

Abstract
Introduction
Disseminated fibrin deposition in the microvasculature such as in disseminated intravascular coagulation (DIC) arises from uninhibited activated coagulation secondary to sustained systemic inflammation. Currently there is no treatment for DIC. Treating the underlying trigger and supportive care are the current recommendations to manage DIC. This study aims at using recombinant von Willebrand factor (VWF) A2 domain polypeptide to inhibit VWF–mediated platelet adhesion to fibrin and prevent DIC.
Materials and Methods
We use flow chamber assay to test the capacity of purified A2 protein to inhibit platelet adhesion to immobilized fibrin(ogen) and platelet-fibrin clot formation. We use a murine model of lipopolysaccharide-induced DIC to examine the effect of A2 protein on DIC.
Results
The A2 protein blocked flow-dependent platelet adhesion to fibrin, delayed fibrin polymerization, and inhibited platelet-fibrin clot formation in vitro. The infusion of the purified A2 protein to the endotoxin-treated mice prevented fibrin-rich microthrombi formation in brain, lung, kidney, and liver. It also attenuated levels of inflammatory mediators, and markedly reduced mortality rates at 96 hours.
Conclusions
The A2 protein inhibited platelet interaction with fibrin(ogen). Furthermore, A2 prevented disseminated fibrin-rich microthrombi and decrease mortality in a lipopolysaccharide-induced DIC murine model. A2 could provide a novel therapeutic approach in critically ill patients with uninhibited activated coagulation and disseminated fibrin deposition such as DIC.
Keywords: microvascular thrombosis, endotoxemia, disseminated intravascular coagulation (DIC), platelet adhesion, and fibrin
Introduction
Most of critically ill patients have some evidences of activated coagulation. However, with sustained systemic inflammation the activated coagulation may become uncontrollable and cause tissue damage. Uninhibited activated coagulation will lead to thrombotic microangiopathy, which is a family of syndromes associated with disseminated microvascular thromboses. Disseminated intravascular coagulation (DIC) is an entity in the spectrum of thrombotic microangiopathy which can contribute to multiple organ dysfunction syndrome and death [1]. DIC can occur in 50–60% of septic patients [2,3], in 14–40% of new onset thrombocytopenic critically ill patients [4–6], and in 8% of all critically ill patients [7]. Conditions associated with triggering DIC include sepsis, trauma, burn, vasculitis, obstetric complications, and toxin exposure. Autopsies performed in patients who died from DIC reveal fibrin-rich microthrombi in small and mid-size vessels in all organs [8–10]. Reported mortalities associated with DIC range from 22–75% [2–4,7]. Currently, the recommended managements for DIC are 1) treat the underlying trigger and 2) provide supportive care [11]. Multiple mono-therapeutic agents have been tried to treat DIC without conclusive success including heparin [12–14], antithrombin III [15], recombinant tissue factor pathway inhibitor [16], recombinant human activated protein C [2,17,18], protein C concentrate [19,20], and recombinant human soluble thrombomodulin [21].
DIC is characterized by a wide spread fibrin deposition, which may contribute in mediating platelet adhesion and thrombus formation. The interaction between circulating platelets and the deposited fibrin is primarily via the fibrinogen receptor glycoprotein (GP)IIb/IIIa [22,23], and the secondary mechanism via the von Willebrand factor (VWF)-GPIbα interaction [24–26]. The mature VWF consists of a 2,050-residue subunit that contains multiple copies of A, C, and D type domains [27]. The central portion of the VWF subunit contains three homologous A domains. While characterizing the isolated A2 domain of human VWF in our laboratory [28], we noticed that this recombinant A2 domain (A2 protein) had a significant binding activity for fibrin. Based on this novel observation, we proposed to examine the significance of this interaction in vitro under shear conditions, and in vivo using in a mouse endotoxemia-induced DIC model.
Materials and methods
Antibodies and reagents
Antibody 6D1 was a gift from Dr. Barry Coller (The Rockefeller University, New York, NY). Human fibrinogen was obtained from Calbiochem (Gibbstown, NJ), D-dimer and fragment E were purchased from Hyphen Biomed (Mason, Ohio). Lipopolysaccharide (LPS, 0111:B4) was obtained from Sigma. Fibrin monomer was prepared as previously described [29]. Purified plasma VWF, A domain proteins (A21481-1668, and A31671-1874) were obtained as previously described [30,31].
