

Abstract
Meditopes are cyclic peptides that bind in a specific pocket in the antigen-binding fragment of a therapeutic antibody such as cetuximab. Provided their moderate affinity can be enhanced, meditope peptides could be used as specific non-covalent and paratope-independent handles in targeted drug delivery, molecular imaging, and therapeutic drug monitoring. Here we show that the affinity of a recently reported meditope for cetuximab can be substantially enhanced using a combination of yeast display and deep mutational scanning. Deep sequencing was used to construct a fitness landscape of this protein-peptide interaction, and four mutations were identified that together improved the affinity for cetuximab 10-fold to 15 nm. Importantly, the increased affinity translated into enhanced cetuximab-mediated recruitment to EGF receptor-overexpressing cancer cells. Although in silico Rosetta simulations correctly identified positions that were tolerant to mutation, modeling did not accurately predict the affinity-enhancing mutations. The experimental approach reported here should be generally applicable and could be used to develop meditope peptides with low nanomolar affinity for other therapeutic antibodies.
Keywords: antibody, cyclic peptide, directed evolution, epidermal growth factor receptor (EGFR), protein engineering
Introduction
The development of new antibody conjugation strategies is a fast expanding field with applications in targeted drug delivery and tumor imaging. Classical non-site-specific coupling of drugs or dyes to primary amines or thiols carries the disadvantage of generating a heterogeneous mixture of antibodies with different numbers of attached molecules as well as different sites of attachment. Therefore, in recent years, several interesting new types of biorthogonal chemistry have been explored for antibody conjugation (1–3). These include the ribosomal incorporation of non-canonical amino acids, chemo-enzymatic conjugation of payloads to the conserved N-glycan of native mAbs (4), the use of SNAP tags and other self-labeling protein and peptide tags (5, 6), or light-controlled covalent attachment of protein G (7, 8). In addition to these covalent antibody conjugation strategies, Donaldson et al. (9) developed a non-covalent affinity tag for therapeutic antibodies, which they coined a “meditope” (10). A meditope is a peptide that binds in a large pocket between the four immunoglobulin domains in the antigen-binding fragment (Fab)2 of antibodies (Fig. 1A). They identified a disulfide-constrained cyclic peptide (sequence, CQFDLSTRRLKC) that binds in this manner to the therapeutic anti-epidermal growth factor receptor (EGFR) antibody cetuximab (trade name, ErbituxTM). Because the binding site that this meditope recognizes is distinct from the pockets found in natural human antibodies, the tag serves as a specific non-covalent and paratope-independent handle that can be used to specifically bind cetuximab. Intriguingly, the site could be grafted onto another humanized therapeutic antibody (trastuzumab) by mutating 13 key interacting residues into their cetuximab counterparts.
FIGURE 1.
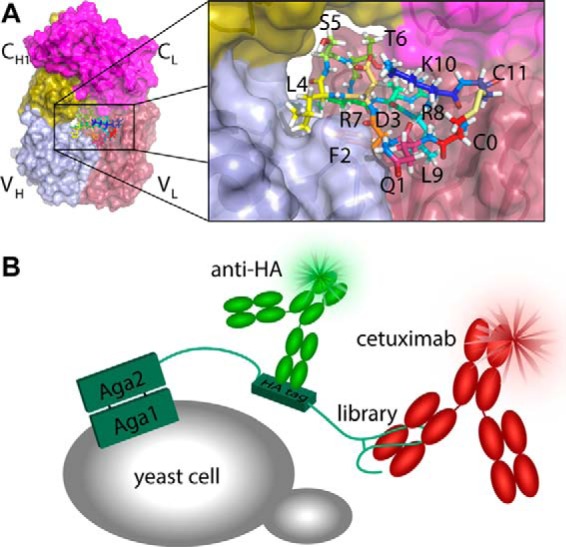
Improving cyclic peptide binding to cetuximab using yeast display. A, structure of the Fab fragment of cetuximab indicating the position of the meditope in the central cavity. B, schematic representation of the yeast display system. Simultaneous labeling of the HA tag and cetuximab with different fluorophores allows selection for binding to be normalized for variations in expression.
At present, many of the potential applications for the cetuximab meditope peptide are limited by its moderate affinity. The original meditope was identified in a phage display screen of random 10-mer peptides flanked by cysteines (11). As the sequence space of such a library is 106 times larger than the library that was used (12), systematic exploration of the local sequence space is likely to reveal affinity enhancing mutations. Yeast surface display (Fig. 1B) is particularly suitable for such affinity maturation (13). In contrast to in vitro molecular display technologies such as phage- or ribosome display, which require panning as a means of selection, Fluorescence Activated Cell Sorting (FACS) is used in yeast display, which offers more refined control of the selection pressure (14). Yeast display is also less vulnerable to unintended selection of the most infectious or fastest-replicating clones (15). Finally, the yeast secretory pathway acts as a filter, ensuring that only well folded full-length proteins and peptides will be displayed (16).
Because subtle affinity enhancements may escape detection in classical screens for a select number of “hit” sequences, recently high-throughput protein display technologies have been used in conjunction with deep sequencing before and after selection, a combination loosely referred to as “deep mutational scanning” (17–19). This strategy allows the construction of fitness landscapes of the selected property (i.e. affinity) in which subtle effects of individual mutations can be discerned by the degree to which they are enriched or depleted by the selection. Deep mutational scanning has been applied successfully in combination with computational protein design for the engineering of enzyme inhibitors (20) and antibody binding proteins (21), for the evolution of antibodies (22–24) and T-cell receptors (25, 26), for epitope mapping (27, 28), for engineering immune co-receptors (29), as well as for establishing protein structure-function relationships (30, 31).
In this work, we used yeast display and deep mutational scanning to improve the affinity of the meditope peptide for the therapeutic antibody cetuximab by screening all single and a select set of double amino acid substitutions. The effects of these mutations were quantified using fluorescence anisotropy titration experiments, and the best variant was tested for improved cancer cell targeting. The experimental approach was also compared with in silico mutagenesis with Rosetta using the crystal structure of the meditope-cetuximab complex to determine whether subtle affinity-enhancing mutations could also be identified computationally.
Results
Display of the Cetuximab Meditope on the Yeast Cell Surface
The phage display screen that identified the cetuximab meditope sequence CQFDLSTRRLKC (hereafter called Md1) also reported three other binding sequences, CVWQRWQKSYVC (Md2), CQYNLSSRALKC (Md3), and CMWDRFSRWYKC (Md4) (11). Because the cetuximab affinities of these other three peptides were not available, we synthesized fluorescently labeled versions of all four peptides and determined their affinity for cetuximab in a fluorescence polarization assay, yielding Kd values of 134.6 ± 5.6 nm for Md1, 371 ± 58 nm for Md2, and 739 ± 115 nm for Md3 (Fig. 2B). For Md4, no significant binding was observed up to 10 μm cetuximab. Next, we tested whether these disulfide-constrained peptides could be efficiently displayed and cyclized by the yeast secretory machinery. Correct folding and display were analyzed for each of the four peptide sequences by using FACS analysis to study binding of fluorescently labeled cetuximab at various concentrations. Remarkably, cetuximab binding was only observed for the original meditope sequence and not for any of the other three sequences (Fig. 2B). The affinity of the interaction between cetuximab and the meditope peptide displayed on the yeast surface seemed to be slightly weaker than that determined in the fluorescence polarization assay, but an accurate determination of Kd was precluded by the lack of a plateau. Especially for weaker interactions, dissociation of the bound cetuximab during washing may result in an underestimation of the affinity. The lack of cetuximab binding observed for Md2 and Md3, which did respond in the fluorescence polarization assay, suggests that these peptides were not cyclized efficiently by the yeast secretory machinery. Yeast display has been used extensively on different types of proteins (25, 32–36) as well as cysteine knot cyclic peptides (37), but those molecules possess a well defined tertiary structure that will favor correct disulfide pairing during oxidative folding in the endoplasmic reticulum. Simple disulfide-constrained cyclic peptides lack such a stabilizing scaffold, and therefore they may form incorrect disulfide bridges with other proteins.
FIGURE 2.
