

Abstract
Conformational changes in proteins can lead to disease. Thus, methods for identifying conformational changes in proteins can further improve our understanding and facilitate detection of disease states. Here we combine limited proteolysis (LiP) with Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) to characterize breast cancer-related conformational changes in proteins on the proteomic scale. Studied here are the conformational properties of proteins in two cell culture models of breast cancer, including the MCF-10A and MCF-7 cell lines. The SILAC-LiP approach described here identified ~200 proteins with cell-line dependent conformational changes, as determined by their differential susceptibility to proteolytic digestion using the non-specific protease, proteinase K. The protease susceptibility profiles of the proteins in these cell lines were compared to thermodynamic stability and expression level profiles previously generated for proteins in these same breast cancer cell lines. The comparisons revealed that there was little overlap between the proteins with protease susceptibility changes and the proteins with thermodynamic stability and/or expression level changes. Thus, the large-scale conformational analysis described here provides unique insight into the molecular basis of the breast cancer phenotypes in this study.
Keywords: mass spectrometry, SILAC, limited proteolysis, proteomics, protein folding, breast cancer, MCF-7, MCF-10A
TOC image
Introduction
Proteins play important roles in many biological processes including those associated with diseases states. In many cases, the conformational properties of a protein are closely tied to its biological function or dysfunction. For example, proteins may undergo conformational changes to induce or preclude their interaction with other molecules to ultimately control fundamental biological processes. Modulating the inherent flexibility of proteins can also affect protein function.1–2 Protein conformational changes induced by point mutations, post-translational modifications, or by new and/or altered binding interactions with cellular ligands (e.g., small molecules or other proteins) can also lead to disease. Indeed, inherited or acquired modifications in protein structure have been recognized as the basis of many diseases including neurodegenerative diseases,3–4 type II diabetes5 and certain type of cancers.6 Methods for identifying conformational changes in proteins can thus enhance our understanding of the molecular basis of disease.
X-ray crystallographic and spectroscopic methods have been used to characterize drug-induced and disease-related conformational changes in a number of protein systems previously implicated in disease and drug-action.7–9 However, because they require relatively large amounts of purified protein samples, these techniques are typically not useful for the large-scale and unbiased discovery of drug-induced and disease-related conformational changes in proteins. More recently, a series of mass spectrometry-based proteomic methods have been introduced for the large-scale conformational analysis of proteins in unpurified biological samples including cell lysates10–15 and even intact cells.16 Some of these methods have utilized the denaturant dependence of a chemical modification reaction10 or a proteolytic digestion,11–12 or utilized the temperature dependence of a protein precipitation reaction16 to probe the more global unfolding/refolding properties of proteins. While other methods have utilized limited proteolysis (LiP) strategies under native conditions to probe more subtle structural transitions.13–15 These proteomic methods for the conformational analysis of proteins in unpurified biological samples have been useful for identifying the protein targets of drugs14, 17–18 and other biologically important molecules.10–11, 15, 19–22
Most of the proteome-wide conformational analyses of proteins in complex biological samples, to-date, have focused on understanding the conformational properties of proteins induced by specific ligands. In these studies, the ligand of interest is typically spiked into the protein mixture of interest and the conformational properties of proteins in the ligand-containing sample are compared to those in the protein mixture in the absence of ligand. Investigated here is the use of LiP to probe the conformational properties of proteins in different biological states. To our knowledge, there are only two such global studies of protein conformational changes associated with different biological states. One study utilized a LiP approach to study the structural transitions of proteins from yeast cultures upon a metabolic transition,13 while the other study utilized the Stability of Proteins from Rates of Oxidation (SPROX) technique to investigate breast cancer-related conformational changes using cell culture models of the disease.23
As part of this work, a recently established LiP protocol13 is used in combination with the Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) technique24 to study the conformational changes of proteins involved in breast cancer pathogenesis. The protein conformational changes in the non-tumorigenic MCF-10A breast cell line and the non-invasive, estrogen receptor positive MCF-7 breast cancer cell line were investigated. These same two cell lines were recently the subject of a study in which the SILAC-SPROX technique was used to profile the chemical denaturant-induced equilibrium unfolding properties of the proteins in these cell lines.23 The SILAC-SPROX study successfully differentiated the two breast cancer cell lines and identified novel molecular signatures of breast cancer.