Binding assays
The analyses of the interaction of A2, or A3 protein with fibrin monomer or fibrin(ogen), which exposes fibrin-specific sequences upon surface adsorption, were performed by enzyme-linked immunosorbent assay (ELISA) as described elsewhere [30,31]. Briefly, the wells of microtiter plate were coated with either fibrin monomer or fibrinogen (5 μg/ml) in 50 mM carbonate buffer, pH 9.6, and blocked with 3% (w/v) bovine serum albumin (BSA). Following incubation and washing, the bound A2, or A3 protein was detected by using monoclonal anti-histidine-horseradish peroxidase conjugate (Sigma). In other assays, microtiter wells were coated with the A2 protein (5 μg/ml) and increasing concentrations of fibrin monomer, fibrinogen, D-dimer, or fragment E were added to the wells. The fibrinogen-related proteins were detected using anti-human fibrinogen antibody (Dako). Surface Plasmon Resonance (SPR) binding studies were performed similar to our previous studies with some modifications using a BIAcore 3000 system (BIAcore, Piscataway, NJ)[32,33]. The human fibrinogen (50 μg/ml in 50 mM sodium acetate pH-5.0) was covalently coupled via amine coupling to sensor chip CM5 as directed by the supplier. The binding assays were performed in 10 mM Tris-HCl, 150 mM NaCl, 0.001% (v/v) Tween-20, pH-7.4 at 25 °C at a flow rate of 10 μl/min. The binding of A domain proteins to fibrinogen was corrected for non-specific binding to the control channel. Fibrinogen binding at equilibrium was determined at different protein concentrations (50 – 2000nM). As previously described [32], the equilibrium dissociation constant (KD) were calculated by curve fitting with the BIAevaluation software (version 4.1.1) supplied by the manufacturer. A 1:1 Langmuir interaction model was used. After measuring the A2 binding to fibrinogen, the chip was regenerated by injection of 10 mM Glycine, pH-3.0, and 1 M NaCl.
Fibrin polymerization
Polymerization of fibrin was evaluated by measuring change in absorbance at λ390 nm using a spectrophotometer (SynergyMX (BioTek Instruments) in a 96 well microtiter format. Polymerization was initiated by the addition of thrombin (0.1 U/mL) at time 0 to the reaction mixture containing fibrinogen (0.1 mg/mL or 0.3 μM) and A2 or A3 protein (0.13 mg/ml or 4.5 μM) in a pH 7.5 buffer containing 10 mM Tris, 0.15 M NaCl, 1 mM CaCl2. Measurements were made at room temperature and at interval of 20 seconds.
Preparation of protein-coated surfaces
Dishes coated with fibrinogen were prepared as we previously described [31]. Fibrinogen was diluted to 100 μg/ml in 65 mM sodium phosphate buffer, pH 6.5, and incubated for 1 h at 37 °C. After washing with phosphate-buffered saline, pH-7.4 (PBS) the dishes were blocked with 3% BSA in PBS before using in the flow assays.
Flow assays
Citrated blood from healthy volunteers was obtained by venipuncture following an informed consent approved by the committee for the protection of human subjects at the Baylor College of Medicine. Perfusion assays were carried out as we described elsewhere [32]. One ml of citrated whole blood was perfused over the fibrin(ogen)-coated coverslip at a shear stress of 1,500 s−1 and tethered platelets were observed with phase contrast objectives, recorded by video-microscopy, and analyzed as previously described [31]. Experiments were performed in triplicate using different blood donors. In some experiments, fibrinogen-coated dish was pre-incubated with A2, A3 protein [5.0 μM] or buffer before perfusion. In some experiments, A2 protein [0.4 μM] and fibrin monomer (20 μg/ml) were added to blood before the perfusion over the fibrinogen surface. In experiments with blood containing fibrin monomer, the platelet-clot formed during the perfusion was instantly arrested to the surface, and several view fields (~10–12) were recorded.
Mice
All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. Mice (C57BL/6, 10–12 weeks old) were injected with LPS (25–40 mg/kg) via intraperitoneally (I.P.) to maximize the manifestations of endotoxin. The A2 (4 mg/kg) or saline was injected via I.P. 1.5 hour after the LPS injection. In some experiments, saline was substituted by the A3 protein (4 mg/kg).
Histology
As described before [34], mice were perfused with phosphate-buffered formaldehyde followed by the removal of brain, liver, kidneys, and lungs after 24 hours of LPS injection in sham and LPS- A2 treated mice. These organs were processed using the services of the Comparative Pathology Laboratory (CLP) of Baylor College of Medicine. Microvascular fibrin-rich thrombi in paraffin embedded brain, liver, kidney, and lung tissues were analyzed with polyclonal fibrinogen antibody (Dako, Carpinteria California). In addition, the levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and urea were determined by using the services of the CPL. Lastly, Bio-Plex Multiplex Immunoassay kit (BioRad) was use to measure the levels of cytokines and chemokines in mice. Bleeding time of mice treated with A2 or saline was performed as previously described [35]. P-values were calculated with student’s t-test.