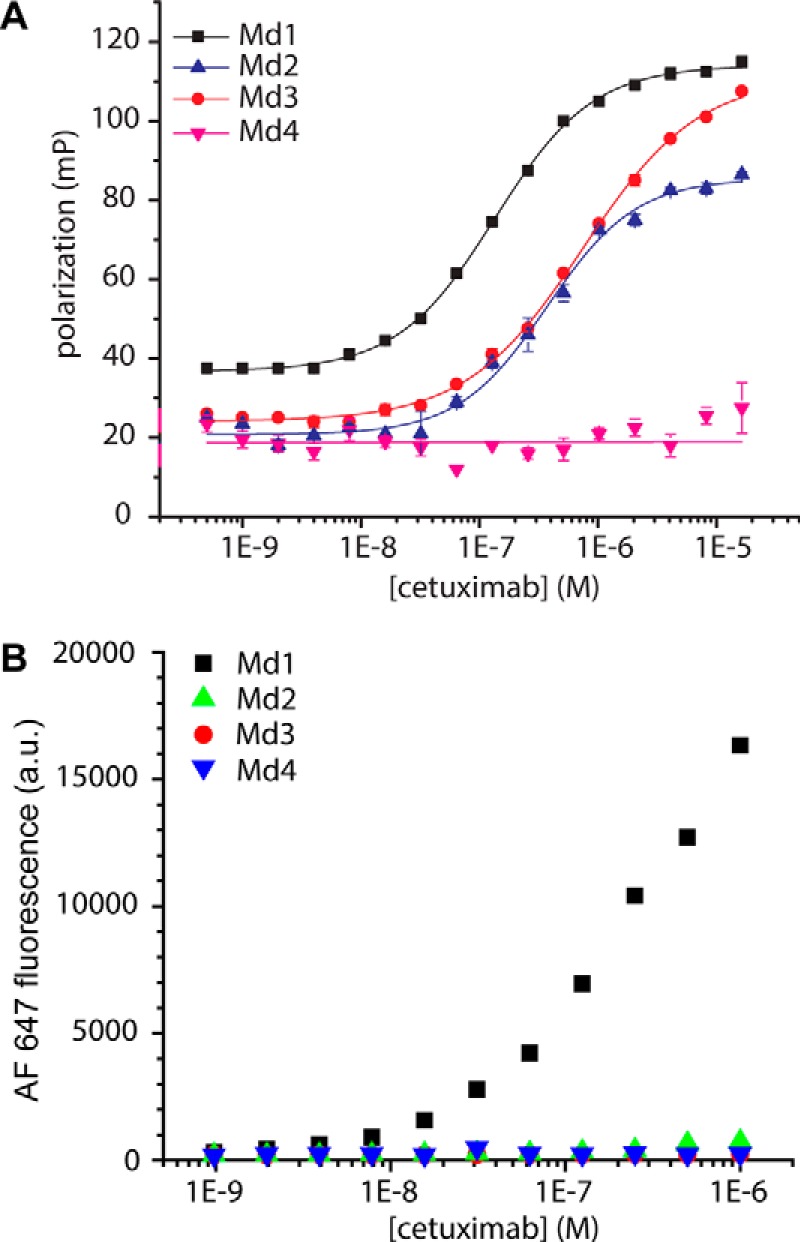
Display of cyclic meditope peptide on yeast cells. A, cetuximab titration to 10 nm FITC-labeled peptides (supplemental Table S2, numbers 1–4) in PBS solution, pH 7.4, with 1 mg ml−1 BSA. Fluorescence polarization was used as a readout. Data represent mean ± S.D. from duplicate experiments. B, titration of cetuximab to yeast displaying the indicated cyclic peptides. Cells were labeled with the indicated concentrations of cetuximab conjugated to Alexa Fluor 647 as well as mouse anti-HA and an Alexa Fluor 488-conjugated goat anti-mouse secondary antibody. The flow cytometric Alexa Fluor 647 mean fluorescence of the Alexa Fluor 488-positive cells was used as a readout.
Systematic Screen of Single Amino Acid Substitutions
Having established that Md1 has the highest cetuximab affinity and is correctly displayed on the yeast surface, we made 10 libraries of Md1 each harboring a single degenerate NNK codon (the cysteines were not mutated). The pooled single NNK libraries were sorted in three consecutive rounds for cetuximab binding. Two different selection stringencies were used in parallel using either 100 or 30 nm cetuximab (Fig. 3A). Sequence analysis of 12 single clones obtained after the third sorting round revealed three mutations that were enriched, Q1V, S5G, and K10R. These three mutant clones were cultured individually and analyzed by FACS, showing a modest increase in cetuximab binding (Fig. 3B). To quantify the effect of each mutation, we synthesized fluorescently labeled meditope peptides containing either a single mutation or harboring combinations of two or three of these mutations. After verification of correct cyclization by mass spectrometry, the dissociation constants for cetuximab-peptide complexes were determined in a fluorescence polarization assay (Fig. 3C and Table 1). The effects of the individual mutations on cetuximab binding were modest, with affinity increases of 1.2–1.8-fold. Fortunately, these subtle effects were fully additive, with the Q1V/S5G/K10R triple mutant showing a 4-fold lower dissociation constant of 30.3 ± 0.9 nm.
FIGURE 3.

Selection of improved mutants from single amino acid substitution libraries. A, FACS sorting of pooled libraries. Libraries were pooled prior to sorting and incubated with either 30 or 100 nm Alexa Fluor 647-conjugated cetuximab, mouse anti-HA, and an Alexa Fluor 488-conjugated goat anti mouse secondary antibody. Plots show the library and sorting gate used in the first (left), second (middle), and third (right) sorting rounds. B, FACS histograms of cells expressing either the original meditope or mutants Q1V, S5G, or K10R. Cells were labeled with 30 nm Alexa Fluor 647-conjugated cetuximab. Populations on the left are non-expressing cells, whereas populations on the right are expressing cells. C, fluorescence polarization assay. Cetuximab was titrated to 10 nm of the indicated synthetic peptides in 50 mm sodium phosphate, pH 7.0, 100 mm sodium chloride, 1 mg ml−1 BSA. Indicated mutations are with respect to Md1. For exact sequences with linkers and flanking residues see supplemental Table S2 peptides 5–12. Error bars represent mean ± S.D. from duplicate measurements.
TABLE 1.
Dissociation constants of mutant peptides
| Sequencea | Kd b |
|---|---|
| nm | |
| Meditope 1 | 129.4 ± 4.7 |
| Q1V | 111.5 ± 3.6 |
| S5G | 113.9 ± 3.1 |
| K10R | 81.3 ± 1.9 |
| Q1V/S5G | 69.4 ± 2.2 |
| Q1V/K10R | 63.2 ± 1.2 |
| S5G/K10R | 41.1 ± 1.2 |
| Q1V/S5G/K10R | 30.3 ± 0.9 |
a Exact peptide sequences (including labels, linkers, and flanking residues) are in supplemental Table S2, numbers 5–12.
Constructing a Local Fitness Landscape with Next Generation Sequencing
Because the three identified mutations showed only minor improvements in affinity, we wondered whether other subtle mutations had evaded detection because of the limited number of analyzed clones. To determine the optimal amino acid at every position, we applied deep mutational scanning, in which the amino acid frequencies at every position are determined by deep sequencing both before and after selection. The degree of enrichment or depletion of an amino acid reflects its positive or negative effect on the binding affinity. Using this information, a “fitness landscape” can also be created to identify positions that tolerate diversity better than others, which is useful for designing double mutation libraries.
Fig. 4A shows the deep mutational scanning strategy used in this study (see under “Materials and Methods” for a detailed description). Briefly, the 10 single NNK libraries were sorted separately for two consecutive rounds using 100 nm cetuximab. In the first round, a rectangular sorting gate was drawn to enrich all functional binders, regardless of their specific affinities (supplemental Fig. S1a). In the second sorting round, a triangular sorting gate was drawn such that half of the expressing population of a control sample displaying Md1 fell within this gate (supplemental Fig. S1b). In this way, any favorable mutants would be enriched more than the original, as a larger fraction of their expressing population would fall within the sorting gate.
FIGURE 4.

Deep sequencing analysis of separately sorted single NNK libraries. A, schematic overview of the deep mutational scanning strategy. B, heat map of robust enrichment ratios (see under “Materials and Methods”) of all single amino acid substitutions of the original cetuximab meditope. All RERs above 1 are indicated in the respective squares. At each position, the original residue is encircled.
DNA was extracted from samples of each unsorted and each twice-sorted population. The cyclic peptide-encoding regions were amplified by PCR, during which Multiplex IDentifier (MID) barcode sequences were incorporated. Using six different forward and six different reverse primers containing unique MIDs, each sub-library (e.g. “NNK10 unsorted”) was tagged with an unambiguous combination of MIDs (Fig. 4A). Only after each sub-library was uniquely tagged were they combined into a single sequencing sample. Sequencing was done using the Illumina MiSeq platform. Paired-end sequencing was used, because the read-length of the Illumina method was too short to capture the cyclic peptide sequence and both MIDs in a single read (see under “Materials and Methods”).