One goal of the current work was to compare the protein hits with altered thermodynamic stabilities identified in the SILAC-SPROX study to those protein hits identified in the SILAC-LiP experiment described here. The types of protein conformational changes probed by the SPROX and LiP techniques are different. Thus, the identified protein hits in the LiP experiment are expected to create novel molecular signatures of breast cancer and provide additional insight into the molecular basis of the disease.
Materials and Methods
Cell Culture and Cell Lysate Preparation
The breast epithelial cell line MCF-10A was cultured following the American Type Culture Collection (ATCC) guidelines. Heavy SILAC-labeled breast cancer cell line MCF-7 was cultured using heavy-labeled lysine and arginine according to the established protocols.25 The cells were lysed by mechanical disruption in the presence of 1 mm diameter zirconia/silica beads (Biospec) using a Disruptor Genie (Scientific Industries). This involved 10 cycles consisting of 20 s of disruption and 1 min of cooling on ice. The lysis buffer was 20 mM HEPES buffer (pH 7.5) containing 150 mM KCl, 10 mM MgCl2 and a cocktail of protease inhibitors that included the following: 1 mM 4-(2-Aminoethyl) benzenesulfonyl uoride hydrochloride (AEBSF), 500 μM Bestatin, 15 μM E-64, 20 μM Leupeptin, and 10 μM Pepstatin A (Thermo Pierce). Lysed cells were centrifuged at 14,000 × g for 15 min at 4°C and the supernatant was used for subsequent analyses. The total protein concentration in each supernatant was determined using a Bradford assay and normalized to 2 mg/mL.
SILAC-LiP Analyses
The SILAC-LiP technique was applied in a comparative study involving heavy-labeled MCF-7 and light-labeled MCF-10A cell lines. The study was performed with five biological replicates. The heavy-labeled MCF-7 and light-labeled MCF-10A cell lysates generated for each biological replicate were each divided into two 50 μL aliquots. One aliquot of each cell lysate was subjected to limited proteolysis under native conditions followed by tryptic digestion under denaturing conditions (double digestion), while the other aliquot of each cell lysate was only subjected to tryptic digestion (single digestion). The proteolysis conditions (i.e., enzyme concentrations, reaction times and temperatures) were the same as those previously described.13 Specifically, in the double digestion group, each of the two cell lysates was treated with proteinase K (pK) (Sigma) at an enzyme/substrate ratio of 1/100 (w/w) for 5 min at room temperature. The reaction was quenched with guanidine hydrochloride (GdmCl) (EMD Millipore) crystals (final concentration 7.6 M) at 99°C for 3 min before combining. In the single digestion group, both the light- and heavy-labeled cell lysates were denatured by GdmCl without pK treatment before combining. The light- and heavy-labeled lysates that were singly digested were combined before proceeding to the proteomic sample preparation as described below, as were the light- and heavy-labeled lysates that were doubly digested.
Proteomic Sample Preparation
The combined light and heavy lysates were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Thermo Fisher, Inc.) for 1 hour at 60°C. The samples were treated with 10 mM methylmethane thiosulfonate (MMTS) (Sigma) for 10 min at room temperature. Samples were diluted with 0.5 M triethylammonium bicarbonate (TEAB) buffer (Sigma) such that the final concentration of GdmCl was less than 2 M. The protein samples were digested with trypsin using an enzyme/substrate ratio of 1/50 (w/w) at 37°C with overnight incubation. The proteolytic digestion reaction was quenched upon acidification (pH ~ 2–3) with trifluoroacetic acid (TFA) (Sigma).
LC-MS/MS Analyses
The peptide mixtures were desalted using C18 resin (The Nest Group) according to the manufacturer’s protocol. LC-MS/MS analyses were performed on a Q-Exactive Plus high-resolution mass spectrometer (Thermo Scientific, Inc.) with a nanoAcquity UPLC system (Waters Corp.) and a nano-electrospray ionization source. Samples were trapped on a Symmetry C18 300 mm × 180 μm trapping column for 3 min at 5 μL/min (99.9/0.1 v/v water/acetonitrile 0.1% formic acid), and separated on a 75 μm × 250 mm column packed with 1.7 μm Acquity HSST3 C18 stationary phase (Waters Corp.). Peptides were separated using a gradient of 3 to 30% acetonitrile with 0.1% formic acid over 90 min at a flow rate of 0.4 μL/min. Data collection was performed in a data-dependent acquisition (DDA) mode with a resolution of 70,000 (at m/z 200) for full MS scan from m/z 375 ~ 1600 with a target AGC value of 1 × 106 ions, followed by 20 product ion scans at a resolution of 17,500 (at m/z 200), using an AGC target value of 1 × 105 ions, a max fill time of 60 ms, and normalized collision energy of 30 V. Each sample was analyzed in triplicate.
