

ABSTRACT
We performed ultra-deep methylation analysis at single molecule level of the promoter region of developmentally regulated D-Aspartate oxidase (Ddo), as a model gene, during brain development and embryonic stem cell neural differentiation. Single molecule methylation analysis enabled us to establish the effective epiallele composition within mixed or pure brain cell populations. In this framework, an epiallele is defined as a specific combination of methylated CpG within Ddo locus and can represent the epigenetic haplotype revealing a cell-to-cell methylation heterogeneity. Using this approach, we found a high degree of polymorphism of methylated alleles (epipolymorphism) evolving in a remarkably conserved fashion during brain development. The different sets of epialleles mark stage, brain areas, and cell type and unravel the possible role of specific CpGs in favoring or inhibiting local methylation. Undifferentiated embryonic stem cells showed non-organized distribution of epialleles that apparently originated by stochastic methylation events on individual CpGs. Upon neural differentiation, despite detecting no changes in average methylation, we observed that the epiallele distribution was profoundly different, gradually shifting toward organized patterns specific to the glial or neuronal cell types. Our findings provide a deep view of gene methylation heterogeneity in brain cell populations promising to furnish innovative ways to unravel mechanisms underlying methylation patterns generation and alteration in brain diseases.
KEYWORDS: Brain DNA methylation, brain cells, epipolymorphism, epialleles, epigenetic dynamics
Introduction
Epigenetic profiles are sculpted during development; in particular, the DNA methylation landscape of mammalian brain cells is dynamically reconfigured through development.1-3 Such reconfiguration occurs at brain specific genes prevalently during the late stages of embryogenesis and early post-natal developmental period, leading to critical changes in the gene expression program that, once the process comes to an end, is thought to be maintained throughout life. A progressive shaping of DNA methylation patterns occurs in a regulated manner, both at CpG and CpH sites, during development and first weeks or years of life, possibly providing an epigenetic memory specific for each type of brain cells.3 Such phenomenon, crucial to the completion of embryonic development, is the result of a dynamic interplay between DNA methylation and demethylation events assisted by different DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b), which may convert cytosine to methylcytosine,4-7 or by Ten-11 translocation enzymes (TETs), which promote 5mC demethylation.8 At the genomic level, as a result of these processes, each type of brain cells acquires a cell type-specific genomic DNA methylation landscape that may provide an ‘identity card’ for different brain cells and govern and stabilize an elected gene expression program. A recent study9 showed that different types of glial cells and neurons display very distinctive methylation signatures, suggesting that the epigenomic landscape reflects neuronal diversity. Appropriate patterns of DNA methylation in the brain play an important role in neurodevelopment and neuropsychiatric conditions,10-20 pointing out to the importance of correct formation and preservation of DNA methylation patterns in brain cells. However, the vast majority of these studies, regardless of the techniques employed, took in consideration the average amount of CpG methylation in specific genomic regions or their genome-wide distribution with only relative high resolution. In principle, each cell may bear a specific combination of methylated CpGs at specific loci that may reflect the origin of the cell and/or the functional state of a given gene (allele) in that cell. This introduces the concept of “epipolymorphism,” which goes far beyond the simple identification of differentially methylated regions, by means that brain cells may be considered a population of epigenetically heterogeneous cells in which each combination of methyl CpGs at a given locus represents a specific epiallele. Such information is lost when the average methylation, even at single CpG sites, is evaluated. Using genome-wide approaches, recent studies based on the comparison of the methylation of few adjacent CpGs have addressed the issue of heterogeneous methylation, accounting for cell-to-cell methylation variability in liver,21 leukemias,22 and immortalized fibroblasts,23 describing the stochastic and clonal evolution of epialleles during carcinogenesis.
In this work, we investigated whether cell-to-cell methylation variability exists in brain and if this variability represents the result of methylation and demethylation events occurring stochastically at individual CpG sites or derives from a well orchestrated process. In order to gain insight into this matter, we performed ultra-deep methylation single molecule analysis of a selected locus, the D-Aspartate oxidase (Ddo) gene,24,25 in different brain developmental stages, areas, cell types, and embryonic stem cells (ESCs) neural differentiation process. The choice of Ddo as a model gene was mainly based on the facts that this gene is developmentally regulated in brain by DNA methylation changes,25 the average methylation levels range between 60 and 30% in different stages, and the promoter is relatively poor in CpG content,25 which we considered ideal conditions to study variation of intermediate epiallele composition at different times during development and in different brain cell types.
Results
Epiallele analysis principles
Amplicon bisulfite sequencing of a given genomic region enables us to determine whether each included CpG dinucleotide, in each single molecule, is methylated or unmethylated. Upon high-coverage bisulfite sequencing, it is possible to determine, with high precision, either the average methylation at each CpG site or the asset of methylated and unmethylated CpG sites present in each amplicon-derived sequence. As an example, a region that includes 4 CpG sites may give origin to 16 possible combinations that may be potentially found in a mixed population of cells (Fig. 1). These different combinations will be here referred to as “epialleles;” the number of different exhibited epialleles provides a measure of the level of “epipolymorphism” (see Fig. 1 for details and examples). We applied this kind of methylation analysis to investigate the epiallele combinations and evolution of the Ddo gene, as example of a locus undergoing methylation changes during brain development.25
Figure 1.
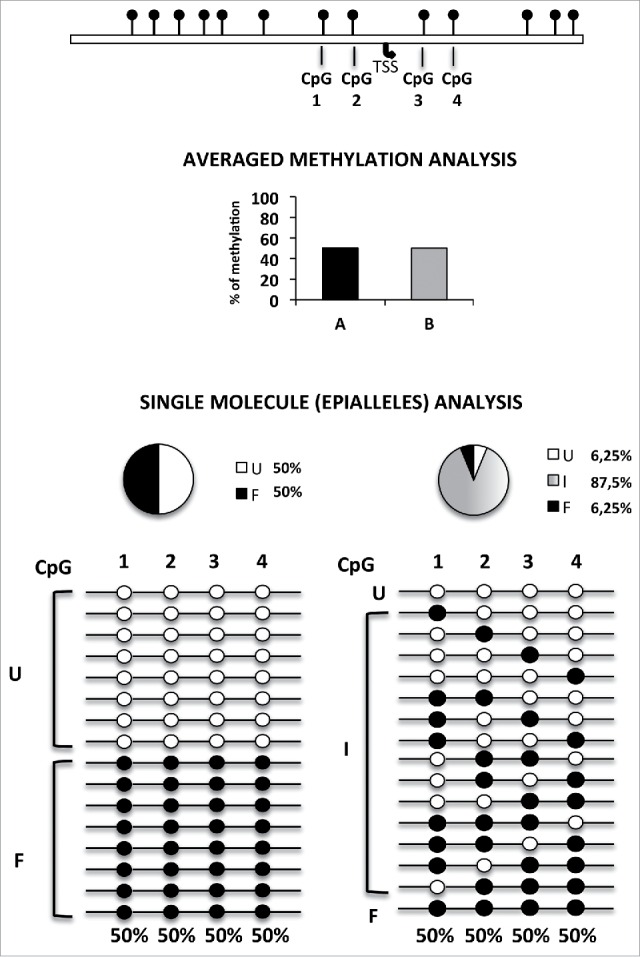
Principle of epipolymorphism and epiallele analysis. Averaged methylation degree, even at single base resolution, does not give any information on cell-to-cell methylation variability. In the example reported in the Figure, which shows analysis of 4 adjacent CpG sites, 50% methylation degree may correspond to completely different methylation scenarios. These range from the lowest degree of epipolymorphism (bottom left) to the highest level (bottom right). However, reliable analysis of the relative frequency of each epiallele must rely on a high number of analyzed sequences. Compared to genomic approaches previously used to measure the general epipolymorphism degree, the here adopted amplicon bisulfite sequencing, although limited to targeted genomic regions, allowed us to magnify the details of cell-to-cell epiallele variability at single loci, thanks of the very high sequencing coverage (about 210,000 in this study) and to comprehensively establish the methylation pattern of several adjacent CpG sites of longer regions (up to 600–1000 bp) in which all CpGs within individual reads are effectively phased and may represent the epigenetic haplotype. TSS: transcription start site. The pie charts represent the percentage of fully unmethylated epialleles (U = white), fully methylated epialleles (F = black), and the 62 remaining possible methyl CpG combination or intermediate epialleles (I = gray gradient).