From the sequence analysis, enrichment ratios were calculated. To minimize the effect of sampling error on the enrichment ratios, we defined the “robust enrichment ratio” (RER) as a metric, which in effect is equal to the lower 95% confidence limit of the enrichment ratio (see under “Materials and Methods” for the definition). The robust enrichment ratios were represented as a heat map (Fig. 4B). The only mutations with higher RERs than the original residues at their respective positions were the three previously identified beneficial mutations, Q1V, S5G, and K10R. This confirmed that our deep mutational scanning strategy was able to correctly identify mutations with enhanced affinities, and this suggested that no other beneficial single mutations had been previously overlooked. In addition to identifying beneficial mutations, the analysis also revealed that some positions were more tolerant to mutation than others. Notably, at positions 1 and 3 and to some extent 5 and 6, many residues had RERs above 1, meaning that these mutants retained at least significant binding. In contrast, substitutions at positions 2, 7, 8, and 9 abrogated binding completely. At position 4, only one mutation (L4M) had an RER above 1, but otherwise this position was also intolerant to mutation.
Affinity Maturation Using Double Amino Acid Substitution Libraries
The effects of the three mutations that had been identified thus far were largely additive, but screening multiple amino acid substitution libraries could identify mutations that are cooperative, i.e. the effect of one mutation is enhanced by the presence of another mutation. Deep mutational scanning of the single mutant libraries revealed that positions 1, 3, 5, and 6 tolerated diversity. We therefore created five double NNK libraries harboring degenerate codons at positions 1 + 3, 1 + 5, 1 + 6, 3 + 5, and 5 + 6. These double amino acid substitution libraries were prepared, sorted, and sequenced in the same way as the single NNK libraries (supplemental Fig. S1). Analysis of the sequencing data confirmed that mutations Q1V and S5G were more strongly enriched compared with the respective original amino acids in the libraries NNK 1 + 3, 1 + 5, 1 + 6, and 3 + 5 (Fig. 5A). Only in NNK 5 + 6 was the original serine at position 5 more enriched than glycine. Two additional mutations, D3N and T6I, were found that had high RERs. In both the NNK 1 + 3 and NNK 3 + 5 libraries, an aspartate was more enriched at position 3 than the original asparagine, which suggested that the D3N mutation is only favorable for binding in the context of other mutations at positions 1 and/or 5. Other potentially beneficial mutations that only appeared in the double amino acid substitution libraries are T6I and T6V. These both had higher RERs (2.5 and 2.4, respectively) than the original threonine (RER 1.4) in the NNK 1 + 6 library. In the NNK 5 + 6 library, the threonine at position 6 had a higher enrichment ratio (8.5) than isoleucine (3.5) or valine (2.4). This suggests that mutating threonine 6 to a hydrophobic valine or isoleucine is favorable only in the context of a second mutation at position 1, but it is disfavored to some extent in the context of another mutation at position 5. In addition to the enrichment of amino acids, we also assessed the enrichments of specific pairs of mutations, to determine which residues were frequently selected together (supplemental Figs. S2–S6). From these analyses, mutations D3N and T6I also emerged as likely candidates that could further improve the affinity.
FIGURE 5.

Deep sequencing analysis of separately sorted double NNK libraries. A, heat map of robust enrichment ratios (see under “Materials and Methods”) of double amino acid substitutions of the original cetuximab meditope. All RERs above 1 are indicated in the respective squares. At each position, the original residue is encircled. B, cetuximab titration to 10 nm FITC-conjugated synthetic peptides in PBS, pH 7.4 1 mg ml−1. Fluorescence polarization was used as a readout. Data points represent mean ± S.D. of triplicate measurements. The indicated mutations are with respect to Md1. For exact peptide sequences, see supplemental Table S2 peptides 13–16.
To assess whether the D3N and T6I could further enhance the affinity of the Q1V/S5G/K10R meditope variant, three fluorescently labeled peptides were synthesized, carrying D3N, T6I, or both in addition to the Q1V, S5G, and K10R mutations. Fluorescence polarization assays showed that each of the single mutations enhanced cetuximab binding by a factor of 2, yielding Kd values of 14.5 ± 0.8 and 16.8 ± 2.1 nm for the tetramutants Q1V/D3N/S5G/K10R and Q1V/S5G/T6I/K10R, respectively. A pentamutant peptide carrying Q1V/D3N/S5G/T6I/K10R had a dissociation constant of 15.8 ± 3.9 nm. Apparently, the two additional mutations indeed improved the affinity further by a factor 2, but their effects were not additive. These combined mutations thus yielded an almost 10-fold increase in affinity.
Comparing the Local Fitness Landscape with in Silico Rosetta Simulations
The availability of the crystal structure of the complex between the original meditope peptide (Md1) and cetuximab (9) provided an opportunity to compare the fitness landscape determined using deep mutational sequencing with one obtained based on in silico mutagenesis. Such a comparison would serve as a rigorous test to assess whether in silico modeling could substitute for experimental screening. The well established Rosetta protein design software was used for in silico modeling, which employs an empirical score function to find the lowest energy conformation of mutant proteins or peptides (38, 39). Using the published crystal structure (Protein Data Bank code 4GW1) as a starting point, all single amino acid substitutions were introduced in the peptide, and the binding of each mutant was compared with the original meditope. This strategy is effectively a computational counterpart of the single NNK screen carried out with deep mutational scanning.
Fig. 6A shows the results of the computational screen as a heat map depicting the difference in binding scores between the mutants and the original peptide. In this heat map, negative values (Fig. 6A, black or dark gray areas) represent binding scores that were larger than that of the non-mutated peptide, suggesting affinity-improving mutations. Highly positive values (Fig. 6A, white or light gray areas), however, indicated a decreased affinity. The simulations globally reflected the experimental observations that positions 1, 3, 5, and 6 are most tolerant to mutation. The computational modeling suggested that especially positions 4, 7, and 8 and to a lesser extent positions 2 and 9 were intolerant to mutation, which is also in agreement with experimental results. In the model position 10 was more tolerant to mutation than observed experimentally.
FIGURE 6.

In silico modeling of meditope mutations. A, heat map of interaction free energies of cetuximab with single amino acid substitutions of the meditope generated by Rosetta modeling. Negative values (improved binding) is represented with dark shades of gray, and positive values (weaker binding than the original meditope) are represented with light shades of gray. Positions are shown on the horizontal axis. B, heat maps of interaction free energies of cetuximab with amino acid substitutions at position 6 of the meditope where position 5 is mutated to tryptophan (left), phenylalanine (middle), or tyrosine (right), generated by Rosetta modeling. Values are normalized to the respective single amino acid substitution at position 5. C, model of double mutant S5Y/T6M showing the position of the side chains that fill the empty space in the binding pocket. D, model of double mutant S5Y/T6M showing the possible hydrogen bond between tyrosine 5 of the meditope and valine 152 of the antibody. E and F, model of mutant D3R showing how the side chain of arginine 3 of the mediotope may fill empty space within the binding pocket (E) and possibly form hydrogen bonds with glutamine 150 and alanine 176 of the antibody (F). G, fluorescence polarization assay. Cetuximab was titrated to 10 nm of the indicated mutant peptides in PBS, pH 7.4, 1 mg ml−1 BSA. Error bars represent mean ± S.D. of triplicate measurements. The indicated mutations are with respect to Md1. For exact sequences see supplemental Table S2 peptides 13, 14, and 18–21.
Of the three beneficial mutations that were experimentally selected, only K10R was correctly identified as an improving mutation by Rosetta, whereas Q1V and S5G did not have higher interface scores than the original peptide. The model suggested several other mutations with high interface scores, in particular mutations of serine 5 into any of the three aromatic residues. Given the fact that positions 5 and 6 are both tolerant to mutation, adjacent, and situated deeply within the pocket where there is some empty space, mutations at position 6 in combination with aromatics at position 5 were also explored (Fig. 6B). This analysis suggested that methionine at position 6 could improve the affinity in the context of an additional S5Y or S5F mutation but not in the original background. Aromatic residues at position 5, especially in combination with the methionine at position 6, would better fill the empty space in the back of the binding pocket and pack against the constant domain of cetuximab (Fig. 6C). The side chain of a tyrosine at position 5 can even come into hydrogen-bonding distance of the backbone carbonyl of valine 152 (Kabat numbering) (40) of the CH1 domain (Fig. 6D). Besides aromatics at position 5, the model also predicted favorable binding for mutation D3R. The side chain of an arginine mutation at position 3 seemed to occupy a considerable amount of empty space and pack against the constant domains (Fig. 6E), forming hydrogen bonds with the side chain of glutamine 150 and with the carbonyl of alanine 176, both in the CH1 domain of cetuximab (Fig. 6F).