Proteomic Data Analysis
The resulting 30 raw LC-MS/MS data files (double and single digestion, five biological replicates, three technical replicates for each sample) were searched using MaxQuant 1.5.2.826 against the 20265 human proteins in the 2014-04 release of the UniProt Knowledgebase (downloaded on 5/16/2014 at ftp://ftp.uniprot.org/pub/databases/uniprot/current_releases/release-2014_04/knowledgebase/). Searches were performed with fixed MMTS modification on cysteine and SILAC labeling of lysine and arginine, variable oxidation of methionine, deamidation of asparagine and glutamine, and acetylation of the protein N-terminus. Trypsin was set as enzyme with semi-specificity and up to two missed cleavages allowed. The mass tolerance for precursor ions was set to 20 ppm for the first search where initial mass recalibration was performed, and a 10 ppm precursor mass tolerance was used for the main search. The mass tolerance for fragment ions was set to 0.02 Da. Also included were a match between runs and re-quantification of the searched peptides. The rest of the parameters were set at the default settings. In cases where a given peptide was matched to multiple protein isoforms or multiple members of a protein family, the peptide was assigned to the leading razor protein listed by MaxQuant algorithm. Peptides and proteins identified with false discovery rates < 1% and positive heavy to light (H/L) ratios were used for subsequent data analysis. The mass spectrometry proteomics data will be deposited to the ProteomeXchange (http://www.proteomexchange.org) via the PRIDE partner repository27 with the data set identifiers to be released after publication.
For each biological replicate, the data in the technical replicates were averaged to generate a single H/L ratio for each charge state of each identified peptide. A median H/L ratio was determined for each protein based on all the identified peptides from a given protein in the single digestion group. The H/L ratios of the peptides from a given protein were subsequently divided by the median H/L ratio for that protein, which effectively normalized for protein abundance differences in the two different cell lines (i.e., all the H/L ratios were normalized to 1). These normalized H/L ratios were transformed to log2 values, and the resulting values obtained for a given peptide at all charge states and from five biological replicates were used in subsequent analyses to select peptide hits. Peptide hits were only selected from those that were identified at least twice in both the double and single digestion groups. For peptide hit selection, Student’s two tailed t-test was used with R (https://www.r-project.org/) to identify hit peptides with significantly different H/L ratios in the doubly and singly digested samples. Hit peptides were selected as those with a p-value less than 0.05. Excel spreadsheets containing all the peptides and proteins identified and effectively assayed in this study are included in the Supporting Information (Table S-1–2).
Results and Discussion
Experimental Design
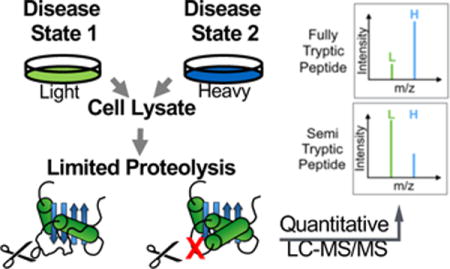
The experimental workflow used in this study is shown in Figure 1. The cell culture models of breast cancer analyzed here included a model of normal mammary epithelial cells (MCF-10A) and a breast cancer cell line (MCF-7). The MCF-7 cell line is estrogen receptor positive, and reflective of the luminal subtype of breast cancer. The conformational properties of proteins in the MCF-10A cell lysate were compared to those in the MCF-7 cell lysate where the MCF-7 cell line was heavy-labeled.
Figure 1.

Schematic representation of the SILAC-based limited proteolysis workflow used in this work.