Results
Recombinant A2 domain of von Willebrand factor has binding activity for fibrin(ogen)
The A2 protein effectively bound to fibrin(ogen) in saturable manner with half-maximal binding at 90 ± 20 nM (KD of 125 ± 30 nM by SPR), while the homologous A3 domain did not have any significant binding (Fig. 1). Same result was obtained when the A2 protein bound to immobilized fibrin monomer on microtiter wells (half-maximal binding at 60 ± 10 nM, Fig. S1C). Since immobilized fibrinogen exposes the neoepitopes of fibrin, we analyzed the binding of soluble fibrin monomer and fibrinogen to immobilized A2 protein in ELISA. The soluble fibrin monomer bound to insoluble A2 protein with a half-maximal binding occurring at ~2.4 ± 0.5 μg/ml (or ~7.0 nM), whereas poor binding was observed with soluble fibrinogen (Fig. 2). We further tested the capacity of the A2 protein to block the binding of VWF to fibrin monomer, and Figure S1A shows that the A2 protein reduced ~60% the VWF-fibrin interaction. To localize the binding domain in fibrinogen, we examined the binding of two fibrin degradation products, D-dimer, and fragment E to immobilized A2 protein. Soluble D-dimer had a binding activity for insoluble A2 domain higher than that of fragment E, with an apparent binding affinity of ~3.1 ± 0.6 μg/ml (or 17.0 nM) (Fig. S1B). These results show that the purified recombinant A2 domain of VWF exhibits a binding site preferably for fibrin.
Fig. 1. Interaction between isolated A domain proteins and fibrinogen.
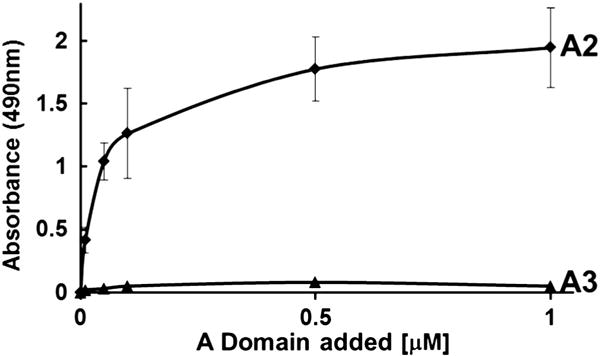
Various concentrations of A2 or A3 protein were incubated with fibrin(ogen) coated wells and the bound proteins were determined by ELISA as described in the methods. The graph represents one of three separated experiments. Each point denotes the mean ± SD of values obtained from a triplicate assay. The A2 protein had binding activity for fibrin(ogen) but not the A3 protein (p < 0.05).
Fig. 2. Fibrin binds to immobilized A2 protein.
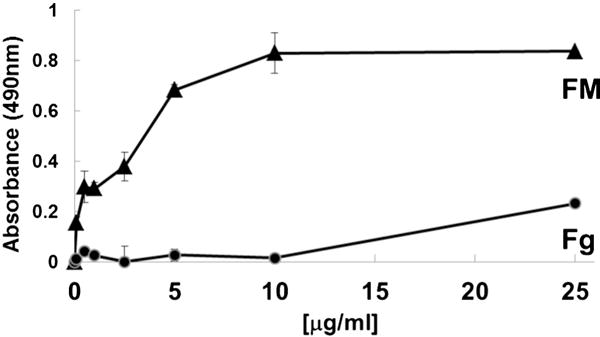
Various concentrations of soluble fibrin monomer (FM) or fibrinogen (Fg) were added to immobilized A2 domain (5 μg/ml) and the bound proteins were determined by peroxidase-labeled anti-fibrinogen antibody. Bound proteins were determined by ELISA as described before. The graphs represent one of two separated experiments. Each point represents the mean ± SD of values obtained from a triplicate assay.
The A2 protein inhibited flow-dependent platelet adhesion to fibrin(ogen)
Because plasma VWF mediates platelet adhesion to fibrin(ogen) [36], we determined the significance of the A2-fibrin(ogen) interaction in VWF-mediated platelet adhesion under flow conditions. Fibrin(ogen)-coated dish was pre-incubated with A2, or A3 domain protein [5.0 μM] or buffer for one hour. Subsequently, whole blood was perfused over the fibrin(ogen)-surface at a shear rate of 1,500 s−1. The A2 protein significantly inhibited platelet adhesion to fibrin(ogen) while under similar conditions, the A3 domain and buffer only had no significant effect (Fig. 3). The magnitude of the inhibitory effect of A2, is similar to anti-GPIbα antibody, 6D1 (Fig. 3), which was used as a control to show the involvement of the GPIbα-VWF interaction in platelet adhesion to fibrin(ogen) at high shear stress. Thus, blocking the interaction of plasma VWF with immobilized fibrin(ogen) results in diminishing the number of VWF molecules available to capture flowing platelets [25].
Fig. 3. The A2 domain blocks flow-dependent platelet adhesion to fibrinogen.