Peptides carrying these single mutations or combinations of two or three mutations (in the background of the Q1V/S5G/K10R triple mutation) were made, and their affinities were measured by fluorescence polarization (Fig. 6G). None of these peptides had improved affinities, however. The peptide carrying S5Y had a Kd of 86.3 ± 6.9 nm. Compared with 28.2 ± 1.8 nm for the Q1V/S5G/K10R peptide or to 63.2 ± 1.2 nm for the peptide carrying only Q1V and K10R (Fig. 3D), this was clearly not the improvement that the Rosetta simulations suggested. Similarly, the T6M mutant had a Kd of 162 ± 7 nm, and the combination of these two mutations (which the modeling predicted to be highly cooperative) resulted in a Kd of 487 ± 149 nm. The D3R mutation had a reduced affinity (Kd of 186 ± 29 nm), and for a peptide with Q1V/D3R/S5Y/T6M/K10R, no binding was observed. Taken together, the in silico modeling was able to identify positions that were amenable to mutation but much less successful in predicting mutations that improved the affinity. Several other studies also found that although in silico design methods can be used effectively to provide a global insight into mutable residues and even provide the first important step in the de novo design of interaction interfaces, these designs invariably require experimental affinity maturation to fully optimize the binding strength (20, 21). Our results show that this need for experimental affinity maturation persists even for concave binding pockets with structurally well characterized peptide ligands.
Meditope Targeting to EGFR Overexpressing Cancer Cells in a Cetuximab-dependent Manner
Having improved the affinity of the cetuximab meditope peptide from 130 to 15 nm, we assessed whether the increased affinity also translates into improved cetuximab-mediated EGFR targeting on cancer cells. A431 skin epidermis carcinoma cells, which show highly elevated EGFR expression levels, were incubated with cetuximab and FITC-conjugated meditope peptides (Fig. 7). Cells stained with the tetra-mutant Q1V/D3N/S5G/K10R were almost an order of magnitude more fluorescent than those stained with the original meditope (Fig. 7A). In agreement with observations by Donaldson et al. (9), significant binding of the original meditope peptide was only observed at a peptide concentration of 500 nm. In contrast, the mutant meditope showed significant binding at 50 nm, and the fluorescence intensity at 100 and 500 nm was consistently higher than for the original peptide (Fig. 7B). The substantially lower concentration of meditope peptide that is required enhances the feasibility of the non-covalent cetuximab “labeling” approach for applications in molecular imaging and targeted drug therapies.
FIGURE 7.

Cetuximab-mediated targeting of meditopes to EGFR-overexpressing cancer cells. A, A431 cells were incubated with 5 nm unlabeled cetuximab and 50 nm FITC conjugated peptide (supplemental Table S2, numbers 13 and 15) and analyzed by flow cytometry. Mutations are with respect to Md1. B, concentration-dependent meditope binding. A431 cells were incubated with cetuximab, and the indicated concentrations of cyclic peptides as in A and analyzed by flow cytometry. Data represent mean ± S.D. from duplicate experiments. The control consisted of 500 nm of the respective meditope peptides without cetuximab and represents background binding to the cells due to the hydrophobic nature of the peptide.
Discussion
In this study, we showed that the affinity of a cyclic peptide meditope for the therapeutic antibody cetuximab, originally obtained from phage display screening, could be improved 10-fold using a combination of yeast display and deep mutational scanning. Construction of an accurate map of the fitness landscape of the meditope-antibody interaction allowed identification of four mutations that by themselves increased the affinity only subtly, but together increased the affinity from Kd 130 to 15 nm. This affinity increase also translated into an order of magnitude more efficient cetuximab-mediated targeting of EGFR-overexpressing tumor cells. The strategy developed in this work can be more generally applied for affinity maturation of protein-protein or protein-peptide interactions, for example to develop peptide meditopes targeting other antibodies.
Most previously described studies have used targeted library screening and deep sequencing for epitope mapping or to identify which positions in a protein are functionally important (27, 28, 30). Some also used deep mutational scanning with the explicit goal of improving the affinity, however. Forsyth et al. (22) used mammalian display and 454 (Roche Applied Science) sequencing to improve the affinity of a humanized version of cetuximab for EGFR, realizing a 4-fold improvement and correctly identifying many affinity-improving mutations. Reich et al. (14) successfully used yeast display and Next Generation Sequencing to rank over a thousand BH3 peptides for their affinity to the anti-apoptotic protein Bcl-Xl. Koenig et al. (24) have used phage display with deep mutational scanning of single and triple mutation libraries to generate a Fab fragment with sub-nanomolar affinities to two unrelated antigens.
In previous studies that use systematic single site saturation mutagenesis, sub-libraries were typically pooled before the selection (17–19, 22, 23, 28, 29). This approach causes a problem in identifying which amino acid is the best choice at each position, however. Sequences harboring mutations at positions that tolerate diversity will be more strongly enriched than the corresponding wild-type residues simply because they will not harbor deleterious mutations at intolerant positions. Therefore, if libraries are pooled before selections, mutations with higher enrichment ratios than the wild-type residue do not necessarily confer increased affinity. This effect becomes especially pronounced if the number of randomized positions in the library is small. To work around this problem, we performed the selections of the different single substitution libraries separately and employed a DNA barcoding scheme for identification of sequencing reads. This strategy carried an additional advantage as it allowed us to quickly determine which positions tolerated diversity better than others by a simple FACS analysis. This information is important for designing multiple amino acid substitution libraries. Separate selections are admittedly more labor intensive than pooled ones, especially if the number of single-site libraries increases, but adjustment of the barcoding strategy could help to remedy this issue. A possible strategy for larger proteins could be to incorporate unique barcode sequences within the plasmid to identify post-sequencing from which library each read was derived.
Another methodological issue that we addressed is how to deal with sampling error. Enrichment ratios are usually calculated by dividing the frequency of a particular mutation in the selected population by the corresponding frequency in the unselected library (17–19). However, it is easier to enrich rare amino acids than frequent ones. If a mutation of interest is rare in the unselected library (e.g. counted only 30 times), sampling error can lead to relatively large overestimations of the true enrichment ratio. We found the effect of sampling error to be particularly strong when analyzing the enrichment of pairs of amino acids in the double NNK libraries. Many pairs occurred less than 100 times, and although the normal enrichment ratio gave a qualitative indication of which pairs were enriched, quantitatively it was unreliable. Some pairs had absurdly high enrichments of over 100-fold. We therefore defined a new parameter called the “robust enrichment ratio,” which corrects for sampling errors (see under “Materials and Methods”). In effect this metric is the enrichment ratio's lower 95% confidence limit. The robust enrichment ratio is a useful metric in all cases where sampling error plays an important role (i.e. when individual clones are read less than 100 times). This is the case when studying the effects of pairs of mutations in double NNK libraries, when scanning large proteins, and when different deep sequencing technologies are used (e.g. 454 sequencing, which yields longer reads, but substantially lower coverage).
Several interesting applications could be explored using the meditope technology. Conjugation of the peptide to a dye or MRI contrast agent could be used for in vivo (tumor) imaging in patients undergoing antibody treatment. Non-covalent antibody conjugates have been explored by several groups (41–43), mostly making use of generic Fc-binding protein domains (e.g. Staphylococcus aureus protein A). Contrary to covalently attached agents, a non-covalent agent would exhibit fast pharmacokinetic clearance rate in the free form, whereas the rate of clearance of the complex would essentially be dictated by the antibody. Thus, the conjugated form would remain in the body for a more extended time period, which would be desirable for imaging. Using a peptide handle specific for the therapeutic antibody, rather than protein A, would prevent the dye or contrast agent from binding to endogenous IgG1 in the bloodstream before it can reach the target site.
Another application that could benefit from an antibody handle is testing patients for the presence of anti-drug antibodies (ADAs). The presence of ADAs indicates an immunogenic response to the drug and usually results in the drug being ineffective. To detect ADAs, the drug-ADA complexes must be captured intact from plasma, which is rich in background IgGs. Although the target protein of the drug can be used as a bait for this purpose, many ADAs are raised against the antigen-binding site. These will dissociate during the capture of the drug and evade detection.
Avery et al. (44) recently reported the construction of a very potent non-covalent cetuximab ligand by fusing the meditope peptide Md1 to a fragment of protein L, a domain originating from Finegoldia magna that interacts specifically with immunoglobulin κ-light chains (45). Fusion to protein L increased the affinity for cetuximab by 3 orders of magnitude. Although the affinity increase realized in our study was smaller, in our case the affinity enhancement did not rely on fusions with bacterial proteins. In addition to the fact that protein L fusions will also bind to endogenous IgGs, protein L is highly immunogenic (46–48), and fusing it to the meditope will probably be detrimental for in vivo applications. The meditope technology could be a general strategy for non-covalent site-specific antibody binding. It has already been shown that the meditope-binding site of cetuximab can be grafted on other therapeutic antibodies (9). Moreover, pockets of similar dimensions are present in the Fab regions of all IgGs. Having shown that cyclic peptides with low nanomolar affinity can be developed for the cetuximab-binding site, it is likely that meditope sequences specific for other therapeutic antibodies can be developed using the approach presented here.