Averaged methylation and epiallele frequency distribution analysis of a 3 kb region in the Ddo gene in whole brain during development
Using mouse brain at stages embryonic day 15 (E15), post-natal day 0 (P0), and post-natal day 30 (P30), we performed high-coverage targeted bisulfite sequencing including 7 amplicons (R1-R7) covering an extended large genomic region (about 3 kb) surrounding the Ddo transcriptional start site (TSS) (Fig. 2A). Primers used in this study are reported in Table 1. Note that we previously reported averaged quantitative methylation data for regions R4 and R5,25 which we have here integrated into Fig. 2B in order to provide a comprehensive view of averaged methylation changes at the extended Ddo locus. Then, we performed single molecule analysis of the R4 region by determination of epiallele frequency (Fig. 3A). Sixty-four (26) possible epialleles were expected. Fig. 3B shows the results from the analysis of whole brains (n = 3) at E15 stage. Several interesting aspects were worth of note. First, we found, with different frequencies, the occurrence of almost all the theoretically predicted epialleles. At E15, fully methylated molecules represented about 13% (the average methylation for each site was evaluated at about 56%), the unmethylated molecules totaled about 10%, while the remaining 77% of the cells harbored one of the 62 intermediate epialleles, which, although with different frequencies, were all represented in the brain cell mixture. However, we observed that some specific epialleles were represented with higher than expected frequency. Such frequency distribution was strikingly conserved among individuals. These findings highlighted either a high degree of epipolymorphism of the Ddo promoter in the brain or a high interindividual conservation of epiallele frequency distribution.
Figure 2.
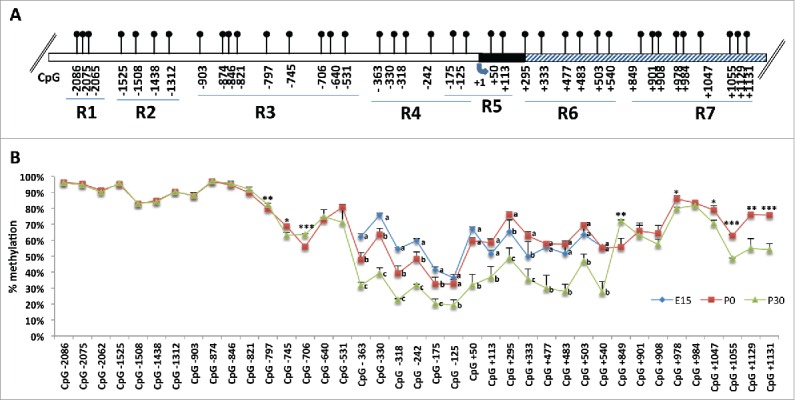
Quantitative methylation analysis of a 3 kb genomic region of Ddo gene. A) Structure of the putative mouse Ddo gene promoter. Blue arrow indicates the transcription start site (+1). White box represents the putative regulatory upstream region. Black box represents exon 1. Gray box represents first intron. Positions of CpG sites are indicated as relative to TSS. The analyzed 3 kb genomic portion is divided in 7 regions (R1-R2-R3-R4-R5-R6-R7) corresponding to different amplicons. B) Graph represents methylation average at each CpG site analyzed by Illumina MiSeq Sequencer. Each color line represents the average of 3 mice at a given developmental stage. Blue = E15; red = P0; green = P30. Error bars show standard deviations. For each CpG site, the points labeled with different letters on top are significantly different (P < 0.05) based on post-hoc ANOVA analysis (Tukey test, performed on each CpG site). Where two developmental times were considered, Student t test was applied with *P < 0.05, **P < 0.01, ***P < 0.001.
Table 1.
List of primers used to amplify Ddo promoter regions (R1-R7) and the M13 mp18 control plasmid. For Ddo regions, the positions refer to TSS; for M13 mp18 the positions refer to sequence entry X02513.1 (NCBI, GenBank).
| GENE | AMPLICON | FW PRIMER | RV PRIMER |
|---|---|---|---|
| Ddo R1 | −2170/−1960 | TttggtttttattTtTaaatTTtgata | AactAActatacatctcacttccctA |
| Ddo R2 | −1578/−1184 | GgttagtggtttTtttgTagTTtttat | AAcattAtcccttcaAtccacaat |
| Ddo R3 | −927/−499 | GTttttTTaTatgtTttggagTTt | acctccctAaaaAtcatttAattcta |
| Ddo R4 | −468/−63 | GtgtgtttTtgaggaggtgaTaTtTa262 3 | aActtaccctccattAAtccatAcc |
| Ddo R5 | −229/+144 | GgTtggtggTaagTtgaagttTttg | acccctaaaatcccaAaAtAcatac |
| Ddo R6 | +267/+671 | TTtagtgttaaTttattagagTtgtgg | AAtacaatcccttcttAcaacaAAca |
| Ddo R7 | +801/+1201 | GagggagttgggTatggagTaTaTata | AactctaAAAaAcaAacacaAaAAtc |
| M13 mp18 | 5946 / 6294 | Ggtgaagggtaattagttgttgtt | ccaataccaaacttacatacct |
The capital letters in the primers sequences indicate the original C or G.
Figure 3.

All possible methyl CpG combinations (epialleles) at the Ddo R4 region. A) Representation of all 64 theoretically possible epialleles at region R4 of Ddo promoter containing 6 CpG sites. Circles represent the CpG sites analyzed (−363, −330, −318, −242, −175, and −125); black circles = methylated CpG; white circles = unmethylated CpG. B) Percent frequency of each epiallele in whole brain from 3 mice (M1, M2, and M3) at E15 stage.
The interindividual conservation of epialleles patterns was strikingly retained in brains at each of the analyzed developmental stages from E15 to P30 (0.786 ≤ Pearson R ≤ 0.992, P < 0.001) (Fig. 4, Table 2). Simulation approach confirmed the non-randomness of the observed epialleles distribution showing a consistent deviation from the simulated frequency for each of the epialleles (Supplemental Fig. S1). Thus, the observed phenomenon appears to be a consequence of a precise mechanism governing the formation and maintenance of predetermined methylation profiles in each of the different brain cells. The same analyses were performed on other regions of the Ddo gene (R1-R7). Although each of the analyzed regions showed variable numbers of CpGs and different methylation averages (ranging from 96 to 20%) and variation over developmental stages, the data clearly confirmed the existence of a strong interindividual conservation for all the regions (Supplemental Fig. S2 and data not shown). However, lower epiallele polymorphism and no changes over time were observed in the upstream regions R1 and R2, as expected by the constant high rate of average methylation in these regions (85–95%) (Supplemental Fig. S2).
Figure 4.