As part of the experimental workflow (Figure 1) each cell line in the comparison was subjected to a double and a single digestion. Proteinase K and trypsin were used in the double digestion, and only trypsin was used in the single digestion. The initial step in the workflow involved performing a limited proteolytic digestion with proteinase K under native conditions on both the light- and heavy-labeled cell lines. The experimental conditions of the limited proteolytic digestion with proteinase K, which were similar to those previously described,13 were controlled such that the extent of primary cleavages is largely determined by the protein conformation. After the proteinase K digestion was quenched, the light- and heavy-labeled samples were combined, and the resulting protein mixture was subjected to a trypsin digestion under denaturing conditions to generate peptides suitable for bottom-up proteomic analysis. The single digestion group, which included proteins subjected to the trypsin digestion, served as a control for endogenous protease cleavages, incomplete labeling in heavy-labeled cells, and protein abundance differences across samples.
Proteins with different conformations in the two cell lines were identified based on the proteinase K induced H/L ratio changes (p < 0.05) of the peptides identified and quantified in the LC-MS/MS readout. A semi-tryptic peptide with an increased H/L ratio, or a fully tryptic peptide with a decreased H/L ratio, indicated greater protease susceptibility in the heavy-labeled MCF-7 cell line. Conversely, a semi-tryptic peptide with a decreased H/L ratio, or a fully tryptic peptide with an increased H/L ratio, suggested protease protection in the heavy-labeled MCF-7 cell line.
Summarized in Table 1 is the proteomic coverage obtained in the comparative analysis performed in this work. In total, approximately 4300 peptides from ~550 proteins were assayed for their proteinase K sensitivity. A total of 489 peptides from 203 proteins were identified as hits with statistically significant (p-value < 0.05), proteinase K-induced H/L ratio changes in the MCF-7 vs. MCF-10A comparison (see Figure 2A and Table S3). The peptide hits accounted for ~11% of those assayed. In order to assess the false positive rate of peptide hit discovery using the SILAC-LiP protocol described here a control experiment was performed using light- and heavy-labeled MCF-7 cells (see Supporting Information for details about the false positive rate determination for hit discovery). Approximately 3300 peptides from ~480 proteins were assayed in the control experiment and 201 peptides from 113 proteins were identified as hits in the control experiment. A total of 32 peptide hits in the control experiment also displayed significantly altered (p-value < 0.05) H/L ratios in the MCF-7 vs. MCF-10A comparison. The proteinase K induced H/L ratio changes in these peptides are not likely to be disease-related. Thus, they were excluded from the final hit lists used in the bioinformatics analyses described below. The fold-change values between the double and single digestions for the final hit peptides spanned a relatively wide range from 1.1-to 8.5-fold (Figure 2A). However, the median fold-change value of the hit peptides identified in this work was relatively small at 1.3-fold. This relatively small value suggests that the protease susceptibility differences of the proteins to which these hit peptides mapped were relatively small.
Table 1.
Proteomic coverage obtained in the comparative analysis performed in this work
| Double digestion | Single digestion | |
|---|---|---|
| Total peptides (proteins) identified | 7408 (812) | 10994 (979) |
| Assayed peptides (proteins)a | 4342 (556) | |
| Peptide (protein) hitsb | 489 (203) | |
Assayed peptides are the peptides that were identified at least twice in both the double and single digestion groups (e.g., a peptide was identified at different charge states, or a peptide was identified in multiple technical and/or biological replicates).
Hit peptides are the peptides with significant differences in their H/L ratios between the double and single digestion groups (p < 0.05).
Figure 2.