Materials and Methods
Molecular Biology
The vector pCTCON2 was a kind gift from Prof. Dr. Dane Wittrup (49). A double-stop codon was introduced between the NheI and BamHI sites within this vector using the QuikChange II site-directed mutagenesis kit (Agilent Genomics) and the oligonucleotides pCT-Stop-F and pCT-Stop-R (exact sequences in supplemental Table S1) according to the manufacturer's instructions. The vector was linearized by digesting 3 μg of DNA with 60 units of NheI-HF (New England Biolabs) and 60 units of BamHI (New England Biolabs) in 60 μl of CutSmart buffer (New England Biolabs) at 37 °C for 1 h. Linear vector DNA was purified by electrophoresis using a 1% agarose/TAE gel and extracted from the gel using the QIAquick gel extraction kit (Qiagen). Inserts for the Md1, Md2, Md3, and Md4 peptides were made as follows: 200 nm (final concentration) of each of four primers was mixed in a total volume of 50 μl, containing 25 μl of 2× Phusion High Fidelity PCR master mix with HF buffer (New England Biolabs). Two of the primers, Constant-F and Constant-R, were used for all inserts, and two primers were insert-specific (e.g. “Md1-F” and “Md1-R”). The reaction was performed by initially heating at 98 °C for 2 min, five subsequent cycles of denaturation at 98 °C for 10 s, and annealing/extension at 72 °C for 30 s, and a final extension at 72 °C for 10 min. 1 μl of the product was used as a template in a 50-μl reaction containing 25 μl of 2× Phusion High Fidelity PCR master mix with HF buffer (New England Biolabs) and 500 nm Constant-F and Constant-R primers. Thermo-cycling was done as described above, but 35 cycles were used instead of 5. The products were purified using the QIAquick PCR purification kit (Qiagen). Inserts of libraries were prepared in a two-step manner as follows: 1 μm primers Constant-F, Constant-R, and one library-specific primer (e.g. “NNK1”) were mixed in a 50-μl reaction containing 25 μl of 2× Phusion High Fidelity PCR master mix with HF buffer (New England Biolabs). The reaction was done in 10 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s, extension at 72 °C for 10 s, and a final elongation step at 72 °C for 1 min. 1 μl of the reaction was used as a template for a second reaction containing 1 μm Constant-F and Constant-R primers and 50 μl of 2× Phusion High Fidelity PCR master mix with HF buffer (New England Biolabs) in a total volume of 100 μl. Cycling was done as for the first reaction, but 35 cycles were used instead of 10. These inserts were purified using the QIAquick PCR purification kit (Qiagen).
Yeast Transformation
pCTCON2 plasmids and inserts were transformed into yeast strain EBY100 using the LiAc/PEG/ssDNA method as described (50–52). For clonal inserts and single NNK libraries, the protocol for transformation of frozen yeast was followed (50, 51). EBY100 was cultured in YPD medium, 2% d-(+)-glucose (Sigma), 2% Peptone from casein (Merck Millipore), and 1% yeast extract (Merck Millipore) at 30 °C for 16 h. The pre-culture was diluted in 400 ml of fresh 2×YPD medium at an OD600 of 0.5 and cultured at 30 °C with shaking at 220 rpm until the OD600 reached 2.0. Cells were then pelleted for 30 min at 3000 × g, washed twice, first with 200 ml of ice-cold sterile water and then with 80 ml ice-cold sterile water, pelleting at 3000 × g each time. The cells were resuspended in 10% dimethyl sulfoxide (DMSO), 5% glycerol in sterile water. 50-μl aliquots (containing ∼3 × 1010 cells) were frozen at −80 °C for long term storage. For each transformation reaction, an aliquot was thawed and centrifuged at 13,400 × g for 30 s, and the supernatant was replaced with 346 μl of ice-cold Transformation Buffer. Yeast was kept on ice until heat-shock. Transformation Buffer was always prepared on the day of use by mixing (per reaction) 260 μl of a 50% (w/v) sterile aqueous solution of polyethylene glycol (PEG) Mr 3350 (Sigma), 36 μl of sterile 1 m LiAc (Sigma), and 50 μl of 2 mg ml−1 single-stranded DNA solution. DNA from salmon testes (Sigma) was dissolved in 10 mm Tris-HCl (Sigma), 1 mm EDTA (Sigma), pH 8.0, filter-sterilized, denatured at 95 °C for 10 min, chilled on ice, and immediately used or stored at −20 °C to prevent re-annealing. 100 ng of linearized vector DNA and 30 ng of insert DNA were mixed with the yeast and Transformation Buffer, and water was added to make a total volume of 360 μl. Yeast were then heat-shocked at 42 °C for 50 min with regular inversion to prevent settling of the cells. After the heat-shock, the cells were pelleted at 13,400 × g for 2 min, resuspended in 200 μl of sterile water, and plated on SD-CAA agar (2 g liter−1 d-(+)-glucose (Sigma), 6.7 g liter−1 Difco yeast nitrogen base without amino acids (BD Biosciences), 5 g liter−1 BactoTM casamino acids (BD Biosciences), 5.4 g liter−1 Na2HPO4 (Calbiochem), 8.56 g liter−1 NaH2PO4·H2O (Calbiochem), 15 g liter−1 agar (Merck Millipore). Colonies were counted after 3 days of incubation at 30 °C. For clonal cultures, a single colony was picked, whereas for single NNK libraries, colonies were gently dislodged with a sterile Drigalski spatula, and the complete libraries were kept in liquid culture. For double NNK libraries, the high efficiency transformation protocol was followed (52). EBY100 was cultured and washed as described before but was not frozen. Instead, 3 × 1010 cells were immediately resuspended in 346 μl of Transformation Buffer. 1 μg of linear vector DNA and 30 ng of the respective inserts was added, and the volume was made up to 360 μl with sterile water. From there, the transformation was done as described above. Double NNK libraries were not plated after transformation, but were resuspended in 5 ml of liquid SDCAA medium, pH 4.5 (as the SDCAA plates described above, but without agar and with 3.92 g of anhydrous citric acid (Amresco), and 6.07 g of trisodium citrate dehydrate (Calbiochem) instead of the phosphate salts). 0.1% of each library was plated onto SDCAA agar to determine the library size, and colonies were counted after 3 days at 30 °C.
Yeast Display and FACS
For yeast display, the protocol by Chao et al. (49) was used as a reference. Clonal yeast cultures and libraries were inoculated at an initial OD600 of 0.01 in 5 ml of SDCAA medium in sterile 14-ml Falcon culture tubes with snap-caps (BD Biosciences) and cultured for 16–20 h at 30 °C and shaking at 220 rpm. Cultures were then diluted to an OD600 of 0.1 in fresh SDCAA medium and cultured until the OD600 reached 0.4. Then the cells were centrifuged at 3000 × g and resuspended in 5 ml of SGCAA medium, which was prepared as SDCAA, but with the glucose replaced by 2% d-(+)-galactose (Sigma). The yeast was then cultured for another 2 days at 20 °C and shaking at 220 rpm to induce peptide display. Cetuximab (Erbitux, Merck) was obtained from the Catharina Hospital (Eindhoven, the Netherlands) and conjugated with Alexa Fluor 647 using an amine-reactive N-hydroxysuccinimide ester. The coupling reaction was done in PBS at room temperature using a 20-fold molar excess of the dye. Excess of dye was removed using a PD-10 buffer exchange column (GE Healthcare), and the conjugate was concentrated using an Amicon centrifuge concentrator with a molecular mass cutoff of 50 kDa. The concentration and dye-to-antibody ratio were determined by UV-visible absorbance at 280 and 633 nm using a NanoDrop ND1000 spectrophotometer and the molar extinction coefficients of 210,000 m−1 cm−1 at 280 nm and 264,000 m−1 cm−1 at 651 nm. The dye-to-antibody ratio was ∼4. Yeast displaying the cyclic peptides were washed with 1 ml of PBSF (PBS, pH 7.4, with 1 mg ml−1 BSA) and incubated with 1:250 mouse anti-HA monoclonal antibody (Thermo Fisher Scientific) diluted in PBSF for 30 min at room temperature. The cells were then washed with 1 ml of ice-cold PBSF and incubated for 20 min on ice with 30 or 100 nm Alexa Fluor 647-conjugated cetuximab and 1:100 Alexa Fluor 488-conjugated goat anti-mouse monoclonal IgG1 (Thermo Fisher Scientific) in PBSF. Cells were washed once more with 1 ml of ice-cold PBSF and kept on ice until FACS analysis or sorting. Cells were analyzed and/or sorted using a FACS Aria III fluorescence-activated cell sorter (BD Biosciences) with a 70-μm nozzle and FACS flow sheath fluid (BD Biosciences) at a pressure of 70 p.s.i. Forward and side scatter (FSC and SSC) as well as Alexa Fluor 488 were excited using a 488-nm laser. SSC was detected using a 488/10 BP filter, and Alexa Fluor 488 was detected using a 530/30 BP filter. Alexa Fluor 633 was excited using a 633-nm laser and detected using a 660/20 BP filter. Yeast cells were selected in a scatter plot, and doublet discrimination was done in both an FSC and SSC plot of signal width versus height. First round sorting was done using the purity mode “yield,” although subsequent sorting was done in “4-way purity” mode. For the separate selections of single and double NNK libraries described in Figs. 4 and 6, at least 500,000 cells were selected in each round for libraries NNK 2, 4, and 7–10, and at least one million cells were selected each round for all other libraries. Selected cells were centrifuged at 3000 × g for 5 min to remove the sheath fluid and then cultured at 30 °C for 1 or 2 days in SD-CAA, after which DNA was extracted for deep sequencing (see below). For the selections of the pooled libraries described in Fig. 3, 5 × 105 and 3 × 105 cells were selected, respectively, in the first and second round, both in 4-way purity mode. A third selection was done with the purity mode set to “single cell,” in which 100 cells were spotted directly on an SD-CAA agar plate, with only a single cell spotted per position on the plate. Colonies were allowed to grow at 30 °C for 3 days. 12 colonies were picked and grown in 5 ml of liquid SD-CAA medium for 1 day. The cells were lysed, and plasmid DNA was extracted by using the Zymoprep yeast plasmid DNA miniprep II (Zymo Research). To improve the quality of the DNA, it was transformed into NovaBlue (Merck Novagen) bacteria, plated on LB agar with 100 mg liter−1 ampicillin. DNA was isolated from the bacteria using the QIAprep miniprep kit, and the DNA was sequenced by StarSEQ GmbH (Mainz, Germany) using the primer Seq-pCON-F.