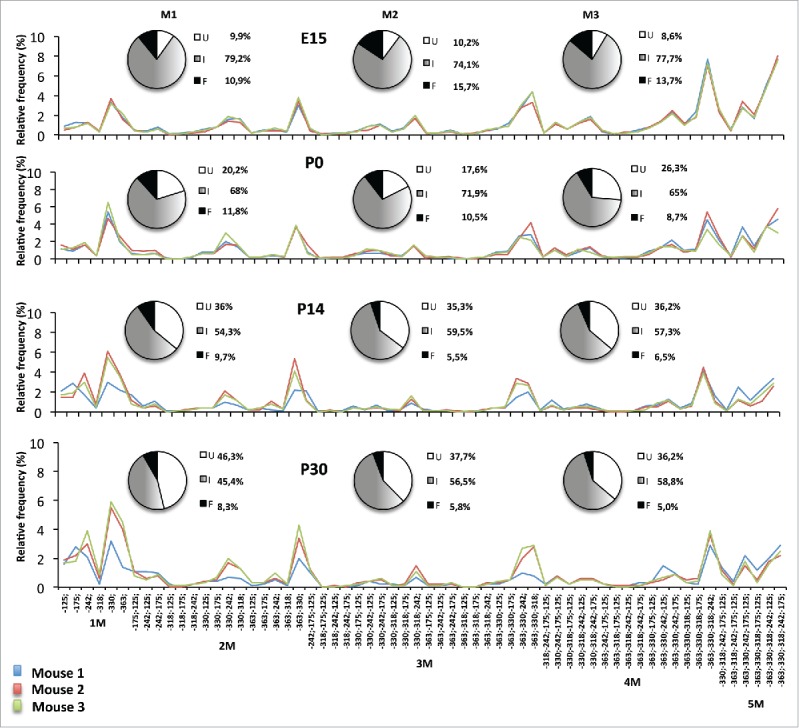
Epialleles frequency distribution at Ddo R4 region during different stages of mouse development (E15, P0, P14, P30). The pie charts represent the percentage of unmethylated epialleles (U = white), fully methylated epialleles (F = black), and all 62 intermediate epialleles (I = gray gradient). The line graph shows the percentage of each of the intermediate epialleles; each color line identifies one mouse. Blue = mouse 1; red = mouse 2, green = mouse 3. At the bottom of the last graph, the specific methyl CpG combinations of the 62 intermediate epialleles are reported. 1M identifies the group of monomethylated molecules; 2M = dimethylated; 3M = trimethylated; 4M = tetramethylated; 5M = pentamethylated epialleles. Pearson correlation R reported in Table 2.
Table 2.
Relationship among the epialleles distribution within each stages group. The analyses were performed by correlating mice of each developmental stage in twos. Pearson's correlation coefficient R and P-value are reported.
| Pearson correlation test of epialleles distribution Ddo R4 region | |||
|---|---|---|---|
| STAGE | Mice | Pearson R | P-value |
| E15 | M1/M2 | 0.986067197 | 1.89637E–48 |
| E15 | M1/M3 | 0.992391755 | 2.71669E–56 |
| E15 | M2/M3 | 0.9868598 | 3.30893E–49 |
| P0 | M1/M2 | 0.959438583 | 1.08587E–34 |
| P0 | M1/M3 | 0.944346038 | 1.15656E–30 |
| P0 | M2/M3 | 0.88145423 | 3.27535E–21 |
| P14 | M1/M2 | 0.786309702 | 3.66001E–14 |
| P14 | M1/M3 | 0.846990179 | 4.13024E–18 |
| P14 | M2/M3 | 0.981271547 | 1.26582E–44 |
| P30 | M1/M2 | 0.833321788 | 4.37257E–17 |
| P30 | M1/M3 | 0.801413887 | 5.13155E–15 |
| P30 | M2/M3 | 0.984769846 | 2.69114E–47 |
Different brain areas display distinct epiallele configurations
The eventuality that distinctive epiallele patterns were recognizable in different brain areas was investigated. Methylation average and epiallele frequency were analyzed in hippocampus, cortex, prefrontal cortex (PFC), cerebellum, and striatum at R4 in the Ddo promoter region. By averaged methylation analysis the highest methylation levels (about 50%) were observed in striatum (Fig. 5A and Fig. 5B). Conversely, in hippocampus, the lowest methylation level (about 15%) was present. Cerebellum, cortex, and PFC showed intermediate levels (about 20–25%) that resembled those observed in whole brain. We analyzed the frequency of intermediate epialleles and observed high epiallele heterogeneity within each area (Fig. 5C). Interestingly, although the profiles were clearly distinguishable among the different brain areas, these mainly differed in the relative abundance of a few major preferential CpG combinations (see peaks in the bottom graph in Fig. 5C). Moreover, it is worth noting that even in highly methylated striatum (average methylation level of about 50%), more than 30% of the cells bear fully unmethylated alleles, against 18% of cells bearing fully methylated ones.
Figure 5.
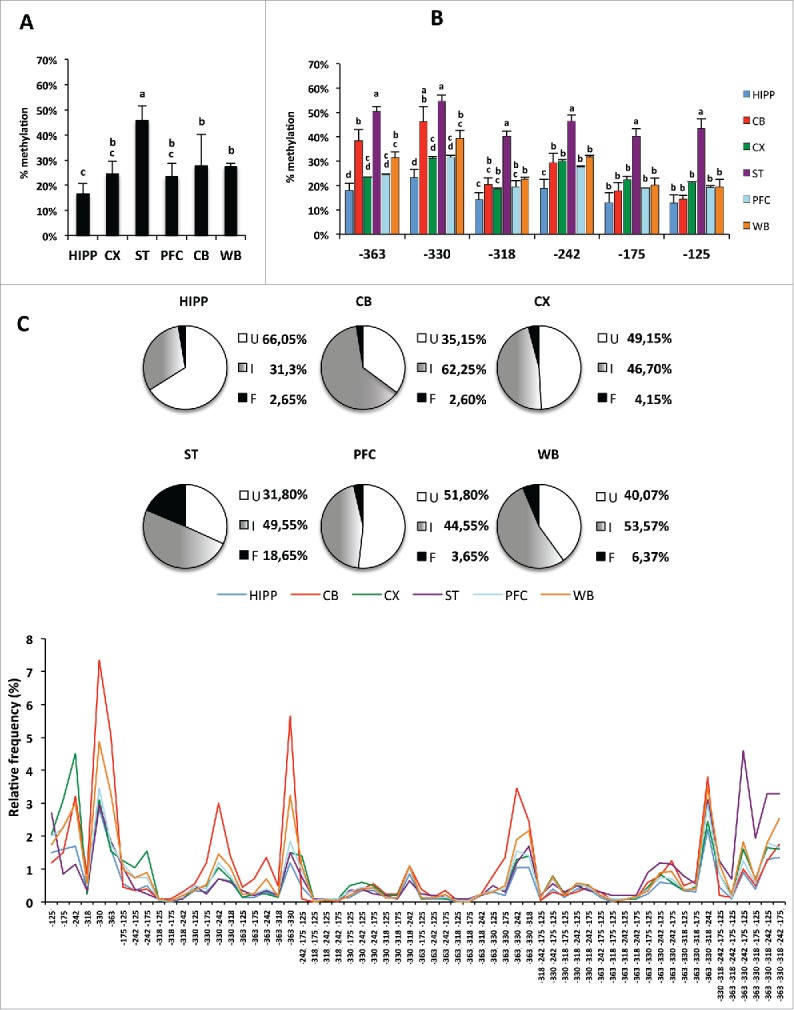
Epiallele composition analysis of Ddo R4 in separated areas of mouse brain (Hipp: hippocampus; CB: cerebellum; CX: Cortex; ST: striatum; PFC: prefrontal cortex) and in whole brain (WB) at P30 stage. A) Average methylation at the Ddo R4 region in each separated brain area. The bars labeled with different letters on top are significantly different based on post-hoc ANOVA statistical analysis (Tukey test). B) Average methylation at each CpG site (−363, −330, −318, −242, −175, and −125). The bars labeled with different letters on top are significantly different based on post-hoc ANOVA analysis (Tukey test) performed for each CpG site. C) Ddo R4 region epiallele frequency distribution. The pie charts represent the percentage of unmethylated epialleles (U = white); fully methylated epialleles (F = black); and all 62 intermediate epialleles (I = gray gradient). Each color line represents the distribution of 62 intermediate epialleles in one brain area, as indicated. At the bottom of the graph, the specific methyl CpG combinations of each of the 62 intermediate epialleles are reported. A P-value ≤ 0.05 was considered statistically significant.
