000) Vasoactive intestinal polypeptide and pituitary adenylate cyclase-activating polypeptides stimulate mitogen-activated protein kinase in the pituitary cell line GH4C1 by a 3′,5′-cyclic adenosine monophosphate pathway. Neuroendocrinology 72, 46–56 [DOI] [PubMed] [Google Scholar]
- 11. Romano D., Magalon K., Ciampini A., Talet C., Enjalbert A., and Gerard C. (2003) Differential involvement of the Ras and Rap1 small GTPases in vasoactive intestinal and pituitary adenylyl cyclase activating polypeptides control of the prolactin gene. J. Biol. Chem. 278, 51386–51394 [DOI] [PubMed] [Google Scholar]
- 12. Kovalovsky D., Refojo D., Liberman A. C., Hochbaum D., Pereda M. P., Coso O. A., Stalla G. K., Holsboer F., and Arzt E. (2002) Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol. Endocrinol. 16, 1638–1651 [DOI] [PubMed] [Google Scholar]
- 13. Li Y., Dillon T. J., Takahashi M., Earley K. T., and Stork P. J. (2016) PKA-independent Ras activation cooperates with Rap1 to mediate activation of ERKs by cAMP. J. Biol. Chem. 291, 21584–21595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murphy L. O., Smith S., Chen R. H., Fingar D. C., and Blenis J. (2002) Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 4, 556–564 [DOI] [PubMed] [Google Scholar]
- 15. Dumaz N., and Marais R. (2003) Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J. Biol. Chem. 278, 29819–29823 [DOI] [PubMed] [Google Scholar]
- 16. Li Y., Takahashi M., and Stork P. J. (2013) Ras-mutant cancer cells display B-Raf binding to Ras that activates extracellular signal-regulated kinase and is inhibited by protein kinase A phosphorylation. J. Biol. Chem. 288, 27646–27657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carey K. D., Watson R. T., Pessin J. E., and Stork P. J. (2003) The requirement of specific membrane domains for Raf-1 phosphorylation and activation. J. Biol. Chem. 278, 3185–3196 [DOI] [PubMed] [Google Scholar]
- 18. Vossler M. R., Yao H., York R. D., Pan M. G., Rim C. S., and Stork P. J. (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89, 73–82 [DOI] [PubMed] [Google Scholar]
- 19. Takahashi M., Dillon T. J., Liu C., Kariya Y., Wang Z., and Stork P. J. (2013) Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J. Biol. Chem. 288, 27712–27723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edreira M. M., Li S., Hochbaum D., Wong S., Gorfe A. A., Ribeiro-Neto F., Woods V. L. Jr., and Altschuler D. L. (2009) Phosphorylation-induced conformational changes in Rap1b: allosteric effects on switch domains and effector loop. J. Biol. Chem. 284, 27480–27486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Altschuler D., and Lapetina E. G. (1993) Mutational analysis of the cAMP-dependent protein kinase-mediated phosphorylation site of Rap1b. J. Biol. Chem. 268, 7527–7531 [PubMed] [Google Scholar]
- 22. Insel P. A., and Ostrom R. S. (2003) Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell. Mol. Neurobiol. 23, 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seifert R., Lushington G. H., Mou T. C., Gille A., and Sprang S. R. (2012) Inhibitors of membranous adenylyl cyclases. Trends Pharmacol. Sci. 33, 64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alleaume C., Eychène A., Caigneaux E., Muller J. M., and Philippe M. (2003) Vasoactive intestinal peptide stimulates proliferation in HT29 human colonic adenocarcinoma cells: concomitant activation of Ras/Rap1-B-Raf-ERK signalling pathway. Neuropeptides 37, 98–104 [DOI] [PubMed] [Google Scholar]
- 25. Dugan L. L., Kim J. S., Zhang Y., Bart R. D., Sun Y., Holtzman D. M., and Gutmann D. H. (1999) Differential effects of cAMP in neurons and astrocytes: role of B-raf. J. Biol. Chem. 274, 25842–25848 [DOI] [PubMed] [Google Scholar]
- 26. Qiu W., Zhuang S., von Lintig F. C., Boss G. R., and Pilz R. B. (2000) Cell type-specific regulation of B-Raf kinase by cAMP and 14-3-3 proteins. J. Biol. Chem. 275, 31921–31929 [DOI] [PubMed] [Google Scholar]
- 27. Schmitt J. M., and Stork P. J. (2000) β2-Adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. J. Biol. Chem. 275, 25342–25350 [DOI] [PubMed] [Google Scholar]
- 28. Yamaguchi T., Wallace D. P., Magenheimer B. S., Hempson S. J., Grantham J. J., and Calvet J. P. (2004) Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 279, 40419–40430 [DOI] [PubMed] [Google Scholar]
- 29. Zeiller C., Blanchard M. P., Pertuit M., Thirion S., Enjalbert A., Barlier A., and Gerard C. (2012) Ras and Rap1 govern spatiotemporal dynamic of activated ERK in pituitary living cells. Cell Signal. 24, 2237–2248 [DOI] [PubMed] [Google Scholar]
