

Acute myeloid leukemia (AML) is a heterogeneous group of haematopoietic neoplasms driven partly by the loss of differentiation and the blockade of cell death. AML is sustained by leukemia-initiating cells (LICs) that arise from pre-leukemic haematopoietic stem and progenitor cells (HSPCs) that carry genetic alterations being selected for during leukemogenesis.1 The resistance of LICs to standard chemotherapies presents a major clinical challenge as they eventually cause disease relapse and death.1
Understanding the mechanisms that protect LICs from cell death therefore provide an ideal target for future anti-leukemia therapy. The ability of LICs and pre-leukemic HSCs to evade cell death has been attributed to several pathways that are distinct from normal HSPCs. LICs for example have constitutive NF-κB activity while normal HSPCs do not. In addition, analysis of patient-derived cells showed that in some cases the apoptotic cell death pathway in LICs is skewed.2
However, work in mature myeloid cells has shown that these cells are exquisitely sensitive to undergoing cell death driven by receptor-interacting serine-threonine kinase 3 (RIPK3).3 In addition to inducing apoptosis, RIPK3 can also induce regulated necrosis, a form of necrotic cell death marked by cell and organelle swelling and plasma membrane rupture, which, in contrast to apoptosis, is accompanied by inflammation due to the release of cytokines and DAMPs from dying cells. We found that in a similar fashion LICs are also sensitive to RIPK3-dependent cell death and that loss of RIPK3 expression underlies the developmental block and oncogenic process in many subtypes of AML.4
In our study, we focused on one of the recurring genetic alterations found in AML that gives rise to internal tandem duplications of the tyrosine kinase receptor FLT3 (FLT3-ITD). FLT3-ITD mutatuions are found in 30–40% of cytogenetically normal AML patients and define a poor prognostic subgroup. Using a model of adoptive transfer of bone marrow (BM) cells transduced with FLT3-ITD we demonstrated that genetic loss of Ripk3 greatly accelerated FLT3-ITD-induced leukemia and mortality. The enhanced disease was marked by an expansion of common myeloid progenitors (CMPs) and short-term haematopoietic stem cells (ST-HSCs), being the cellular compartment to contain LICs. In addition, RIPK3 deletion rendered FLT3-ITD splenocytes serially transplantable, indicating an enhanced ability of transformed HSPCs to survive and propagate. RIPK3 functioned to induce cell death of ST-HSCs and CMPs and acted as a tumor suppressor.
Further, consistent with their roles in RIPK3 activation, we showed that deletion of TNF receptors 1 and 2 (Tnfr1/2−/−) or inhibition of RIPK1 phenocopied the leukemic disease induced by Ripk3−/− BM. However, Mlkl deficiency did not abrogate cell death in vitro, and disease progression in vivo was indistinguishable between WT and Mlkl−/−, suggesting that RIPK3 may also induce apoptosis of LICs, a function that is consistent with recently published results.5
Importantly, WT FLT3-ITD-transformed cells produced substantial amounts of the inflammatory cytokine IL-1β, while Ripk3 or Mlkl deficiency severely blunted expression of IL-1β. The role of IL-1 β in promoting LIC differentiation and thus restricting leukemogenesis was confirmed with Pycard−/− and Il1r1−/− HSPCs. Hence, Ripk3 signals through 2 complementary pathways, cell death and IL-1β production, to restrict LICs and to promote myeloid cell differentiation, respectively (Fig. 1).
Figure 1.
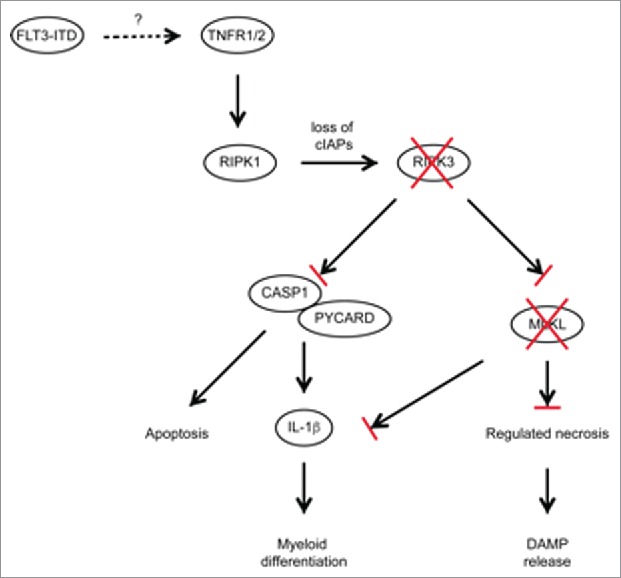
Schematic diagram of RIPK3-dependent cell death and inflammasome activation in FLT3-ITD expressing LICs. Activation of TNF receptors 1/2 activates RIPK1/RIPK3, together with loss of cIAPs resulting in the induction of regulated necrosis and inflammasome formation, propagating proinflammatory signaling and substantial cytokine secretion. Several AML subtypes including FLT3-ITD show reduced expression of RIPK3 and MLKL, preventing LIC cell death and IL-1β production.