Specific epiallele patterns distinguish different purified brain cell populations
The high degree of epipolymorphism found both in total brain and separated brain areas could be potentially explained by cellular heterogeneity. Therefore, we decided to investigate whether different purified brain cells, namely neurons, oligodendrocytes, astrocytes, and microglial cells, displayed cell type-specific methylation features. First, we performed quantitative methylation analysis and showed that different cell types exhibited different average methylation degree, with astrocytes being the less methylated and microglial cells the most methylated (Fig. 6A). Analysis of epiallele frequency distribution indicated that, as was also observed in separated brain areas, that higher average methylation levels were associated with higher percentage of hypermethylated epihaplotypes [see, as an example, microglia (green line) vs. astrocytes (purple line) in Fig. 6B]. Conversely, lower average methylation was associated with higher frequency of specific mono- and di-methylated conformations. However, specific epialleles, although enriched at different degrees, showed peaks that were clearly coincident across cell types and, for the most part, also with peaks observed in whole brain tissue samples (compare Figs. 4 and 5). These observations again point to the existence of a hierarchy among epialleles that is highly conserved across different brain derived cells. In summary, we confirmed that most of the methylation rules observed in brain tissues applied also for isolated brain cells, including the high percentage of intermediate epialleles, high degree of epipolymorphism, and high conservation of epialleles frequency among the same type of cells among individuals. Our data strongly suggest that, within a cell population, either in whole brain, separated brain areas, or isolated primary cells, the epiallele frequency distribution occurs in a non-stochastic manner. In particular, we observed a spatial specificity by means of some CpG sites (−363, −330, −242) being prone and some (−318, −125, −175) being more resistant to methylation. However, in a subset of cells for each lineage, in a proportion consistent with the level of average methylation, methylation spread from susceptible sites by recruiting nearby CpGs, increasing the number of molecules in which resistant CpGs became methylated. In the future, it would be particularly intriguing to explore the potential contribution of epiallele distribution analysis to the reliable evaluation of cell function and cell type-specific enrichment in given areas, during lifetime and/or in pathological conditions.
Figure 6.
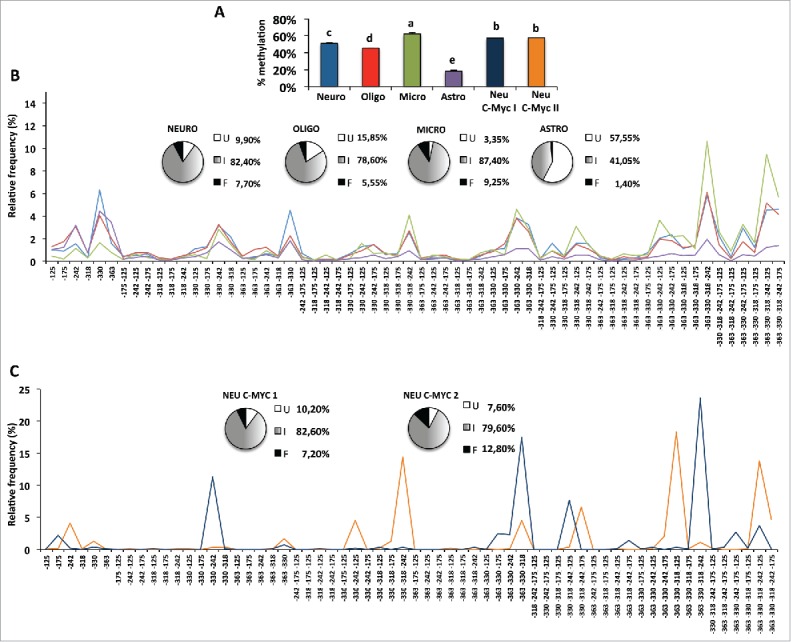
Averaged and single molecule methylation analysis of the Ddo promoter region in isolated primary brain cells and in cultured c-myc immortalized neurons. A) Average methylation in different indicated brain cells. The bars labeled with different letters on top are significantly different based on post-hoc ANOVA statistical analysis (Tukey test). B) Frequency distribution of intermediate epialleles in primary cells. C) Epiallele distribution in c-myc cultured immortalized neurons. Neuro: primary neurons, blue bars in A and blue lines in B. Oligo: primary oligodendrocytes, red bars in A and red lines in B. Micro: primary microglial cells, green bars in A and green lines in B. Astro: primary astrocytes cells, violet bars in A and violet lines in B. Neu C-Myc I and Neu C-Myc II: A1 C-Myc immortalized neurons, dark blue and orange, respectively, in A (bars) and C (lines). The pie charts represent the percentage of unmethylated epialleles (U = white); fully methylated epialleles (F = black); and all 62 intermediate epialleles (I = gray gradient). At the bottom of each graph, the specific methyl CpG combinations of each of the 62 intermediate epialleles are reported.
Primary vs. immortalized neuronal cells
Interestingly, when we analyzed epiallele frequencies in A1 cells, c-myc immortalized neurons at different culture passages (Fig. 6C), despite observing an average methylation similar to the one observed in primary neurons (see Fig. 6A and pies in 6B and 6C), an opposite methylation scenario was observed in terms of epiallele frequency distribution. In fact, few epialleles were highly prevalent, most were absent, and epiallele profiles were discordant both among different plates (Pearson R = 0.083) and when compared with those derived by primary cells. These data suggest that immortalized neurons, by contrast with tissue and primary cells, show lower epiallele heterogeneity (clonal-like behavior) and a likely stochastic epiallele frequency distribution.
Dynamic remodeling of Ddo epiallele profiles in ESCs upon induction of neural differentiation
Evolution of epiallele frequency distribution at the Ddo promoter was then tracked in ESCs upon neural differentiation. Although average methylation of the 6 CpG sites at the Ddo R4 region was almost identical in undifferentiated and differentiated ESCs (Fig. 7A), and minor differences were observed at single CpG sites (Fig. 7B), when analysis of epiallele frequency distribution was applied we observed a striking and reproducible methylation drift in 3 independent experiments (Fig. 7C). Undifferentiated ESCs displayed an apparently disorganized, rather scattered, distribution of the possible 62 combinations, with a slight prevalence of monomethylated molecules. Upon neural differentiation toward neural cells, we observed a specific and highly reproducible rearrangement of the epiallele frequency distribution profile (Pearson R = 0.492, P < 0.001) (Fig. 7C and Fig. 7D) that strongly supported the existence of a programmed combinatorial code and a methylation drift within the cell population. In particular, differentiated ESCs showed sharp organized peaks corresponding to constant enrichment of few specific intermediate epialleles and concomitant impoverishment of most of the others and their derivatives (e.g., molecules containing methylated −242 and/or −125). Noticeably, 7 (out of 62) intermediate epialleles that bloomed upon differentiation (−330; −363; −330/−318; −363/−330; −363/−330/−318; 363/−330/−318/−242, 363/−330/−318/−242/−175) mostly corresponded to the major peaks observed in developing whole brain as well as neurons and glial cells, with different relative abundance. In fact, we performed an analysis of the epigenetic correlation based either on the type of displayed epialleles and on the relative amount of each possible methyl CpG arrangement found in each analyzed sample. This was performed by principal component analysis (PCA). Results reported in Fig. 8A and Fig. 8B confirmed the reproducibility of the Ddo epigenetic drift between undifferentiated and differentiated state and showed that differentiated ESCs dramatically shifted toward mature neural and glial patterns. Taken together, these data are compatible with a model in which in the undifferentiated state the CpG methylability at the Ddo promoter mainly depends on the primary sequence and occurs in a random manner at one or more CpG sites. Conversely, in the differentiated state, possibly due to newly generated structured chromatin conformation constraints, a coordinated interplay between contiguous CpGs differing in methylation susceptibility underlies specific epiallele frequency and dynamics in a spatial-specific manner, where the methylated state of more susceptible sites (−330 and −363) increases the likelihood that adjacent resistant sites become methylated. These basic rules are then retained in the mature glia and neurons. In support of this model, Fig. 8C shows the relative contribution of each CpG to the formation of different epialleles.
Figure 7.