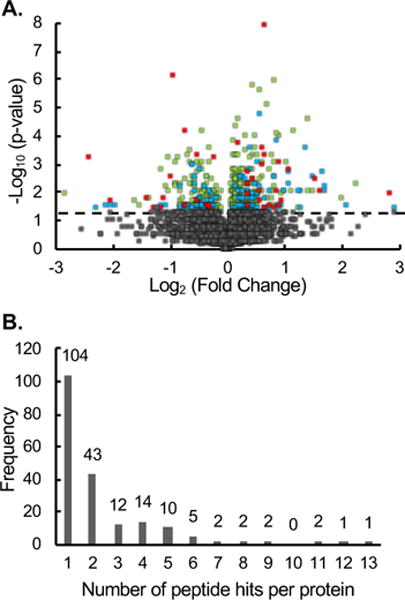
Global analysis of the H/L ratios generated for the assayed peptides in the MCF-7 vs MCF-10A comparison. (A) Volcano plot of statistical significance against fold change between single and double digestion. The horizontal dashed line represents a p-value = 0.05. The grey points (n = 3853) represent non-hit peptides (p-value > 0.05). The green points (n = 304) represent peptides hits (p-value < 0.5) observed in four or five of the biological replicates. The blue points (n = 114) represent peptide hits (p-value < 0.05) observed in two or three of the biological replicates. The red points (n = 39) represent peptide hits (p-value < 0.05) observed in one biological replicate. (B) Bar graph showing the number of protein hits that were identified with multiple peptides
One limitation of the current control experiment is that it does not account for potential error introduced to protein abundance changes. The false discovery of peptide hits in the SILAC-LiP experiment could result from errors in the determination of protein median H/L ratios, which are ultimately used for the normalization of protein abundance differences. To assess the impact of this potential error on our hit discovery, the fraction of hits with no significant (i.e., less than 2-fold) protein expression level changes (69%) was compared to the fraction of assayed proteins with no significant protein expression level changes (67%). This analysis indicated the protein hits were not enriched in proteins that had significant expression level changes (i.e., those would be more prone to errors in the determination of a median H/L ratio). Also, the relative standard deviations (RSD) associated with the protein median H/L ratios were similar in the MCF-7 vs. MCF-10A comparison (median RSD = 0.18) and the control experiment (median RSD = 0.15).
Hit selection at the peptide level is prone to error. While all the peptide hits identified in this work had p-values < 0.05, not all were identified in all five biological replicates. Approximately 9% of the peptide hits were identified based on data from only one biological replicate, and another 25% of the peptide hits were identified based on data from two or three biological replicates. The strongest peptide hits were a subset of approximately 66% of the peptide hits that were determined to be hits based on data from four or five independent biological replicates (see Figure 2A and Table S3).
Also strong were those protein hits that result from multiple peptide hits. Since the protease-induced cleavage in LiP only occurs at sites in a protein where conformational changes occur, not all peptides assayed from a hit protein are expected to be a hit. However, in cases where different peptides from the same protein show altered protease susceptibility, the confidence in the given protein hit increases. In this study, approximately half of the protein hits were determined based on two or more peptide hits (Figure 2B). For example, the ribosomal protein RPS12 was a protein hit as determined from 3 different peptide hits. Among these 3 peptides, a semi-tryptic peptide SNCDEPMYVK (residues 54–63) showed an increased H/L ratio, while the fully tryptic peptide QAHLCVLASNCDEPMYVK (residues 46–63) encompassing the semi-tryptic peptide showed a decrease H/L ratio. Together, these peptides suggest a conformational change at this site of RPS12 between the MCF-10A and MCF-7 cell lines.
The observation of a semi-tryptic peptide and its corresponding fully tryptic peptide showing opposite changes in H/L ratios is especially useful, because such peptide pairs can act as “internal controls” for the quantitative changes in their proteolytic susceptibility. Unfortunately, the detection and quantitation of such peptide pairs is a challenge using the SILAC-LiP approach described here. This is largely because semi-tryptic peptides tend to be singly charged during electrospray ionization, which makes their sequencing difficult. In fact, such singly-charged ions are not generally selected for sequencing in shot-gun proteomics experiments. Moreover, many semi-tryptic peptides do not contain lysine or arginine, which not only further reduces their ionization efficiency but also makes them difficult to quantify using SILAC.
Of the 4342 peptides assayed in our experiments only 13% of them were semi-tryptic peptides. The percentage of hit peptides identified here that were semi-tryptic was also similar (i.e., 12%). Not surprisingly, only a small number of the tryptic and semi-tryptic peptide hits identified in this work (39 and 38, respectively) had corresponding semi-tryptic and tryptic peptides that were successfully assayed. In total only seven pairs were identified as hits based on both tryptic and semi-tryptic peptides (i.e., the tryptic and semi-tryptic peptides showed opposite and significant (p-value < 0.05) changes in their H/L ratios, Table S3).