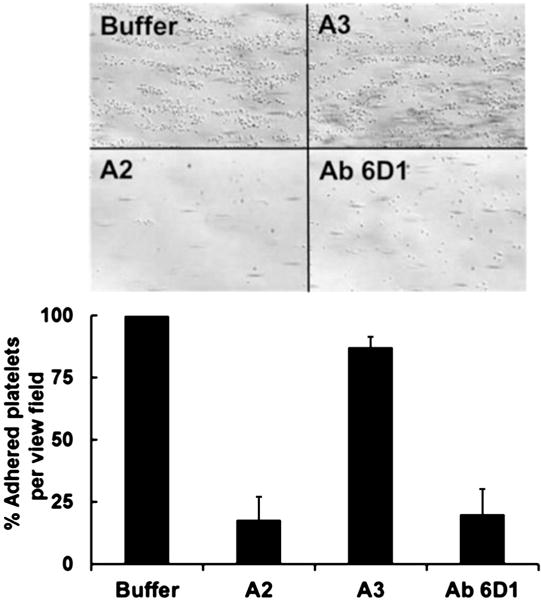
Surfaces coated with fibrinogen were incubated with buffer, A2, or A3 domain (5.0 μM) for one hour. Citrated whole blood was perfused over each surface at 1,500 s-1 shear rates. After a 2-min perfusion, the plates were washed with buffer, and the adherent platelets were recorded in several frames. The photomicrographs depict the platelets adhered to the surface after 2 min of perfusion. The monoclonal antibody against human GPIbα, Ab 6D1, was added (15 μg/ml) to the blood prior to the perfusion over fibrin(ogen) pre-incubated with buffer. The bar graph shows the % of platelets attached per view field (6–8 fields) of two separated experiments with blood from two different donors. In comparison to buffer only, the A2 domain significantly reduced platelet adhesion to fibrin(ogen) (p < 0.05).
We further analyzed whether the interaction of the purified A2 domain with fibrin monomer has functional consequences. As shown in Fig. 4A, the isolated A2 domain protein (4.0 μM) modestly delayed the polymerization of fibrin as compared to the effect of A3 domain. These results show interaction of the A2 domain specifically with fibrin monomer delays fibrin polymerization. It has been described that the interaction of soluble fibrinogen with fibrin monomer instantly provokes fibrin polymerization and formation of a clot in a thrombin independent manner [37,38]. Following those studies, we added a small amount of fibrin monomer (20 ug/ml) to citrated whole blood just before its perfusion over a surface coated with fibrin(ogen) at high shear stress (1,500 s−1). The quickly formed clots, containing platelets and red blood cells, were arrested firmly to the fibrin(ogen) surface and had a surface area of about two-three view fields. When, whole blood was incubated with A2 domain (0.4 μM) and fibrin monomer (20 μg/ml) followed by perfusion over the immobilized fibrin(ogen), the formation of platelet-clot was blocked or the size was markedly reduced (Fig. 4B, right panels).
Fig. 4. The effect of the A2 domain on fibrin polymerization.
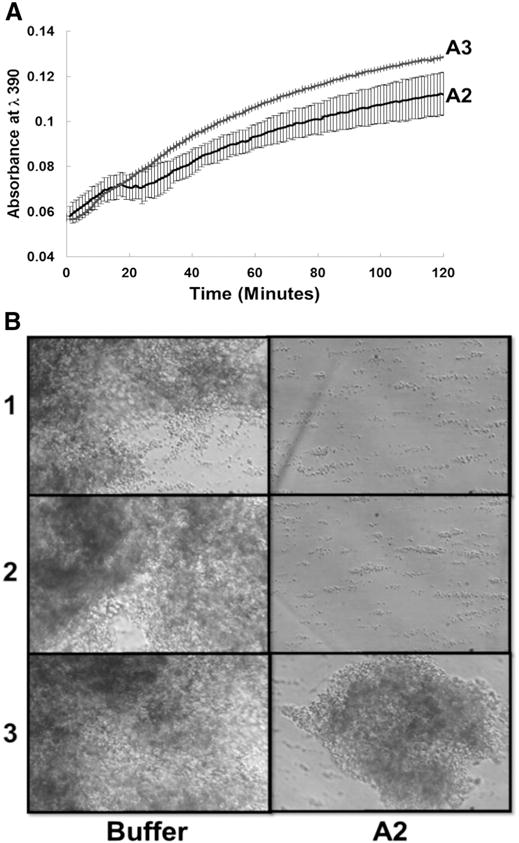
(A) A2 or A3 protein (0.13 mg/ml) was incubated with fibrinogen (0.1 mg/mL) in the presence of 1 mM CaCl2 for 30 minutes and polymerization was initiated by the addition of thrombin (0.1 U/mL) at time 0 and measured by monitoring absorbance at λ390. Measurements were made at room temperature and at interval of 20 seconds. Contrary to A3, the A2 protein modestly delayed fibrin polymerization (p < 0.05). (B) Whole blood containing buffer (left panel) or A2 domain (0.4 μM, right panel) was mixed with fibrin monomer (20 μg/ml) and perfused over the fibrinogen surface at 1,500 s-1 shear rates for 2 minutes. After a perfusion, the plates were washed and several fields were recorded to observe the size of the platelet-clot formed. The A2 protein markedly reduced the formation of the platelet-clot. Shown are photomicrographs of three separated experiments using two different blood donors.