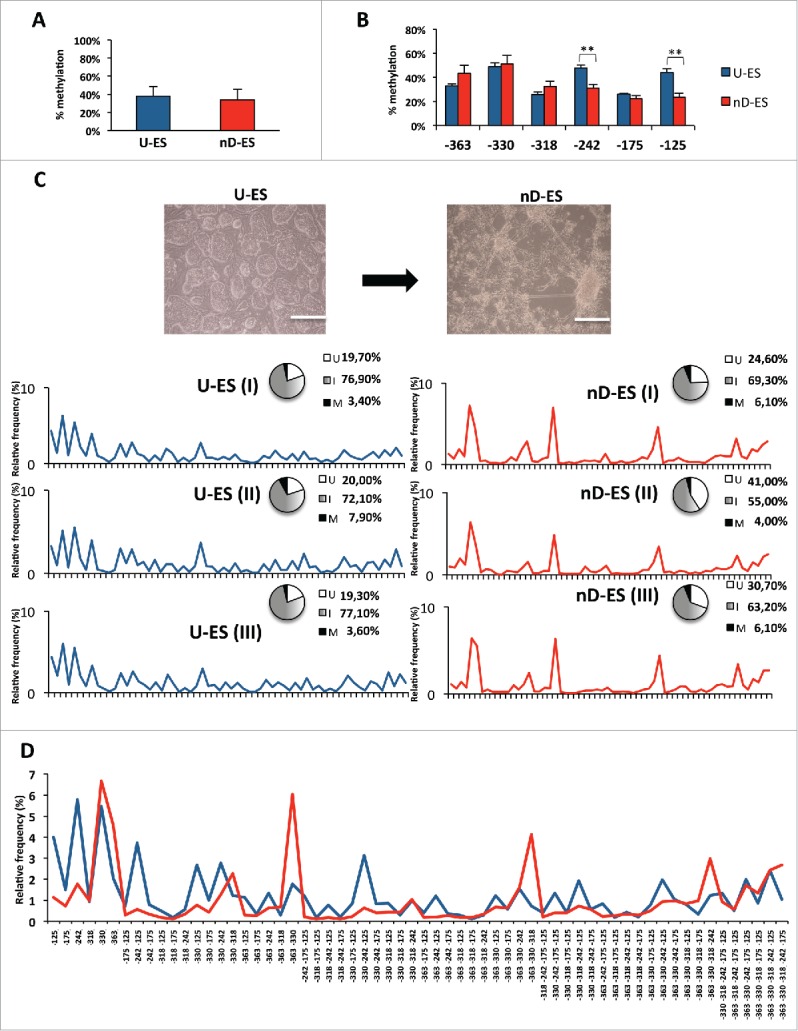
Single molecule methylation analysis in ESC neural differentiation model. A) Averaged methylation at Ddo R4 region in undifferentiated ESCs (U-ES) and in differentiated ESCs (nD-ES) from 3 independent experiments. B) Averaged methylation of each CpG site (−363, −330, −318, −242, −175, and −125) at Ddo R4 region (data are showed as mean ± standard deviation; **P < 0.01). C) Microscope images of ESCs in undifferentiated state (U-ES) and upon differentiation toward neuronal phenotype (nD-ES). Bar = 200 µm. Epiallele frequency distribution in 3 independent ESCs differentiation experiments (I, II, and III). The pie charts represent the relative percentage of fully unmethylated epialleles (U = white), fully methylated epialleles (M = black), and the 62 intermediate epialleles (I = gray gradient). The line plots represent the frequency distribution of each of the 62 intermediate epialleles (blue: undifferentiated ESC; red: differentiated ESCs). D) Comparison between average frequency distribution of each of the 62 Ddo intermediate epialleles, from 3 independent experiments, in undifferentiated (blue line) vs. differentiated (red line) ESCs. The specific methyl CpG combinations of each of the 62 intermediate epialleles are indicated on the x-axis.
Figure 8.
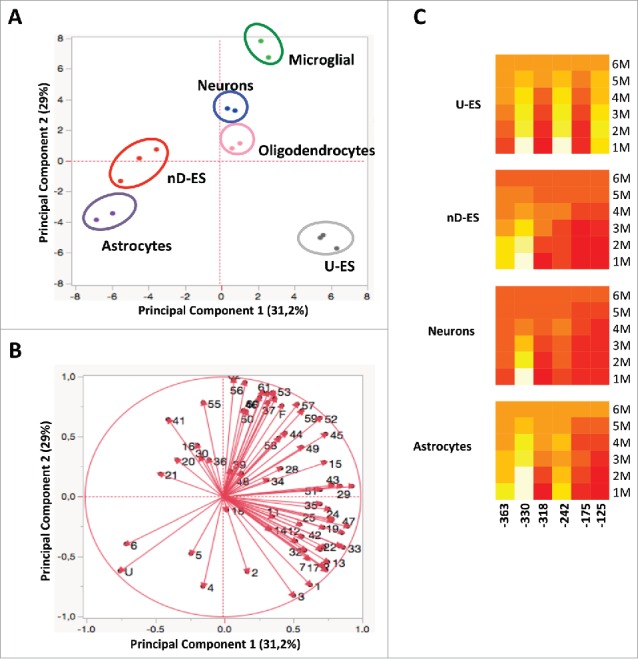
Epigenetic correlation of Ddo R4 epialleles distribution in ES and different purified brain-derived cell types performed by PCA. A) Scores plot represents the distribution of each specific cell type (blue = neurons; pink = oligodendrocytes; green = microglial; violet = astrocytes; gray = U-ES undifferentiated ESCs, red = nD-ES differentiated ESCs). The analysis was based on the qualitative and quantitative influence of each of the 64 epialleles displayed by each cell type. B) Loading plots of PCA. The vectors represented by red arrows show how (the direction) and how much (the length) each epiallelic profile contributes to the individual correlations represented by PC1 and PC2. Note that each epiallele is named with numbers (1–62) or letters (U, F), as reported in Fig. 3B. C) The heatmaps show the contribution of each CpG in different epiallelic classes among 4 cell types (U-ES = undifferentiated ESC; nD_ES = neuronal differentiated ESCs). The color scale from red to yellow (from low to high values) shows the contribution of each CpG to formation of monomethylated (1M), dimethylated (2M), trimethylated (3M), tetramethylated (4M), pentamethylated (5M), and fully methylated (6M) molecules, respectively.