Using several human patient sample cohorts, we observed a reduced expression of both RIPK3 and MLKL in FLT3-mutated AML, and many other AML subtypes. For example, samples from patients harboring the AML-ETO fusion, which is characterized by t(8;21) translocation, also exhibited reduced RIPK3 and MLKL expression. Ripk3 deficiency similarly accelerated leukemic disease in mice transplanted with AML-ETO-transduced BM. By contrast, AML associated with mixed-lineage leukemia (MLL) translocations showed normal RIPK3 and MLKL expression, and Ripk3 deficiency did not alter the myeloproliferative neoplasm caused by MLL-ENL. This finding is consistent with independent studies showing that deletion of Ripk3 did not alter disease progression of MLL-ENL-, MLL-AF9-, NUP98-HoxA9-, and HoxA9/Meis1-induced leukemia.6,7 Furthermore, the authors showed that these leukemia subsets were sensitive to killing by the SMAC mimetic birinapant in a RIPK1/TNFR1-dependent manner. Therefore, our work suggests that alternative therapeutic approaches will be required for AML subsets with reduced RIPK3 expression.
The reduction of RIPK3 expression was specific to AML as we did not observe changes in RIPK3 expression in patient cohorts diagnosed with other myeloid proliferative neoplasms.
Hence, RIPK3 and MLKL have key tumor suppressor roles in a broad, but not all-inclusive, range of AML subtypes. Loss of this signaling pathway acts as a double-edged sword; it first promotes survival of transforming HSPCs and secondly inhibits LIC differentiation along the myeloid lineage. Together, this leads to the accumulation of LICs and thus leukemogenesis. The finding that RIPK3 promotes inflammatory cell death and differentiation of LICs emphasizes the relevance of blocking this pathway for LIC integrity. In order to eradicate the pool of LICs, future concepts aiming at reinstating RIPK3 signaling might therefore harbor therapeutic potential in some AML subtypes.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al.. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506:328-33; PMID:24522528; http://dx.doi.org/ 10.1038/nature13038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, DeAngelo DJ, Frattini MG, Letai A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012; 151:344-55; PMID:23063124; http://dx.doi.org/ 10.1016/j.cell.2012.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB, Kanegane H, Ringelhan M, Kaufmann T, Heikenwälder M, Strasser A, et al.. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell reports 2014; 7:1796-808; PMID:24882010; http://dx.doi.org/ 10.1016/j.celrep.2014.05.008 [DOI] [PubMed] [Google Scholar]
- [4].Höckendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S, Kauschinger J, Magnani G, Reisinger F, Heuser M, et al.. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer cell 2016; 30:75-91; PMID:27411587; http://dx.doi.org/ 10.1016/j.ccell.2016.06.002 [DOI] [PubMed] [Google Scholar]
- [5].Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al.. RIP3 induces apoptosis independent of pronecrotic kinase activity. Molecular cell 2014; 56:481-95; PMID:25459880; http://dx.doi.org/ 10.1016/j.molcel.2014.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brumatti G, Ma C, Lalaoui N, Nguyen N-Y, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N, et al.. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Science translational medicine 2016; 8:339ra69; http://dx.doi.org/ 10.1126/scitranslmed.aad3099 [DOI] [PubMed] [Google Scholar]
- [7].Lalaoui N, Hänggi K, Brumatti G, Chau D, Nguyen N-YN, Vasilikos L, Spilgies LM, Heckmann DA, Ma C, Ghisi M, et al.. Targeting p38 or MK2 enhances the anti-leukemic activity of Smac-mimetics. Cancer cell 2016; 29:145-58; PMID:26859455; http://dx.doi.org/ 10.1016/j.ccell.2016.01.006 [DOI] [PubMed] [Google Scholar]