Isolated A2 domain of human VWF inhibits fibrin-rich microthrombi, reduces inflammatory mediators, and protects against LPS induced death in mice
The ability of the purified A2 domain protein to delay fibrin polymerization and reduce the platelet-clot formation in flowing blood, prompted us to analyze its effect in vivo by using a mouse model for endotoxemia-induced DIC. The DIC model employed in this study used a high dose of LPS to promote fibrin formation and deposition in different organs [34]. Mice were challenged with LPS (25–40 mg/kg) and 1.5 hours later they were injected with either A2 domain (4 mg/kg), or saline. The A2 domain was detected in circulation for the first two hours after its infusion (Fig. S2). Organs were harvested 18 hours after the LPS injection to examine the formation of microvascular thrombosis by immunostaining for fibrin. Mice treated with LPS displayed widespread intravascular fibrin-rich microthrombi in brain, liver, kidneys, and lungs (Fig. 5A). In contrast, intravascular microthrombi were markedly diminished in A2 treated mice (Fig. 5A). Liver injury was assessed by measuring the liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST). Mice treated with the A2 domain (n = 6) had a lower AST and ALT levels at 18 hours after the LPS insult than control mice (n = 3) (34 ± 17 U/L and 100 ± 19 U/L versus with 283 ± 129 U/L and 299 ± 106 U/L, p < 0.05 for ALT and AST). Similarly, mice treated with the A2 domain had lower circulating levels of inflammatory cytokines than control mice (Fig. S3). These observations suggest that the A2 domain may indeed modulate inflammatory responses to endotoxin. In contrast, the platelet count decreased similarly in both groups (Fig. S4). Finally, there were no deaths in A2 domain-treated mice (n = 22) compared to 60% mortality in control mice (n = 17) up to 96 h after the administration of endotoxin (Fig. 5B). In other experiment there were no deaths in A2 domain-treated mice (n = 2) compared to 100% mortality in mice treated with A3 protein (n = 2) up to 72 h after the insult with LPS (not shown).
Fig. 5. The effect of A2 protein on microvascular thrombosis in vivo.
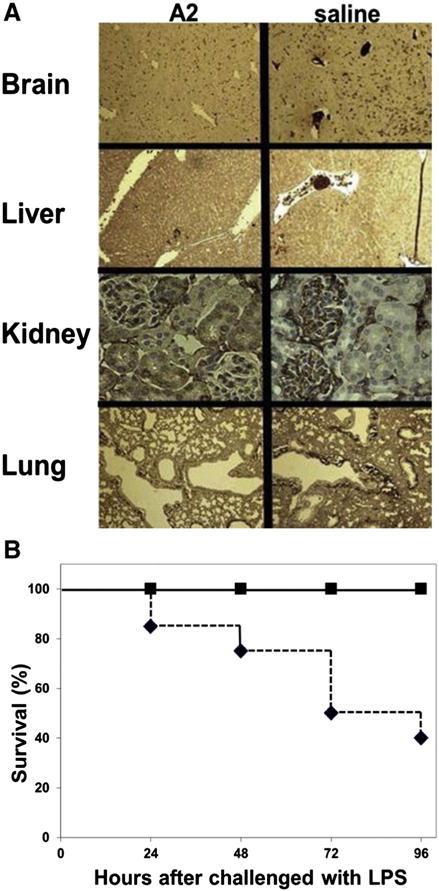
(A)Various organs were harvested at 18 hours after the administration of endotoxin to mice and stained for fibrin. Mice treated with the A2 protein are compared with mice that received saline. Dark brown color depicts the fibrin-rich microthrombi. Slides are representative for 3 mice per group. (B) The effect of A2 protein on survival. Mice treated with A2 protein (closed squares, n = 22) or saline (closed diamonds, n = 17) at 24, 48, 72, and 96 hours after the administration of endotoxin. Differences are statistically significant (p < 0.05).
Discussion
The capacity of the A2 protein to block flow-dependent platelet adhesion to immobilized fibrin(ogen), and platelet-clot formation prompted us to analyze its effect in vivo by using a mouse model for DIC. We tested the hypothesis that the A2 protein may interact with deposited fibrin, blocking platelet adhesion, and reducing the microvascular thrombi formation. In other words, impairing the interaction of plasma VWF with immobilized fibrin(ogen) results in diminishing the number of VWF molecules available to capture flowing platelets via the GPIb receptor [25]. The DIC model employed in this study used a high dose of LPS that promotes fibrin deposition in different organs, and accelerates disseminated microvascular thromboses [34], most probably by a generalized endothelial activation that leads to a prothrombotic and antifibrinolytic state. It is relevant to note that the A2 protein, which is the A2 domain of human VWF, was injected into the peritoneal cavity 1.5 hours after the LPS insult. Notably, no difference was observed in the manifestation of symptoms between the two testing groups after the LPS injection. Both the A2 administered mice and the control mice were less mobile, prostrated, head tucked into abdomen, and breathing fast after LPS administration for 16–20 hours.