Discussion
In this study we have performed an ultra-deep methylation analysis to study the methylation profiles of single genomic regions (surrounding the Ddo gene) taking into account cell-to-cell heterogeneity and quantifying the frequency of each methylation pattern (epiallele) present in mixed and pure populations of brain cells during development and neural differentiation. The results substantiate well-appreciated general phenomena: i) methylation is highly polymorphic and possibly undergoes a continuous turnover; ii) such diversity is deterministic and not stochastic; and iii) the diversity converges in the course of neural development. Furthermore, an epigenetic drift at the Ddo gene, clearly appreciated in terms of epiallele profiles and fully unpredictable by conventional methylation determination, marked ESCs neural differentiation process. Overall, the findings support the hypothesis that the methylation state of each CpG in single cells is not stable; rather, it is subject to periodic fluctuations. This appears to occur in a spatial-specific manner, where the methylated state of more susceptible sites (e.g., −330 and −363 in the R4 region) favors methylation of adjacent otherwise partially resistant sites. These rules are retained in mature brain where the differential average methylation in each cell type mainly derives from the relative percentage of fully methylated and unmethylated molecules and from the relative abundance of specific peaks, within a prevalent fixed set of intermediate epialleles. Whether these rules may apply to different genomic regions or are limited to some specific regions (e.g., CpG-poor promoter) remains to be determined. However, it is intriguing to hypothesize that specific CpGs lying upstream the TSS may serve as a “core” and “seed” by means that their methylation state may directly influence the methylation state of the adjacent regions, giving origin, in a continuous dynamic manner, to specific epiallele profiles distributions. Although a limit of this approach is that it does not allow the direct match of cell-by-cell methylation, mRNA expression, and cell identity data, it may circumvent the difficulties and high costs of single-cell analyses, with which it shares the ability to address cell-to-cell methylation differences and underlying mechanisms in a cell population. We have recently developed a pipeline, namely amplimethprophiler (https://sourceforge.net/projects/amplimethprofiler), that renders such kind of analysis, performed at individual genomic loci, easily approachable by other researchers interested in applying these principles to any biological system and genomic regions. To our knowledge, no study to date has addressed the epiallele diversity in brain and in brain-derived cells. In the recent past, very few studies ingeniously addressed cell-to-cell methylation heterogeneity, prevalently in tumor systems and at genome-wide level,22,23,26-30 supporting general rules that are mostly consistent with the here presented data. Landan et al.23 suggested that a stochastic series of subtle and progressive methylation changes in cancer evolution leads to deterministic methylation profiles. Spatially specific methylation patterns emerged, by means that some CpGs are particularly sensitive to changes in methylation, creating an initiation point for methylation that then spreads over the region. Our data are compatible with this model, according to which, in subsets of cells within each population, methylation profiles take origin by the spreading from susceptible sites that, when methylated, become able to influence nearby CpGs. Moreover, by the reanalysis of reduced representation bisulfite sequencing (RRBS) data from normal and cancerous tissues, Landan et al.23 also observed that 2 methylation patterns of DNA molecules from the same cell population were rather different from each other with exception of H1 ESC and testis, which displayed coherent and homogeneous methylation profiles. By contrast, in developing brain and purified brain cells, we found a striking conservation of epiallele profiles at the Ddo gene within the same type of samples deriving from different mice. Moreover, we found that in undifferentiated ESCs the methylation profiles were rather disorganized and highly polymorphic at the Ddo gene, shifting toward an ordered, non-stochastic, and less polymorphic epiallele profile upon induction of neural differentiation. These apparent discrepancies may derive by the fact that our analysis was based on a higher number of reads per locus and that it was performed on CpG-poor gene regulatory region, while CpG-rich regions are mostly considered, especially in RRBS-based analyses, by genome-wide approaches. In a recent study, Li et al.,22 using the methclone program, were able to map a series of loci (“eloci”), consisting of 4 adjacent CpGs, subjected to epigenetic drift during leukemia progression. This study, based on combinatorial entropy as a measure of epiallele shift, observed at single locus throughout the genome, revealed enrichment of eloci in the genic region, in particular at promoter regions, which is in line with our findings. Moreover, by analyzing eloci during leukemia progression, the authors concluded that the analysis of epiallele composition may reveal clonal shifts at specific loci otherwise not detectable. In agreement with this observation, we found that, upon differentiation of ESCs, the Ddo promoter underwent a methylation shift, as clearly detected by epiallele analysis, which was unpredictable by traditional methylation analysis. However, previous studies addressing epiallele composition were mostly performed in tumor progression systems and, thus, epiallele changes were interpreted as clonal shifts. In the future, it would be very interesting to establish whether the epigenetic drift, here reported at the Ddo gene during differentiation, is due to a clonal effect or, rather and more likely, to a methylation remodeling occurring in the ESC population upon induction to differentiate. Although at this stage no association can be made between each epihaplotype and functional state of the gene, it will be important, in the near future, to establish how individual epihaplotypes impact gene expression.
In support of our data, a recent paper addressed the cell-to-cell epivariation by single cell analysis concluding that epivariation frequency in DNA methylation is at least 2 orders of magnitude higher than mutation frequency.31 Nevertheless, data on single cell analyses are rapidly accumulating, including simultaneous methylome and transcriptome determination on single cells.32 These efforts will likely help us decipher the “epiallele code” that, in turn, could be easily applied to take maximum advantage of, and to interpret more in depth, the data obtained on single genomic regions using the here proposed “affordable” comprehensive approach. The latter retains the unique advantage to allow not only the description of heterogeneity in a cell population but also the determination of whether the heterogeneity is conserved among individuals.
Overall, our findings indicate that epiallele composition dynamics in brain cell population are very finely tuned and strikingly conserved. We suggest that tracking epiallele profiles may help get insight into the mechanisms underlying DNA methylation establishment, changes and, potentially, alterations of these processes that occur in neurodevelopmental diseases, which may not be revealed by conventional analyses of (gene-specific or genome-wide) average methylation.
Materials and methods
Animals
C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbour, ME). In CE.IN.GE's laboratories, mice were housed in groups (n = 4 or 5) in standard cages (29×17.5×12.5 cm) at constant temperature (22 ± 1°C) and maintained on a 12/12 h light/dark cycle, with food and water ad libitum. All research involving animals was performed in accordance with the European directive 86/609/EEC governing animal welfare and protection, which is acknowledged by the Italian Legislative Decree no. 116 (January 27, 1992). Animal research protocols were also reviewed and consented by a local animal care committee. Whole brains were collected from mice at different developmental stages, at time points E15, P0, P7, P14, P21, P30, and P60. Five brain areas were dissected (prefrontal cortex, cortex, hippocampus, cerebellum, and striatum) from 2 mice (P30). All tissue samples were pulverized in liquid nitrogen and stored at −80°C.
Cells
Primary Cortical Neurons. Cortical neurons were prepared from brains of 17-day-old C57BL/6J mouse embryos, as previously described.33 Briefly, mice were first anesthetized and then killed by cervical dislocation to minimize the animals' pain and distress. Dissection and dissociation were performed in Ca2/Mg2-free buffer saline (HBSS). Tissues were incubated with trypsin for 20 min at 37°C and dissociated by trituration in culture medium. Cells were plated at 3.5–5×106 in 60 mm plastic Petri dishes, precoated with poly-D-lysine (20 mg/ml), in MEM/F12 (Invitrogen, Carlsbad, CA) containing glucose, 5% deactivated FBS, and 5% horse serum (Invitrogen), glutamine, and antibiotics. Ara-C (10 mM) was added within 48 h of plating to prevent non-neuronal cell growth. Neurons were cultured at 37°C in a humidified 5% CO2 atmosphere. All the experiments on primary cortical neurons were performed according to the procedures described in experimental protocols approved by the Ethical Committee of the Federico II University of Naples.
Primary microglia, oligodendrocytes, and astrocytes isolation from mixed glial cell cultures
Purified microglia, oligodendrocyte, and astrocyte cells were prepared from primary mouse mixed glial cells as previously described.34,35 Briefly, cerebral cortices isolated from post-natal day 1 mouse brain were first dissociated enzymatically in a solution containing 0.125% trypsin and 1.5 mg/mL DNase (Sigma-Aldrich, St. Louis, MO) and then mechanically in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 2 mmol/L L-glutamine (Invitrogen). Cell pellet was resuspended and plated in tissue culture flasks in normal medium at 37°C in a humidified, 5% CO2 incubator. After 12–15 days, the microglia were separated by mechanical shaking of flasks on a orbital shaker for 60 min at 200 rpm at 37°C. The suspension containing microglia was centrifuged at 1000 rpm for 5 min and the pellet stored at −80°C. This procedure yielded 98% IB4- FITC or OX42-positive cells. To isolate oligodendrocyte lineage cells, the cultures were then subjected to an additional 16 h of shaking at 200 rpm. To minimize contamination by microglial cells, the suspension of detached cells was incubated twice for 40 minutes at room temperature. The non-adhering oligodendrocytes were centrifuged at 1000 rpm for 5 min and the pellet stored at −80°C. This procedure yielded 98% NG2-positive cells. Finally, the remaining confluent astrocytes were washed, scraped, centrifuged, and the pellets stored at −80°C until further analysis. This procedure yielded 98% GFAP-positive cells.
Embryonic stem cells
In vitro differentiation toward neurons and glia cells was performed as previously described.36 Briefly, at day 0, wild type TBV (129/SvP) ESCs were dissociated in a single-cell suspension and 1000 cells/cm2 were plated on gelatin-coated plates. The culture medium was replaced daily during differentiation process.
Immortalized cells
A1 mes c-myc (A1) is a cell line immortalized by means of infection with a c-myc-carrying retroviral vector of a primary mouse mesencephalon derived cells culture prepared from 11-day-old embryos (E11) as previously described.37 These cells proliferate and remain undifferentiated when grown in MEM/F12 (Invitrogen, Milan, Italy) supplemented with 10% FBS (HyClone, Milan, Italy).
DNA extraction from cells and tissues
DNA was extracted from a portion of liquid nitrogen–pulverized tissue or cell pellets. DNA was prepared using DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany), following the manufacturer's instructions. DNA was quality checked by 260/280 absorbance ratio using NanoDrop 2000, (Thermo Scientific) and it was quantified using Qubit® 2.0 Fluorometer with the dsDNA broad range assay kit (Invitrogen, Q32850).