- 30. Dumaz N., and Marais R. (2005) Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways: based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 272, 3491–3504 [DOI] [PubMed] [Google Scholar]
- 31. Wang Z., Dillon T. J., Pokala V., Mishra S., Labudda K., Hunter B., and Stork P. J. (2006) Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 26, 2130–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roskoski R., Jr. (2010) RAF protein-serine/threonine kinases: structure and regulation. Biochem. Biophys. Res. Commun. 399, 313–317 [DOI] [PubMed] [Google Scholar]
- 33. Kuroda S., Ohtsuka T., Yamamori B., Fukui K., Shimizu K., and Takai Y. (1996) Different effects of various phospholipids on Ki-Ras-, Ha-Ras-, and Rap1B-induced B-Raf activation. J. Biol. Chem. 271, 14680–14683 [DOI] [PubMed] [Google Scholar]
- 34. Karhoff D., Sauer S., Schrader J., Arnold R., Fendrich V., Bartsch D. K., and Hörsch D. (2007) Rap1/B-Raf signaling is activated in neuroendocrine tumors of the digestive tract and Raf kinase inhibition constitutes a putative therapeutic target. Neuroendocrinology 85, 45–53 [DOI] [PubMed] [Google Scholar]
- 35. Rajakulendran T., Sahmi M., Lefrançois M., Sicheri F., and Therrien M. (2009) A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–545 [DOI] [PubMed] [Google Scholar]
- 36. Hu J., Stites E. C., Yu H., Germino E. A., Meharena H. S., Stork P. J., Kornev A. P., Taylor S. S., and Shaw A. S. (2013) Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stork P. J., and Schmitt J. M. (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 12, 258–266 [DOI] [PubMed] [Google Scholar]
- 38. Therrien M., Michaud N. R., Rubin G. M., and Morrison D. K. (1996) KSR modulates signal propagation within the MAPK cascade. Genes Dev. 10, 2684–2695 [DOI] [PubMed] [Google Scholar]
- 39. Nguyen A., Burack W. R., Stock J. L., Kortum R., Chaika O. V., Afkarian M., Muller W. J., Murphy K. M., Morrison D. K., Lewis R. E., McNeish J., and Shaw A. S. (2002) Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol. 22, 3035–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith F. D., Langeberg L. K., Cellurale C., Pawson T., Morrison D. K., Davis R. J., and Scott J. D. (2010) AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12, 1242–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McKay M. M., Ritt D. A., and Morrison D. K. (2011) RAF inhibitor-induced KSR1/B-RAF binding and its effects on ERK cascade signaling. Curr. Biol. 21, 563–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu J., Yu H., Kornev A. P., Zhao J., Filbert E. L., Taylor S. S., and Shaw A. S. (2011) Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. U.S.A. 108, 6067–6072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Riou P., Kjær S., Garg R., Purkiss A., George R., Cain R. J., Bineva G., Reymond N., McColl B., Thompson A. J., O'Reilly N., McDonald N. Q., Parker P. J., and Ridley A. J. (2013) 14-3-3 proteins interact with a hybrid prenyl-phosphorylation motif to inhibit G proteins. Cell 153, 640–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fischer T. H., Collins J. H., Gatling M. N., and White G. C. (1991) The localization of the cAMP-dependent protein kinase phosphorylation site in the platelet rat protein, rap 1B. FEBS Lett. 283, 173–176 [DOI] [PubMed] [Google Scholar]
- 45. Jagemann L. R., Pérez-Rivas L. G., Ruiz E. J., Ranea J. A., Sánchez-Jimenez F., Nebreda A. R., Alba E., and Lozano J. (2008) The functional interaction of 14-3-3 proteins with the ERK1/2 scaffold KSR1 occurs in an isoform-specific manner. J. Biol. Chem. 283, 17450–17462 [DOI] [PubMed] [Google Scholar]
- 46. Cacace A. M., Michaud N. R., Therrien M., Mathes K., Copeland T., Rubin G. M., and Morrison D. K. (1999) Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol. Cell. Biol. 19, 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Müller J., Ory S., Copeland T., Piwnica-Worms H., and Morrison D. K. (2001) C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol. Cell 8, 983–993 [DOI] [PubMed] [Google Scholar]
- 48. Zhou M., Horita D. A., Waugh D. S., Byrd R. A., and Morrison D. K. (2002) Solution structure and functional analysis of the cysteine-rich C1 domain of kinase suppressor of Ras (KSR). J. Mol. Biol. 315, 435–446 [DOI] [PubMed] [Google Scholar]
- 49. Ory S., Zhou M., Conrads T. P., Veenstra T. D., and Morrison D. K. (2003) Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr. Biol. 13, 1356–1364 [DOI] [PubMed] [Google Scholar]