The A2 protein decreased the formation of fibrin-rich microthrombi in the liver, brain, lungs, and kidneys in a murine DIC model (Fig. 4). It is well documented that a widespread microvascular thromboses caused by DIC may lead to organ failure, and death [1]. It is likely that the reduced levels of markers for liver injury as well as the improved survival are due to the decrease in fibrin-rich thrombi in A2-treated mice. Note that inhibition of the adhesion of circulating platelets to a developing fibrin clot will not inhibit the interaction of platelets with subendothelial matrix proteins (e.g. collagen). In fact, mice treated only with the A2 protein had a tail bleeding time (152.5 ± 64 s) comparable to that of mice with saline only (170 ± 48 s) (p = 0.7) for up to four hours after the injection of A2 protein or saline.
Furthermore, the levels of some inflammatory mediators were significantly reduced in mice treated with the A2 protein (Fig. S3). This outcome suggests that the recombinant protein might attenuate inflammatory responses by reducing the formation of fibrin-rich microthrombi and/or by impairing the binding of fibrin(ogen) to various integrins and adhesion molecules of inflammatory cells. This is because it is well established that fibrin(ogen) increases the interaction of those cells with endothelium that strongly increase the inflammatory responses [39]. Another potential mechanism is via vimentin since it binds to the A2 domain [35]. Vimentin, which can be found on the cell surface of different cell types including platelets and endothelial cells [40–42], plays a role in the innate immune response to infection [43]. Lastly, we have demonstrated that the interaction of isolated A2 domain with the A1 domain in VWF effectively reduces the adhesion of flowing platelets to immobilized VWF. However, the binding of the human A2 domain to murine A1 domain has not been determined. All these possibilities are being investigated with great interest.
The remarkable observed phenomenon with the A2 protein needs to be reported. There are many potential benefits of decreasing the adhesion of circulating platelets to a developing fibrin clot, especially in inflammation-induced coagulopathy. The adhesion of circulating platelets to a developing fibrin clot may lead to the formation of a life-threatening thrombosis. Moreover, approaches using synthetic biomaterials in vascular surgeries such as coronary artery bypass or cannulas in circulatory assist devices have encountered problems due in part to the adhesion of platelets to the immobilized fibrinogen on these devices, contributing for thrombus formation [44]. Therefore, the binding properties of the A2 protein for fibrin and its capacity in preventing fibrin-rich thrombi in a mouse DIC model indicate that this recombinant protein can be a potential reagent to prevent fibrin-mediated thrombosis without the risk of causing hemorrhage. Additionally, this A2 protein may represent a therapeutic option for clinical conditions associated with sustained systemic inflammation resulting in microvascular thrombosis and multiple organ failure. More experiments are being performed to dissect the mechanism of the novel and interesting observations reported here.
Finally, the purpose of this report goes beyond the description of a potential novel binding site for fibrin in the A2 domain of VWF. This intriguing concept is part of another ongoing investigation.
A limitation of this study is that even though our in vitro and in vivo data showed that A2 protein inhibited fibrin-mediated thrombotic microangiopathy and decreased mortality in a LPS-induced DIC murine model, we did not know whether these phenomena would occur in human. In fact, although we do not know this as a fact, the beneficial effect of the A2 protein could be affected by the VWF-cleaving protease in the human setting [28]. One way to address this potential issue is by mutating the amino acids that form the scissile bond in A2 protein. Finally, our in vitro data showed that the A2 protein specifically bound to fibrin(ogen), however, it is unclear whether this was the only interaction that led to the improved survival in our LPS-induced DIC murine model.
Conclusions
We found that A2 protein could inhibit platelet interaction with fibrin(ogen) and decrease mortality in a LPS-induced DIC murine model. A2 protein could provide a novel therapeutic approach in critically ill patient with uninhibited activated coagulation and disseminated fibrin deposition such as in DIC. Further studies, including large animal and human studies, are warranted to confirm our findings and to further evaluate the mechanism of A2 protein.
Supplementary Material
Acknowledgments
National Institute of Health (NIH) grants GM83212 (T.C.N) and HL72886 (M.A.C), the Alkek Foundation, Veterans Affairs Merit Review Program and Mary Gibson Foundation.
Abbreviations
- VWF
von Willebrand factor
- GP
glycoprotein
- ELISA
enzyme-linked immunosorbent assay
- LPS
lipopolysaccharide
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.thromres.2015.02.033.