Deep Sequencing Library Preparation
From each library between 108 and 109 cells were collected, and DNA was extracted using the Zymoprep Yeast Plasmid DNA Miniprep II kit (Zymo Research). The eluates were diluted 20-fold in autoclaved MilliQ water, and 1 μl of each eluate was used as a template in a separate 50-μl PCR containing 500 nm YD-tag-F and YD-tag-R primers, 200 μm deoxynucleotides (dNTPs; New England Biolabs), and 3 units of Phusion High Fidelity DNA polymerase (New England Biolabs) in 1× Phusion HF buffer (New England Biolabs). Reactions were initially heated to 98 °C for 30 s and then cycled 24 times at 98 °C for 10 s, 64 °C for 30 s, and 72 °C for 10 s, and finally extended at 72 °C for 30 s. 1 μl of each of the products of these reactions was used as template in a second 50-μl PCR in which each library was amplified with a unique pair of primers (see supplemental Table S3). The conditions of this reaction were 500 nm forward and reverse primers, 200 μm dNTPs (New England Biolabs), and 3 units of Phusion High Fidelity DNA polymerase in 1× Phusion HF buffer. These reactions were initially denatured at 98 °C for 30 s and then cycled 25 times between heating at 98 °C for 10 s and annealing/extension at 72 °C for 40 s and finally extended for 1 min at 72 °C. The products of these reactions were separately purified by electrophoresis through a 3% agarose/TAE gel and extracted from the gel using the QIAquick gel extraction kit (Qiagen). 1 μl of each PCR product was used as a template in a third 50-μl PCR containing 500 nm Amp-F and Amp-R primers, 200 μm dNTPs, and 3 units of Phusion High Fidelity DNA polymerase (New England Biolabs) in 1× Phusion HF Buffer (New England Biolabs). This reaction was heated at 98 °C for 30 s and then cycled 25 times between denaturation at 98 °C for 10 s, annealing at 59 °C for 30 s, and extension at 72 °C for 10 s, and finally extended at 72 °C for 30 s. The products of these reactions were purified using the QIAquick PCR purification kit (Qiagen) and eluted in TE buffer supplied in the kit. The concentrations of the eluates were measured by UV absorbance spectroscopy using a NanoDrop ND100 spectrophotometer, and the size and purity of the final products were confirmed by electrophoresis through a 3% agarose/TAE gel. Equimolar amounts of each library were combined into a single sequencing sample. 2 × 150 Paired End sequencing was performed by Eurofins Genomics GmbH (Ebersberg, Germany) using the Illumina MiSeq platform and V3 chemistry. Because of the low complexity of the sample, up to 30% PhiX control had to be added. Sequencing returned a total of 19,750,002 paired-end reads.
Deep Sequencing Data Analysis
Data were processed in several consecutive steps. First, the reads were clustered by their combination of barcode sequences using a dedicated Python script (supplemental file “NGSRead.py”). Then, sequences with insertions, deletions, Phred scores below 30 (within the stretch encoding the cyclic peptide), or with unintended additional mutations were discarded, and the sequences that passed these filters were translated, using two Matlab scripts (supplemental ScoringScript.m and supplemental removeNonWildTypes.m). After subtraction of reads that did not fulfill the criteria, each sorted and unsorted sub-library still contained over 100,000 reads (supplemental Table S3). Subsequently, the amino acids at the mutated position in each library were counted using a Matlab script (supplemental DetermineCounts.m). Amino acid frequency distributions in the unsorted single NNK libraries were compared with the theoretically expected distribution. Certain amino acids were significantly over- or under-represented, which may be caused by bias in the PCR step on the Illumina sequencing chip (53–55). Nevertheless, each individual residue was counted at least 1000 times at every position in the unsorted single NNK libraries (one exception being histidine at position 9, which was counted 738 times). For the double NNK libraries, a more detailed analysis was made where the enrichment of each pair of amino acids was investigated using another Matlab script (supplemental DetermineCountsCombination.m). Amino acid pairs that were counted less than 10 times in the unsorted libraries were excluded from this analysis.
Robust Enrichment Ratio Definition
The enrichment ratio is commonly used as a metric for the degree to which a mutation contributes to the property for which it is selected. It is defined as shown in Equation 1,
| (Eq. 1) |
where fr, xs is the frequency of amino acid r at position x in the selected population, and fr, xu is the corresponding frequency in the unselected population. The frequencies are calculated by dividing the number of times that amino acid r is counted at position x (nr, x) by the total number of counts at that position.
| (Eq. 2) |
This definition can also apply to a pair of amino acids at two positions. The frequency in the unselected population is usually a very small number, particularly if pairs of residues are investigated. When we calculated enrichment ratios of amino acid pairs in this way, sampling error lead to quite drastic overestimation of the true enrichment ratios of some pairs. To determine a relation between the magnitude of the sampling error and the number of counts in the distribution (nr, x), we performed 60 Bootstrap analyses on a set of 20 simulated frequency distributions with a varying number of counts nr, x between 1 and 100, and a total number of reads of 10,000 or 100,000 (supplemental Fig. S7). We determined that the coefficient of variation (CV) of the Bootstrap analyses depended on nr, x as shown in Equation 3.
| (Eq. 3) |
Therefore, we defined the RER as shown in Equation 4.
| (Eq. 4) |
The RER can thus be regarded as the lower 95% confidence limit of the enrichment ratio under the assumption that sampling is unbiased.
Peptide Synthesis
All peptides were synthesized by standard Fmoc solid phase peptide synthesis on a 50 or 100 μmol scale on an automated peptide synthesizer (Intavis Multiprep RS) using a NovaSyn TGR resin (Novabiochem) as a solid support. Each coupling was done two times for 30 min each. Peptide 1 (see supplemental Table S2 for numbering) was fluorescently labeled on resin at the N terminus using a 5-fold molar excess of 5-(6)-carboxyfluorescein two times for 8 h, essentially as described before (56). The other peptides were manually Fmoc-deprotected two times for 3 min in 20% piperidine in N-methyl-2-pyrrolidone (NMP) and washed four times with NMP. An Fmoc-protected pentanoic acid (O1Pen) linker (Iris Biotec) was activated by mixing 2 volumes of 0.2 m linker in NMP with 1 volume of 0.33 m 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate in NMP, incubating for 1 min, and then adding 1 volume of 1.6 m N,N-diisopropylethylamine (DIPEA) in NMP. An 8-fold molar excess of activated O1Pen linker was coupled on resin to the free amine of the peptides for 60 min. Peptides were then washed and Fmoc-deprotected as described above. Fluorescein isothiocyanate (FITC) was coupled to the O1Pen linker using a 7-fold molar excess of FITC (Sigma) and a 14-fold molar excess of DIPEA and reacting overnight. Beads were washed four times with NMP, then five times alternating between diethyl ether and dichloromethane, and then swollen in dichloromethane. Cleavage from the resin was done for 2 h in 95% TFA, 2.5% triisopropylsilane, 2.5% ethanedithiol, 2.5% water. The peptides were precipitated in ice-cold diethyl ether, repeatedly washed, and then dissolved in a water/acetonitrile mixture. Water/acetonitrile ratio was varied between 30 and 50% depending on peptide solubility. The peptides were then lyophilized. Cyclization of peptides 1–12 (supplemental Table S2) was done in 1–3 m guanidinium chloride, 100 mm Tris-HCl, pH 8.0, at a peptide concentration of 1 mg ml−1 for several days stirring in an open tube at 4 °C. Excessive salts were removed using a solid phase extraction column (Strata XL, Phenomenex), from which the peptides were eluted in 70% (v/v) water, 30% (v/v) acetonitrile. Peptides 13–22 (supplemental Table S2) were cyclized in 69% (v/v) phosphate-buffered saline, pH 7.4, 30% (v/v) acetonitrile, containing 1% (v/v) dimethyl sulfoxide (DMSO) as a mild oxidizing agent (57) for several days in an open tube at 4 °C. Reversed phase HPLC was performed on a Shimadzu LC-8A HPLC system by using a VYDAC protein and peptide C18 column.