- 50. McKay M. M., Ritt D. A., and Morrison D. K. (2009) Signaling dynamics of the KSR1 scaffold complex. Proc. Natl. Acad. Sci. U.S.A. 106, 11022–11027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kortum R. L., Johnson H. J., Costanzo D. L., Volle D. J., Razidlo G. L., Fusello A. M., Shaw A. S., and Lewis R. E. (2006) The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol. Cell. Biol. 26, 2202–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Athuluri-Divakar S. K., Vasquez-Del Carpio R., Dutta K., Baker S. J., Cosenza S. C., Basu I., Gupta Y. K., Reddy M. V., Ueno L., Hart J. R., Vogt P. K., Mulholland D., Guha C., Aggarwal A. K., and Reddy E. P. (2016) A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell 165, 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ponting C. P., and Benjamin D. R. (1996) A novel family of Ras-binding domains. Trends Biochem. Sci. 21, 422–425 [DOI] [PubMed] [Google Scholar]
- 54. Wohlgemuth S., Kiel C., Krämer A., Serrano L., Wittinghofer F., and Herrmann C. (2005) Recognizing and defining true Ras-binding domains I: biochemical analysis. J. Mol. Biol. 348, 741–758 [DOI] [PubMed] [Google Scholar]
- 55. Lavoie H., and Therrien M. (2015) Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 16, 281–298 [DOI] [PubMed] [Google Scholar]
- 56. Nassar N., Horn G., Herrmann C., Scherer A., McCormick F., and Wittinghofer A. (1995) The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase cRaf1 in complex with Rap1A and a GTP analogue. Nature 375, 554–560 [DOI] [PubMed] [Google Scholar]
- 57. Light Y., Paterson H., and Marais R. (2002) 14-3-3 antagonizes ras-mediated raf-1 recruitment to the plasma membrane to maintain signaling fidelity. Mol. Cell. Biol. 22, 4984–4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ottmann C., Yasmin L., Weyand M., Veesenmeyer J. L., Diaz M. H., Palmer R. H., Francis M. S., Hauser A. R., Wittinghofer A., and Hallberg B. (2007) Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 26, 902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Enserink J. M., Christensen A. E., de Rooij J., van Triest M., Schwede F., Genieser H. G., Døskeland S. O., Blank J. L., and Bos J. L. (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906 [DOI] [PubMed] [Google Scholar]
- 60. Gao L., Feng Y., Bowers R., Becker-Hapak M., Gardner J., Council L., Linette G., Zhao H., and Cornelius L. A. (2006) Ras-associated protein-1 regulates extracellular signal-regulated kinase activation and migration in melanoma cells: two processes important to melanoma tumorigenesis and metastasis. Cancer Res. 66, 7880–7888 [DOI] [PubMed] [Google Scholar]
- 61. Ribeiro-Neto F., Urbani J., Lemee N., Lou L., and Altschuler D. L. (2002) On the mitogenic properties of Rap1b: cAMP-induced G1/S entry requires activated and phosphorylated Rap1b. Proc. Natl. Acad. Sci., U.S.A. 99, 5418–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McKay M. M., Freeman A. K., and Morrison D. K. (2011) Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases 2, 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hisata S., Sakisaka T., Baba T., Yamada T., Aoki K., Matsuda M., and Takai Y. (2007) Rap1-PDZ-GEF1 interacts with a neurotrophin receptor at late endosomes, leading to sustained activation of Rap1 and ERK and neurite outgrowth. J. Cell Biol. 178, 843–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu C., Takahashi M., Li Y., Song S., Dillon T. J., Shinde U., and Stork P. J. (2008) Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol. Cell. Biol. 28, 7109–7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chadee D. N., and Kyriakis J. M. (2004) MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat. Cell Biol. 6, 770–776 [DOI] [PubMed] [Google Scholar]
- 66. Bhatt K. V., Hu R., Spofford L. S., and Aplin A. E. (2007) Mutant B-RAF signaling and cyclin D1 regulate Cks1/S-phase kinase-associated protein 2-mediated degradation of p27Kip1 in human melanoma cells. Oncogene 26, 1056–1066 [DOI] [PubMed] [Google Scholar]
- 67. Fernandez M. R., Henry M. D., and Lewis R. E. (2012) Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol. Cell. Biol. 32, 3718–3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu C., Takahashi M., Li Y., Dillon T. J., Kaech S., and Stork P. J. (2010) The interaction of Epac1 and Ran promotes Rap1 activation at the nuclear envelope. Mol. Cell. Biol. 30, 3956–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Franke B., Akkerman J. W., and Bos J. L. (1997) Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 16, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]