Footnotes
Disclosures
“None”
References
- 1.Levi M, Van Der Poll T. Disseminated intravascular coagulation: a review for the internist. Intern Emerg Med. 2013;8:23–32. doi: 10.1007/s11739-012-0859-9. [DOI] [PubMed] [Google Scholar]
- 2.Nadel S, Goldstein B, Williams MD, Dalton H, Peters M, Macias WL, et al. Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet. 2007;369:836–43. doi: 10.1016/S0140-6736(07)60411-5. [DOI] [PubMed] [Google Scholar]
- 3.Khemani RG, Bart RD, Alonzo TA, Hatzakis G, Hallam D, Newth CJ. Disseminated intravascular coagulation score is associated with mortality for children with shock. Intensive Care Med. 2009;35:327–33. doi: 10.1007/s00134-008-1280-8. [DOI] [PubMed] [Google Scholar]
- 4.Stephan F, Hollande J, Richard O, Cheffi A, Maier-Redelsperger M, Flahault A. Thrombocytopenia in a surgical ICU. Chest. 1999;115:1363–70. doi: 10.1378/chest.115.5.1363. [DOI] [PubMed] [Google Scholar]
- 5.Vanderschueren S, De WA, Malbrain M, Vankersschaever D, Frans E, Wilmer A, et al. Thrombocytopenia and prognosis in intensive care. Crit Care Med. 2000;28:1871–6. doi: 10.1097/00003246-200006000-00031. [DOI] [PubMed] [Google Scholar]
- 6.Corrigan JJ., Jr Thrombocytopenia: a laboratory sign of septicemia in infants and children. J Pediatr. 1974;85:219–21. doi: 10.1016/s0022-3476(74)80396-3. [DOI] [PubMed] [Google Scholar]
- 7.Gando S, Saitoh D, Ogura H, Mayumi T, Koseki K, Ikeda T, et al. Natural history of disseminated intravascular coagulation diagnosed based on the newly established diagnostic criteria for critically ill patients: results of a multicenter, prospective survey. Crit Care Med. 2008;36:145–50. doi: 10.1097/01.CCM.0000295317.97245.2D. [DOI] [PubMed] [Google Scholar]
- 8.Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–79. doi: 10.1016/0049-3848(85)90180-x. [DOI] [PubMed] [Google Scholar]
- 9.Burke AP, Mont E, Kolodgie F, Virmani R. Thrombotic thrombocytopenic purpura causing rapid unexpected death: value of CD61 immunohistochemical staining in diagnosis. Cardiovasc Pathol. 2005;14:150–5. doi: 10.1016/j.carpath.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Levi M, de Jonge E, van der PT. New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Ann Med. 2004;36:41–9. doi: 10.1080/07853890310017251. [DOI] [PubMed] [Google Scholar]
- 11.Wada H, Thachil J, Di NM, Mathew P, Kurosawa S, Gando S, et al. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. J Thromb Haemost. 2013;11(4):761–7. doi: 10.1111/jth.12155. [DOI] [PubMed] [Google Scholar]
- 12.Aoki N, Matsuda T, Saito H, Takatsuki K, Okajima K, Takahashi H, et al. A comparative double-blind randomized trial of activated protein C and unfractionated heparin in the treatment of disseminated intravascular coagulation. Int J Hematol. 2002;75:540–7. doi: 10.1007/BF02982120. [DOI] [PubMed] [Google Scholar]
- 13.Jaimes F, De La Rosa G, Morales C, Fortich F, Arango C, Aguirre D, et al. Unfractioned heparin for treatment of sepsis: A randomized clinical trial (The HETRASE Study) Crit Care Med. 2009;37:1185–96. doi: 10.1097/CCM.0b013e31819c06bc. [DOI] [PubMed] [Google Scholar]
- 14.Sakuragawa N, Hasegawa H, Maki M, Nakagawa M, Nakashima M. Clinical evaluation of low-molecular-weight heparin (FR-860) on disseminated intravascular coagulation (DIC)–a multicenter co-operative double-blind trial in comparison with heparin. Thromb Res. 1993;72:475–500. doi: 10.1016/0049-3848(93)90109-2. [DOI] [PubMed] [Google Scholar]
- 15.Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–78. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 16.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–47. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 17.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 18.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–64. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 19.de Kleijn ED, de GR, Hack CE, Mulder PG, Engl W, Moritz B, et al. Activation of protein C following infusion of protein C concentrate in children with severe meningococcal sepsis and purpura fulminans: a randomized, double-blinded, placebo-controlled, dose-finding study. Crit Care Med. 2003;31:1839–47. doi: 10.1097/01.CCM.0000072121.61120.D8. [DOI] [PubMed] [Google Scholar]
- 20.Rintala E, Kauppila M, Seppala OP, Voipio-Pulkki LM, Pettila V, Rasi V, et al. Protein C substitution in sepsis-associated purpura fulminans. Crit Care Med. 2000;28:2373–8. doi: 10.1097/00003246-200007000-00032. [DOI] [PubMed] [Google Scholar]
- 21.Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost. 2007;5:31–41. doi: 10.1111/j.1538-7836.2006.02267.x. [DOI] [PubMed] [Google Scholar]
- 22.Endenburg SC, Hantgan RR, Sixma JJ, de Groot PG, Zwaginga JJ. Platelet adhesion to fibrin(ogen) Blood Coagul Fibrinolysis. 1993;4:139–42. [PubMed] [Google Scholar]
- 23.Zaidi TN, McIntire LV, Farrell DH, Thiagarajan P. Adhesion of platelets to surface-bound fibrinogen under flow. Blood. 1996;88:2967–72. [PubMed] [Google Scholar]