Bisulfite treatment and amplicon library preparation
Bisulfite treatment was performed using EZ DNA Methylation Kit (Zymo Research). Genomic DNA (2 μg) was converted with “C/T conversion reagent” and eluted in 50 µl of H2O following the manufacturer's instruction. To evaluate the DNA methylation levels, a double step PCR strategy was used to generate an amplicon library of bisulfite treated DNA that was sequenced by Illumina Miseq Sequencer. In the first PCR reaction, we designed primers to generate tiled amplicons ranging in size between 300–450 bp (all primers pairs are reported in Table 1). The 5′ end of these primers contains overhang adaptor sequences (FW: 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG ‘3; RV: 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG ‘3) that must be used in the second step of PCR to add multiplexing indices and Illumina sequencing adaptors. To obtain first PCR products we used the FastStart High Fidelity PCR System (Roche) at the following thermo cycle condition: one cycle at 95°C for 2 min followed by 32 cycles at 95°C for 30 s, [primer Tm] for 40 s, 72°C for 50 s, followed by a final extension step at 72°C for 6 min. Reactions were performed in 30 μl total volumes: 3 μl 10x reaction buffer, 0.6 μl of 10 mM dNTP mix, 0.9 μl of 5 µM forward and reverse primers, 3.6 μl MgCl2 25 mM, 2–4 μl bisulfite template DNA, 0.25 μl FastStart Taq, and H2O up to a final volume of 30 μl. To eliminate small DNA fragments (primers dimers), we used AMPure purification magnetic Beads (Beckman-Coulter, Brea, CA) following the manufacturer's protocol. The ratio between AMPure Beads volume and PCR products volume was 0.8. Product size was checked on 1.5% agarose gel. Second PCR step was performed in 50 µl total volumes: 5 μl 10x reaction buffer, 1 μl dNTP mix, 5 μl forward and reverse “Nextera XT” primers (Illumina, San Diego, CA), 6 μl 25 mM MgCl2, 5 μl of first PCR product, 0.4 μl FastStart Taq, and H2O up to a final volume of 50 μl. Thermocycler settings were: one cycle at 95°C for 2 min followed by 8 cycles at 95°C 30 s, 55°C for 40 s, 72°C for 40 s, followed by a final extension step at 72°C for 5 min. Another purification step by AMPure Beads was performed and all amplicons were quantified using Qubit® 2.0 Fluorometer. The quality of each amplicon was checked by Agilent 2100 Bioanalyzer using the DNA 1000 Kit (Agilent Technologies, Santa Clara, CA), according to the manufacturer's instructions. Amplicons were pooled at equimolar ratio and then diluted to final concentration of 8 picomolar. Phix control libraries (Illumina) were combined with normalized library [15% (v/v)] to increase diversity of base calling during sequencing. Amplicons library was subjected to sequencing using V3 reagents kits on Illumina MiSeq system (Illumina). Paired-end sequencing was performed in 281 cycles per read (281×2). An average of 210,000 reads/sample were used for further analysis. The rate of bisulfite conversion was estimated as 98–99%, as calculated by spike-in experiments performed by adding fully unmethylated M13mp18 double strand DNA (New England BioLabs) in 10 representative samples (primers pairs are reported in Table 1).
Sequence handling
First, paired-end reads obtained from Illumina Miseq sequencer platform were merged together using PEAR tool38 with a minimum of 40 overlapping residues as threshold and quality filtered was obtained using as threshold a mean PHREAD score of at least 33. FASTQ assembled reads were converted to FASTA format using PRINSEQ tool.39 To analyze the methylation status of each amplicon, we used our pipeline software (Amplimethprofiler), freely available at https://sourceforge.net/projects/amplimethprofiler. This tool is specifically designed for deep targeted bisulfite amplicon sequencing of multiple genomic regions and provides functions to demultiplex, filter and extract methylation profiles directly from FASTA files. All the filters (read length, percent of similarity between end of the read and the sequence primer, bisulfite efficiency, maximum percentage of ambiguously aligned CG sites, percent of aligned read) may be tuned by user specified threshold. In the first step, Amplimethprofiler generates, for each sequenced region a quality filtered FASTA file, whose entries are annotated with the ID of the region and of the sample, then discarding reads that do not match expected length (possibly derived from dimers and/or other artifacts). Next, reads are aligned to the corresponding bisulphite converted reference using BLASTn.40 As output, the pipeline returns: i) a summary and quality statistics file, containing information about the number of reads that pass filtering, the methylation percentage of each C in CpG sites, and the bisulfite efficiency for each C in non-CpG sites; ii) an alignment file where the bisulfite efficiency/read is calculated; iii) a CpG methylation profiles file, containing a matrix that reports the methylation status for each CpG dinucleotide: 0 if the site is recognized as unmethylated, 1 if the site is recognized as methylated, and 2 if the methylation state could not be assessed. Each row of the matrix can be considered as the CpG methylation profile and defines an epiallele in subsequent analyses. Then, we used this output to perform downstream analyses. Quantitative methylation averages for each site are then computed as the number of non-converted bases mapped on that site over the total number of mapped reads. Single molecule CpG methylation arrangement was then performed. Based on R software (R Core Team, 2016, https://www.R-project.org), the abundance of each one of the 2NCpG distinct epialleles (where NCpG stands for the number of CpG sites in the analyzed region) was evaluated for each sample by counting the number of passing filter reads containing that epiallele. Filter was set in order to include only reads with the expected lenght and containing no more that 2% unconverted cytosine outside CpG sites. The number of total reads and the reads used for epiallele analyses are reported in supplemental Table S1.
Statistical analysis
Methylation average data are expressed as means ± standard deviation. Comparisons between 2 groups were performed using the unpaired Student t test. Multiple comparisons were made by 1-way ANOVA followed by Tukey post-hoc test. A P-value ≤ 0.05 was considered statistically significant. Pearson correlation test was used to assess the relationship among the epialleles distribution within each stages group. PCA was performed on the abundance of each 64 epialleles presents in the analyzed cell population. PC1 explained 31.2% and PC2 explained 29% of the observed variance. All statistical analyses were performed using JMP software (SAS, Cary, NC).
Availability of data: Accession Numbers
The data sets generated during the current study are deposited on “European Nucleotide Archive (ENA)” server, www.ebi.ac.uk/ena, under the accession number PRJEB16320.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grant from Epigenomics Flagship Projects —EPIGEN, C.N.R to LC, VEA and GaM and from NARSAD Young investigator grant 2015 to FE.