Fluorescence Polarization Assays
Cetuximab was obtained from the Catharina Hospital Pharmacy, Eindhoven, The Netherlands, and was titrated to 10 nm FITC-labeled peptides in PBS, pH 7.4 (Sigma), containing 1 mg ml−1 BSA (Sigma) in total sample volumes of 50 μl in black 384 wells OptiPlate (PerkinElmer Life Sciences). Polarization was recorded using a TECAN Infinite F500 Plate Reader. Experiments were performed in duplicate or triplicate as indicated in the respective figure legends. Hill equations of the form of Equation 5 were fit through the data using Origin 2015 (OriginLab), using statistical weights for the fit,
| (Eq. 5) |
where P is the polarization, and n is the Hill coefficient. Hill coefficients did not deviate much from one, indicating no cooperativity, which was also expected.
Rosetta Modeling
The crystal structure of cetuximab in complex with the Md1 peptide was obtained from the Protein Data Bank (code 4GW1) (9). The structure was refined using the Rosetta relax application using the score12 score function. A scan of the influence of mutations was executed utilizing the Rosetta backrub application, which allows optimization using a flexible peptide backbone (39). Mutations were systematically introduced at the desired positions, and the conformation was optimized using the Rosetta backrub application. All residues near the interface were taken as pivot residues. For each mutation, the minimal energy structure of an ensemble of 100 structures was taken, and its interface score was compared with the wild-type variant. Structure visualization was done using PyMOL (Schrödinger Software).
Cell Experiments
A431 skin epidermoid carcinoma cells (ATCC number CRL-1555) were cultured in RPMI 1640 medium + glutamine (Thermo Fisher Scientific) supplemented with 10% (v/v) fetal bovine serum (Gibco) and 1% (v/v/) penicillin/streptomycin (Gibco) in Falcon polystyrene cell culture flasks (BD Biosciences). Cells were passaged twice per week by washing three times with sterile PBS, incubating with trypsin/EDTA (Gibco) for 5 min at 37 °C, briefly slapping the flask to dislodge cells, and neutralizing the trypsin by adding fresh medium to the original volume. For each passage, cells were diluted 6-fold. Experiments were done only with cells at passage numbers between 10 and 20. Cells were trypsinized as described above and washed with 1 ml of PBS, pH 7.4, 1 mg ml−1 BSA (PBSF). Cyclic peptides Md1 and Q1V/D3N/S5G/K10R (peptides 13 and 15, supplemental Table S2) were dissolved at a 1 μm concentration in 10% DMSO in PBSF. UV-visible absorbance was measured on a NanoDrop ND1000 spectrophotometer, and the fluorescein molar extinction coefficient of 75,000 m−1 cm−1 at 496 nm (58) was used for determining the peptide concentrations. 2.5 × 105 cells were incubated for 30 min at room temperature in 1 ml of PBSF with 10 nm cetuximab and 0, 50, 100, or 500 nm FITC-labeled cyclic peptides, which had been pre-incubated for 1 h at room temperature before the cells were added. Control samples contained 500 nm peptide, but no cetuximab. After incubation, the cells were centrifuged at 500 × g for 5 min, re-suspended in PBSF, and analyzed by flow cytometry using a FACS Aria III. FITC was excited by the 488-nm laser and detected through a 530/30 BP filter. SSC was excited by the 488-nm laser and detected through a 488/10 BP filter. The cells were gated in an FSC/SSC plot. Doublet discrimination was done using both the forward and side scatter signal width versus height plots. Measurements were performed in duplicate.
Author Contributions
M. v. R. conceived and coordinated the study, carried out the experiments shown in Figs. 4, 5B, 6G, and 7, and wrote the manuscript. B. M. G. J. carried out the experiments shown in Fig. 3C. N. M. H. carried out the experiments in Fig. 3, A and B. A. J. v. d. L. wrote the Python and MATLAB scripts for and performed the bioinformatics on the deep sequencing data. D. W. carried out the experiments in Fig. 2. P. A. P. and T. F. A. d. G. performed the Rosetta screen described in Fig. 6, A and B. M. M. conceived and coordinated the study and revised the manuscript.
This work was supported by European Research Council (ERC) Starting Grant ERC-2011-StG 280255, ERC Proof-of-Concept Grant ERC-2013-PoC 632274, and NanoNext, a micro and nanotechnology consortium of the Government of the Netherlands and 130 partners. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S3, Figs. 1–7, and Python and MatLab scripts.
- Fab
- antigen-binding fragment
- EGFR
- epidermal growth factor receptor
- DIPEA
- N,N-diisopropylethylamine
- RER
- robust enrichment ratio
- SSC
- side scatter
- FSC
- forward scatter
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- O1Pen
- 5-amino-3-oxapentanoic acid
- ADA
- anti-drug antibody
- MID
- Multiplex Identifier
- NMP
- N-methyl-2-pyrrolidone
- BP
- bandpass.
References
- 1. Agarwal P., and Bertozzi C. R. (2015) Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 26, 176–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sochaj A. M., Świderska K. W., and Otlewski J. (2015) Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 33, 775–784 [DOI] [PubMed] [Google Scholar]
- 3. Yao H., Jiang F., Lu A., and Zhang G. (2016) Methods to design and synthesize antibody-drug conjugates (ADCs). Int. J. Mol. Sci. 17, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Geel R., Wijdeven M. A., Heesbeen R., Verkade J. M., Wasiel A. A., van Berkel S. S., and van Delft F. L. (2015) Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug. Chem. 26, 2233–2242 [DOI] [PubMed] [Google Scholar]
- 5. Kornberger P., and Skerra A. (2014) Sortase-catalyzed in vitro functionalization of a HER2-specific recombinant Fab for tumor targeting of the plant cytotoxin gelonin. mAbs 6, 354–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beerli R. R., Hell T., Merkel A. S., and Grawunder U. (2015) Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLoS ONE 10, e0131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hui J. Z., Tamsen S., Song Y., and Tsourkas A. (2015) LASIC: light-activated site-specific conjugation of native IgGs. Bioconjug. Chem. 26, 1456–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee Y., Jeong J., Lee G., Moon J. H., and Lee M. K. (2016) Covalent and oriented surface immobilization of antibody using photoactivatable antibody Fc-binding protein expressed in Escherichia coli. Anal. Chem. 88, 9503–9509 [DOI] [PubMed] [Google Scholar]
- 9. Donaldson J. M., Zer C., Avery K. N., Bzymek K. P., Horne D. A., and Williams J. C. (2013) Identification and grafting of a unique peptide-binding site in the Fab framework of monoclonal antibodies. Proc. Natl. Acad. Sci. U.S.A. 110, 17456–17461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams J. C., Horne D. A., Ma Y., and Chen H. W. (February 25, 2014) U. S. Patent US8658774 B2
- 11. Riemer A. B., Kurz H., Klinger M., Scheiner O., Zielinski C. C., and Jensen-Jarolim E. (2005) Vaccination with cetuximab mimotopes and biological properties of induced anti-epidermal growth factor receptor antibodies. J. Natl. Cancer Inst. 97, 1663–1670 [DOI] [PubMed] [Google Scholar]
- 12. Riemer A. B., Klinger M., Wagner S., Bernhaus A., Mazzucchelli L., Pehamberger H., Scheiner O., Zielinski C. C., and Jensen-Jarolim E. (2004) Generation of peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J. Immunol. 173, 394–401 [DOI] [PubMed] [Google Scholar]
- 13. Boder E. T., and Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 [DOI] [PubMed] [Google Scholar]
- 14. Reich L. L., Dutta S., and Keating A. E. (2015) SORTCERY–a high-throughput method to affinity rank peptide ligands. J. Mol. Biol. 427, 2135–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vodnik M., Zager U., Strukelj B., and Lunder M. (2011) Phage display: selecting straws instead of a needle from a haystack. Molecules 16, 790–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shusta E. V., Kieke M. C., Parke E., Kranz D. M., and Wittrup K. D. (1999) Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 292, 949–956 [DOI] [PubMed] [Google Scholar]
- 17. Fowler D. M., and Fields S. (2014) Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shin H., and Cho B.-K. (2015) Rational protein engineering guided by deep mutational scanning. Int. J. Mol. Sci. 16, 23094–23110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Starita L. M., and Fields S. (2015) Deep mutational scanning: a highly parallel method to measure the effects of mutation on protein function. Cold Spring Harb. Protoc. 2015, 711–714 [DOI] [PubMed] [Google Scholar]