- 24.Loscalzo J, Inbal A, Handin RI. von Willebrand protein facilitates platelet incorporation in polymerizing fibrin. J Clin Invest. 1986;78:1112–9. doi: 10.1172/JCI112668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Endenburg SC, Hantgan RR, Lindeboom-Blokzijl L, Lankhof H, Jerome WG, Lewis JC, et al. On the role of von Willebrand factor in promoting platelet adhesion to fibrin in flowing blood. Blood. 1995;86:4158–65. [PubMed] [Google Scholar]
- 26.Beguin S, Kumar R, Keularts I, Seligsohn U, Coller BS, Hemker HC. Fibrin-dependent platelet procoagulant activity requires GPIb receptors and von Willebrand factor. Blood. 1999;93:564–70. [PubMed] [Google Scholar]
- 27.Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. 2012;120:449–58. doi: 10.1182/blood-2012-01-405134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz MA, Whitelock J, Dong JF. Evaluation of ADAMTS-13 activity in plasma using recombinant von Willebrand Factor A2 domain polypeptide as substrate. Thromb Haemost. 2003;90:1204–9. doi: 10.1160/TH03-06-0398. [DOI] [PubMed] [Google Scholar]
- 29.Gorkun OV, Veklich YI, Weisel JW, Lord ST. The conversion of fibrinogen to fibrin: recombinant fibrinogen typifies plasma fibrinogen. Blood. 1997;89:4407–14. [PubMed] [Google Scholar]
- 30.Martin C, Morales LD, Cruz MA. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. J Thromb Haemost. 2007;5:1363–70. doi: 10.1111/j.1538-7836.2007.02536.x. [DOI] [PubMed] [Google Scholar]
- 31.Auton M, Sowa KE, Smith SM, Sedlak E, Vijayan KV, Cruz MA. Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibalpha and platelet activation under shear stress. J Biol Chem. 2010;285:22831–9. doi: 10.1074/jbc.M110.103358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cruz MA, Chen J, Whitelock JL, Morales LD, Lopez JA. The platelet glycoprotein Ib-von Willebrand factor interaction activates the collagen receptor alpha2beta1 to bind collagen: activation-dependent conformational change of the alpha2-I domain. Blood. 2005;105:1986–91. doi: 10.1182/blood-2004-04-1365. [DOI] [PubMed] [Google Scholar]
- 33.Morales LD, Martin C, Cruz MA. The interaction of von Willebrand factor-A1 domain with collagen: mutation G1324S (type 2 M von Willebrand disease) impairs the conformational change in A1 domain induced by collagen. J Thromb Haemost. 2006;4:417–25. doi: 10.1111/j.1538-7836.2006.01742.x. [DOI] [PubMed] [Google Scholar]
- 34.Levi M, Dorffler-Melly J, Reitsma P, Buller H, Florquin S, Van Der Poll T, et al. Aggravation of endotoxin-induced disseminated intravascular coagulation and cytokine activation in heterozygous protein-C-deficient mice. Blood. 2003;101:4823–7. doi: 10.1182/blood-2002-10-3254. [DOI] [PubMed] [Google Scholar]
- 35.Da Q, Behymer M, Correa JI, Vijayan KV, Cruz MA. Platelet adhesion involves a novel interaction between vimentin and von Willebrand factor under high shear stress. Blood. 2014;123(17):2715–21. doi: 10.1182/blood-2013-10-530428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keuren JF, Baruch D, Legendre P, Denis CV, Lenting PJ, Girma JP, et al. von Willebrand factor C1C2 domain is involved in platelet adhesion to polymerized fibrin at high shear rate. Blood. 2004;103:1741–6. doi: 10.1182/blood-2003-07-2267. [DOI] [PubMed] [Google Scholar]
- 37.Olexa SA, Budzynski AZ. Evidence for four different polymerization sites involved in human fibrin formation. Proc Natl Acad Sci U S A. 1980;77:1374–8. doi: 10.1073/pnas.77.3.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Husain SS, Weisel JW, Budzynski AZ. Interaction of fibrinogen and its derivatives with fibrin. J Biol Chem. 1989;264:11414–20. [PubMed] [Google Scholar]
- 39.Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med. 2011;17:568–73. doi: 10.2119/molmed.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pall T, Pink A, Kasak L, Turkina M, Anderson W, Valkna A, et al. Soluble CD44 interacts with intermediate filament protein vimentin on endothelial cell surface. PLoS One. 2011;6:e29305. doi: 10.1371/journal.pone.0029305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou Y, He L, Huang SH. Identification of a surface protein on human brain microvascular endothelial cells as vimentin interacting with Escherichia coli invasion protein IbeA. Biochem Biophys Res Commun. 2006;351:625–30. doi: 10.1016/j.bbrc.2006.10.091. [DOI] [PubMed] [Google Scholar]
- 42.Podor TJ, Singh D, Chindemi P, Foulon DM, McKelvie R, Weitz JI, et al. Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J Biol Chem. 2002;277:7529–39. doi: 10.1074/jbc.M109675200. [DOI] [PubMed] [Google Scholar]
- 43.Mor-Vaknin N, Legendre M, Yu Y, Serezani CH, Garg SK, Jatzek A, et al. Murine colitis is mediated by vimentin. Sci Rep. 2013;3:1045. doi: 10.1038/srep01045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25:5681–703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.