It is noteworthy that the rest of the tryptic/semi-trypitc peptide pairs are not necessarily false positives. Due to the non-specificity of proteinase K, a given tryptic peptide can be cleaved into several different semi-tryptic peptides, whose relative proportions may be dictated by the protein conformational differences. Thus, it is possible that semi-tryptic peptides that map to tryptic peptide hits did not contain the proteinase K site that was differentially cleaved. Similarly, it is also possible for semi-tryptic peptide hits to map to tryptic peptide non-hits. For this reason, tryptic and semi-tryptic peptide hits mapping to semi-tryptic and tryptic peptide non-hits (respectively) were not removed from the hit list.
Expression Level Analysis
The H/L ratios obtained in the single digestion can be used to assess the relative abundance of proteins in each cell line. The expression level data generated in this work was in general agreement with that previously reported in the literature for the cell lines in this study (see Table S-3).23, 28 For example, the σ isoform of the 14-3-3 proteins has been directly implicated in breast cancer as a negative cell cycle regulator.29 Protein expression level studies have found that 14-3-3σ is down regulated in transformed mammary epithelial cells.30 The expression level information obtained in this work indicates a similar down-regulation of 14-3-3σ in the MCF-7 cells compared with MCF-10A cells. Interestingly, ~69% of the protein hits identified with conformational differences between the two cell lines did not have significant (i.e., less than 2-fold) changes in their expression levels (Figure 3). This is similar to that observed in the SILAC-SPROX analyses of the cell lines used in these studies (i.e., ~50% of the differentially stabilized proteins in the SILAC-SPROX experiments did not have significant changes in their expression levels).23 These results suggest that conformational changes can provide information about disease states that is orthogonal to that obtained in protein expression level analyses.
Figure 3.
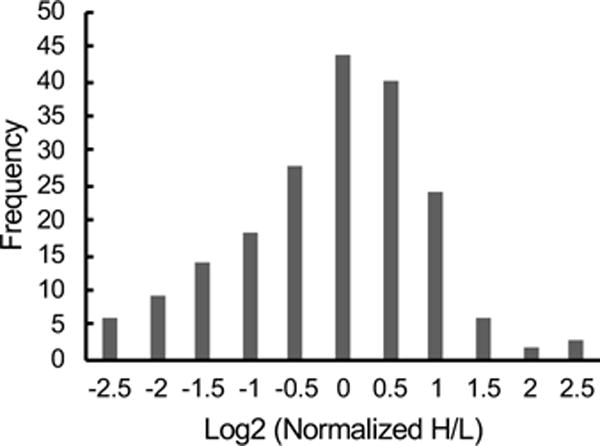
Bar graph showing the distribution of the relative expression levels of the protein hits identified in the MCF-7 vs MCF-10A comparison.
Hit Characterization
Approximately 58% of the hit peptides identified here were more protected from proteolytic cleavage with the rest becoming less protected from proteolytic cleavage in the MCF-7 cells as compared to the MCF-10A cells. The positions of 190 hit peptides in 80 proteins with available structures in the Protein Data Bank were examined. Most of the hit peptides (i.e., 149 out of 190) mapped to loops, turns, or regions with no secondary structure (Figure 4). This is consistent with these regions of protein structure being more susceptible to proteolytic cleavages.31 For example, the loop that connects the third and fourth helix in the 14-3-3σ (i.e., SNEEGSEEKGPEVREYR, residues 69–85) was found to be the site where the altered proteolysis pattern was observed in our work (i.e., decreased protease susceptibility in the MCF-7 cells) (Figure 5A). This loop region appears as an unstructured region in the X-ray crystallographic structure of the dimeric 14-3-3σ.32–34 Yang and coworkers suggest that the conformational flexibility at the dimerization interface enhanced by this loop region could facilitate binding of 14-3-3σ to proteins of various sizes and sequences.32 Our finding that this loop region is more protected from proteolytic cleavage in the MCF-7 cell line than in the MCF-10A cell line suggests that these previously reported protein-protein interactions involving this loop region in 14-3-3σ32 may be disease-related.
Figure 4.