- 20. Procko E., Hedman R., Hamilton K., Seetharaman J., Fleishman S. J., Su M., Aramini J., Kornhaber G., Hunt J. F., Tong L., Montelione G. T., and Baker D. (2013) Computational design of a protein-based enzyme inhibitor. J. Mol. Biol. 425, 3563–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Strauch E.-M., Fleishman S. J., and Baker D. (2014) Computational design of a pH-sensitive IgG binding protein. Proc. Natl. Acad. Sci. U.S.A. 111, 675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Forsyth C. M., Juan V., Akamatsu Y., DuBridge R. B., Doan M., Ivanov A. V., Ma Z., Polakoff D., Razo J., Wilson K., and Powers D. B. (2013) Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. mAbs 5, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doolan K. M., and Colby D. W. (2015) Conformation-dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing. J. Mol. Biol. 427, 328–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koenig P., Lee C. V., Sanowar S., Wu P., Stinson J., Harris S. F., and Fuh G. (2015) Deep sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular degeneration. J. Biol. Chem. 290, 21773–21786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith S. N., Harris D. T., and Kranz D. M. (2015) T cell receptor engineering and analysis using the yeast display platform. Methods Mol. Biol. 1319, 95–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris D. T., Wang N., Riley T. P., Anderson S. D., Singh N. K., Procko E., Baker B. M., and Kranz D. M. (2016) Deep mutational scans as a guide to engineering high-affinity T cell receptor interactions with peptide-bound MHC. J. Biol. Chem. 291, 24566–24578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Blarcom T., Rossi A., Foletti D., Sundar P., Pitts S., Bee C., Melton Witt J., Melton Z., Hasa-Moreno A., Shaughnessy L., Telman D., Zhao L., Cheung W. L., Berka J., Zhai W., et al. (2015) Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J. Mol. Biol. 427, 1513–1534 [DOI] [PubMed] [Google Scholar]
- 28. Kowalsky C. A., Faber M. S., Nath A., Dann H. E., Kelly V. W., Liu L., Shanker P., Wagner E. K., Maynard J. A., Chan C., and Whitehead T. A. (2015) Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J. Biol. Chem. 290, 26457–26470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu Z., Juan V., Ivanov A., Ma Z., Polakoff D., Powers D. B., Dubridge R. B., Wilson K., and Akamatsu Y. (2012) Affinity and cross-reactivity engineering of CTLA4-Ig to modulate T cell costimulation. J. Immunol. 189, 4470–4477 [DOI] [PubMed] [Google Scholar]
- 30. Mishra P., Flynn J. M., Starr T. N., and Bolon D. N. (2016) Systematic mutant analyses elucidate general and client-specific aspects of Hsp90 function. Cell Rep. 15, 588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang H.-J., Jian J.-W., Hsu H.-J., Lee Y.-C., Chen H.-S., You J.-J., Hou S.-C., Shao C.-Y., Chen Y.-J., Chiu K.-P., Peng H.-P., Lee K. H., and Yang A.-S. (2014) Loop-sequence features and stability determinants in antibody variable domains by high-throughput experiments. Structure 22, 9–21 [DOI] [PubMed] [Google Scholar]
- 32. Hackel B. J., Kapila A., and Wittrup K. D. (2008) Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J. Mol. Biol. 381, 1238–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen I., Dorr B. M., and Liu D. R. (2011) A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U.S.A. 108, 11399–11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y.-S., Bhandari R., Cochran J. R., Kuriyan J., and Wittrup K. D. (2006) Directed evolution of the epidermal growth factor receptor extracellular domain for expression in yeast. Proteins Struct. Funct. Genet. 62, 1026–1035 [DOI] [PubMed] [Google Scholar]
- 35. Pavoor T. V., Cho Y. K., and Shusta E. V. (2009) Development of GFP-based biosensors possessing the binding properties of antibodies. Proc. Natl. Acad. Sci. U.S.A. 106, 11895–11900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kadonosono T., Yabe E., Furuta T., Yamano A., Tsubaki T., Sekine T., Kuchimaru T., Sakurai M., and Kizaka-Kondoh S. (2014) A fluorescent protein scaffold for presenting structurally constrained peptides provides an effective screening system to identify high affinity target-binding peptides. PLoS ONE 9, e103397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kimura R. H., Levin A. M., Cochran F. V., and Cochran J. R. (2009) Engineered cystine knot peptides that bind αvβ3, αvβ5, and α5β1 integrins with low-nanomolar affinity. Proteins 77, 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kortemme T., Kim D. E., and Baker D. (2004) Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, pl2. [DOI] [PubMed] [Google Scholar]
- 39. Smith C. A., and Kortemme T. (2008) Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 380, 742–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kabat E. A., Wu T. T., Perry H. M., Gottesman K. S., and Foeller C. (eds) (1991) Sequences of Proteins of Immunological Interest, 5th Ed., p. 662, United States Department of Health and Human Services, National Institutes of Health, Bethesda [Google Scholar]
- 41. Murelli R. P., Zhang A. X., Michel J., Jorgensen W. L., and Spiegel D. A. (2009) Chemical control over immune recognition: a class of antibody-recruiting small molecules that target prostate cancer. J. Am. Chem. Soc. 131, 17090–17092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mazzucchelli S., Colombo M., De Palma C., Salvadè A., Verderio P., Coghi M. D., Clementi E., Tortora P., Corsi F., and Prosperi D. (2010) Single-domain protein A-engineered magnetic nanoparticles: toward a universal strategy to site-specific labeling of antibodies for targeted detection of tumor cells. ACS Nano 4, 5693–5702 [DOI] [PubMed] [Google Scholar]
- 43. Muguruma K., Yakushiji F., Kawamata R., Akiyama D., Arima R., Shirasaka T., Kikkawa Y., Taguchi A., Takayama K., Fukuhara T., Watabe T., Ito Y., and Hayashi Y. (2016) Novel hybrid compound of a plinabulin prodrug with an IgG binding peptide for generating a tumor selective noncovalent-type antibody-drug conjugate. Bioconjug. Chem. 27, 1606–1613 [DOI] [PubMed] [Google Scholar]
- 44. Avery K. N., Zer C., Bzymek K. P., and Williams J. C. (2015) Development of a high affinity, non-covalent biologic to add functionality to Fabs. Sci. Rep. 5, 7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Björck L. (1988) Protein L. A novel bacterial cell wall protein with affinity for Ig L chains. J. Immunol. 140, 1194–1197 [PubMed] [Google Scholar]
- 46. Zouali M. (2007) B cell superantigens subvert innate functions of B cells. Chem. Immunol. Allergy 93, 92–105 [DOI] [PubMed] [Google Scholar]
- 47. Zouali M., and Richard Y. (2011) Marginal zone B-cells, a gatekeeper of innate immunity. Front. Immunol. 2, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosenthal M. E., Rojtman A. D., and Frank E. (2012) Finegoldia magna (formerly Peptostreptococcus magnus): an overlooked etiology for toxic shock syndrome? Med. Hypotheses 79, 138–140 [DOI] [PubMed] [Google Scholar]
- 49. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 [DOI] [PubMed] [Google Scholar]
- 50. Gietz R. D., and Schiestl R. H. (2007) Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 1–4 [DOI] [PubMed] [Google Scholar]
- 51. Gietz R. D., and Schiestl R. H. (2007) Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 35–37 [DOI] [PubMed] [Google Scholar]
- 52. Gietz R. D., and Schiestl R. H. (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 [DOI] [PubMed] [Google Scholar]
- 53. Dohm J. C., Lottaz C., Borodina T., and Himmelbauer H. (2008) Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 39, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aird D., Ross M. G., Chen W. S., Danielsson M., Fennell T., Russ C., Jaffe D. B., Nusbaum C., and Gnirke A. (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakamura K., Oshima T., Morimoto T., Ikeda S., Yoshikawa H., Shiwa Y., Ishikawa S., Linak M. C., Hirai A., Takahashi H., Altaf-Ul-Amin M., Ogasawara N., and Kanaya S. (2011) Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 39, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Janssen B. M., Lempens E. H., Olijve L. L., Voets I. K., Van Dongen J. L., De Greef T. F., and Merkx M. (2013) Reversible blocking of antibodies using bivalent peptide-DNA conjugates allows protease-activatable targeting. Chem. Sci. 4, 1442–1450 [Google Scholar]
- 57. Tam J. P., Wu C.-R., Liu W., and Zhang J.-W. (1991) Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. J. Am. Chem. Soc. 113, 6657–6662 [Google Scholar]
- 58. Sjöback R., Nygren J., and Kubista M. (1995) Absorption and fluorescence properties of fluorescein. Spectrochimica Acta Part Mol. Biolmol. Spectrosc. 51, L7–L21 [Google Scholar]