References
- 1.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron 2008; 60:961-74; PMID:19109904; http://dx.doi.org/ 10.1016/j.neuron.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, Colantuoni C, Weinberger DR, Kleinman JE, Lipska BK. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet 2012; 90:260-72; PMID:22305529; http://dx.doi.org/ 10.1016/j.ajhg.2011.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al.. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341:1237905; PMID:23828890; http://dx.doi.org/ 10.1126/science.1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915-26; PMID:1606615; http://dx.doi.org/ 10.1016/0092-8674(92)90611-F [DOI] [PubMed] [Google Scholar]
- 5.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247-57; PMID:10555141; http://dx.doi.org/ 10.1016/S0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- 6.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999; 27:2291-8; PMID:10325416; http://dx.doi.org/ 10.1093/nar/27.11.2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001; 293:1089-93; PMID:11498579; http://dx.doi.org/ 10.1126/science.1063443 [DOI] [PubMed] [Google Scholar]
- 8.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929-30; PMID:19372393; http://dx.doi.org/ 10.1126/science.1169786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al.. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015; 86:1369-84; PMID:26087164; http://dx.doi.org/ 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007; 8:355-67; PMID:17453016; http://dx.doi.org/ 10.1038/nrn2132 [DOI] [PubMed] [Google Scholar]
- 11.Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, Jia P, Assadzadeh A, Flanagan J, Schumacher A, et al.. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008; 82:696-711; PMID:18319075; http://dx.doi.org/ 10.1016/j.ajhg.2008.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller G. Epigenetics. The seductive allure of behavioral epigenetics. Science 2010; 329:24-7; PMID:20595592; http://dx.doi.org/ 10.1126/science.329.5987.24 [DOI] [PubMed] [Google Scholar]
- 13.Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, Sacchetti S, Lembo F, Angiolillo A, Jovanovic N, et al.. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch Gen Psychiatry 2010; 67:258-67; PMID:20194826; http://dx.doi.org/ 10.1001/archgenpsychiatry.2010.9 [DOI] [PubMed] [Google Scholar]
- 14.Keller S, Sarchiapone M, Zarrilli F, Tomaiuolo R, Carli V, Angrisano T, Videtic A, Amato F, Pero R, di Giannantonio M, et al.. TrkB gene expression and DNA methylation state in Wernicke area does not associate with suicidal behavior. J Affect Disord 2011; 135:400-4; PMID:21802740; http://dx.doi.org/ 10.1016/j.jad.2011.07.003 [DOI] [PubMed] [Google Scholar]
- 15.Keller S, Errico F, Zarrilli F, Florio E, Punzo D, Mansueto S, Angrisano T, Pero R, Lembo F, Castaldo G, et al.. DNA methylation state of BDNF gene is not altered in prefrontal cortex and striatum of schizophrenia subjects. Psychiatry Res 2014; 220:1147-50; PMID:25219617; http://dx.doi.org/ 10.1016/j.psychres.2014.08.022 [DOI] [PubMed] [Google Scholar]
- 16.Akbarian S, Nestler EJ. Epigenetic mechanisms in psychiatry. Neuropsychopharmacology 2013; 38:1-2; PMID:23147478; http://dx.doi.org/ 10.1038/npp.2012.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perroud N, Salzmann A, Prada P, Nicastro R, Hoeppli ME, Furrer S, Ardu S, Krejci I, Karege F, Malafosse A. Response to psychotherapy in borderline personality disorder and methylation status of the BDNF gene. Transl Psychiatry 2013; 3:e207; PMID:23422958; http://dx.doi.org/ 10.1038/tp.2012.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.B Labonté, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, Bureau A, Mechawar N, Szyf M, Meaney MJ, et al.. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry 2012; 69:722-31; PMID:22752237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lutz PE, Almeida D, Fiori LM, Turecki G. Childhood maltreatment and stress-related psychopathology: the epigenetic memory hypothesis. Curr Pharm Des 2015; 21:1413-7; PMID:25564388; http://dx.doi.org/ 10.2174/1381612821666150105124928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic basis of mental Illness. Neuroscientist 2015; 22(5):447-63; pii:1073858415608147; PMID:26450593; http://dx.doi.org/ 10.1177/1073858415608147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu W, Tsukahara T, Nakamura R, Yurino H, Hashimoto S, Tsuji S, Takeda H, Morishita S. Assessing Cell-to-Cell DNA Methylation variability on individual long reads. Sci Rep 2016; 6:21317; PMID:26888466; http://dx.doi.org/ 10.1038/srep21317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Garrett-Bakelman F, Perl AE, Luger SM, Zhang C, To BL, Lewis ID, Brown AL, D'Andrea RJ, Ross ME, et al.. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol 2014; 15:472; PMID:25260792; http://dx.doi.org/ 10.1186/s13059-014-0472-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein DA, Goldfinger N, Zundelevich A, et al.. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet 2012; 44:1207-14; PMID:23064413; http://dx.doi.org/ 10.1038/ng.2442 [DOI] [PubMed] [Google Scholar]
- 24.Errico F, D'Argenio V, Sforazzini F, Iasevoli F, Squillace M, Guerri G, Napolitano F, Angrisano T, Di Maio A, Keller S, et al.. A role for D-aspartate oxidase in schizophrenia and in schizophrenia-related symptoms induced by phencyclidine in mice. Transl Psychiatry 2015; 5:e512; PMID:25689573; http://dx.doi.org/ 10.1038/tp.2015.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Punzo D, Errico F, Cristino L, Sacchi S, Keller S, Belardo C, Luongo L, Nuzzo T, Imperatore R, Florio E, et al.. Age-Related changes in d-Aspartate oxidase promoter methylation control extracellular d-Aspartate levels and prevent precocious cell death during brain aging. J Neurosci 2016; 36:3064-78; PMID:26961959; http://dx.doi.org/ 10.1523/JNEUROSCI.3881-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGowan PO Szyf M. The epigenetics of social adversity in early life: implications for mental health outcomes. Neurobiol Dis 2010; 39:66-72; PMID:20053376; http://dx.doi.org/ 10.1016/j.nbd.2009.12.026 [DOI] [PubMed] [Google Scholar]
- 27.Mikeska T, Candiloro IL, Dobrovic A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010; 2:561-73; PMID:22121974; http://dx.doi.org/ 10.2217/epi.10.32 [DOI] [PubMed] [Google Scholar]
- 28.Candiloro IL, Mikeska T, Dobrovic A. Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics 2011; 6:500-7; PMID:21364322; http://dx.doi.org/ 10.4161/epi.6.4.14853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown AL, et al.. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 2016; 22(7):792-9; PMID:27322744; http://dx.doi.org/ 10.1038/nm.4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wee EJ, Rauf S, Shiddiky MJ, Dobrovic A, Trau M. DNA ligase-based strategy for quantifying heterogeneous DNA methylation without sequencing. Clin Chem 2015; 61:163-71; PMID:25274555; http://dx.doi.org/ 10.1373/clinchem.2014.227546 [DOI] [PubMed] [Google Scholar]
- 31.Gravina S, Dong X, Yu B, Vijg J. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol 2016; 17:150; PMID:27380908; http://dx.doi.org/ 10.1186/s13059-016-1011-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, Krueger F, Smallwood SA, Ponting CP, Voet T, et al.. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 2016; 13:229-32; PMID:26752769; http://dx.doi.org/ 10.1038/nmeth.3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sisalli MJ, Secondo A, Esposito A, Valsecchi V, Savoia C, Di Renzo GF, Annunziato L, Scorziello A. Endoplasmic reticulum refilling and mitochondrial calcium extrusion promoted in neurons by NCX1 and NCX3 in ischemic preconditioning are determinant for neuroprotection. Cell Death Differ 2014; 21:1142-9; PMID:24632945; http://dx.doi.org/ 10.1038/cdd.2014.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boscia F, D'Avanzo C, Pannaccione A, Secondo A, Casamassa A, Formisano L, Guida N, Sokolow S, Herchuelz A, Annunziato L. Silencing or knocking out the Na(+)/Ca(2+) exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ 2012; 19:562-72; PMID:21959935; http://dx.doi.org/ 10.1038/cdd.2011.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boscia F, Esposito CL, Casamassa A, de Franciscis V, Annunziato L, Cerchia L. The isolectin IB4 binds RET receptor tyrosine kinase in microglia. J Neurochem 2013; 126:428-36; PMID:23413818; http://dx.doi.org/ 10.1111/jnc.12209 [DOI] [PubMed] [Google Scholar]
- 36.Fico A, Manganelli G, Simeone M, Guido S, Minchiotti G, Filosa S. High-throughput screening-compatible single-step protocol to differentiate embryonic stem cells in neurons. Stem Cells Dev 2008; 17:573-84; PMID:18576914; http://dx.doi.org/ 10.1089/scd.2007.0130 [DOI] [PubMed] [Google Scholar]
- 37.Colucci-D'Amato GL, Tino A, Pernas-Alonso R, ffrench-Mullen JM, di Porzio U. Neuronal and glial properties coexist in a novel mouse CNS immortalized cell line. Exp Cell Res 1999; 252:383-91; PMID:10527628; http://dx.doi.org/ 10.1006/excr.1999.4636 [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2013; 30:614-20; PMID:24142950; http://dx.doi.org/ 10.1093/bioinformatics/btt593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011; 27:863-4; PMID:21278185; http://dx.doi.org/ 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics 2009; 10:421; PMID:20003500; http://dx.doi.org/ 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.