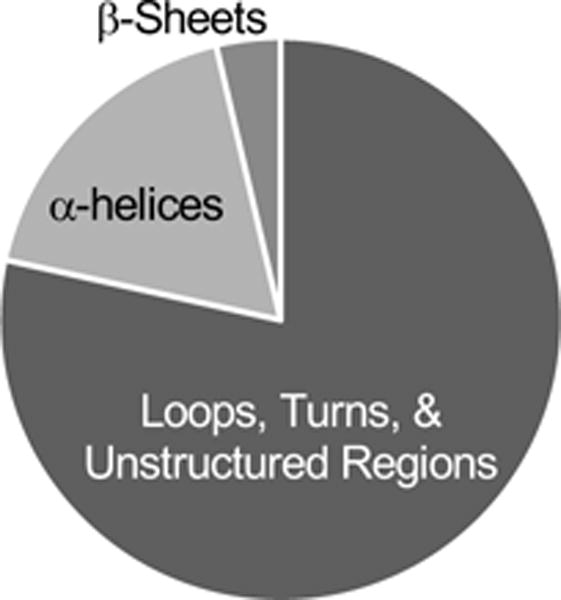
Pie chart showing the distribution of different protein structural elements to which the peptide hits identified in the MCF-7 vs MCF-10A comparison mapped.
Figure 5.
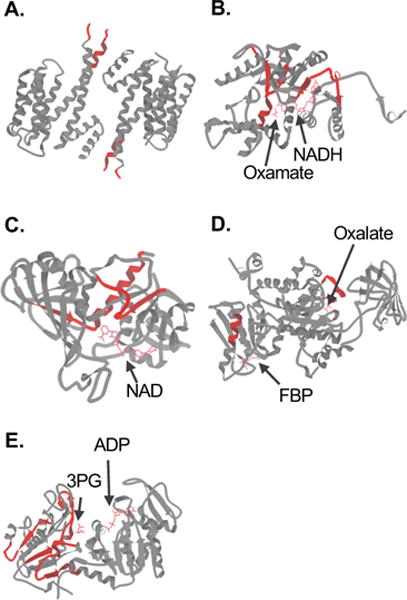
Schematic representations of the folded three-dimensional structures of selected protein hits identified in the MCF-7 vs MCF-10A comparison. The nine antiparallel helices that comprise the 14-3-3σ homodimer (PDB 1YZ5) are shown in (A). The structure of LDHA in complex with NADH and the inhibitor oxamate (PDB 1I10) is shown in (B). The structure of GAPDH in complex with NAD (PDB 1ZNQ) is shown in (C). The structure of PKM2 in complex with Mg2+, K+, the inhibitor oxalate and the allosteric activator fructose 1,6-bisphosphate (FBP) (PDB 1T5A) is shown (D). The structure of PGK1 in complex with 3-phosphoglycerate (3PG) and ADP (PDB 2XE7) is shown in (E). In each case, the regions to which the peptide hits mapped are colored in red. Images were generated in KiNG (Kinemage, Next Generation).60
The limited proteolysis-based approaches generally assume that the differential protease susceptibility is due to alterations in the protein conformation. However, there is the possibility that intrinsic modifications in proteins such as post-translational modifications can prevent the access of proteinase K to its protein substrate by stearic hindrance. In such cases, the different cleavage patterns may result solely from different post-translational modifications on the protein in the different disease states rather than differences in protein conformation. In order to assess this possibility, we matched the peptide hits identified in this work to the known post-translational modification sites in the MCF-10A and MCF-7 cell lines using PhosphoSitePlus knowledgebase35 (http://www.phosphosite.org/). A total of 26 peptide hits from 19 proteins have known phosphorylation sites in the MCF-10A and/or MCF-7 cell lines (see Table S3). These peptides, which account for ~6% of the peptide hits, may be a result of differential post-translational modifications in the two cell lines inducing different proteinase K cleavages. Additional experiments are needed to evaluate the effect of phosphorylation on the proteolysis.
An analysis of the final hit proteins using PANTHER36 revealed that the protein complex biogenesis process was significantly overrepresented by the protein hits (p-value = 0.01). Molecular chaperones are one of the major classes of proteins related to this process. This finding is consistent with the results of previous work that found increased chaperone activity to be characteristic of breast tumorigenesis.37–39 Many heat shock proteins, including HSPA8, HSPA9, HSPD1, HSPB1, HSP90B1, HSP90AB1, HSP90AA1, HSPH1 and HSPA1A, displayed different protease susceptibility in the two different cell lines (Table S-3). In agreement with this observation, it has been suggested that the HSP family members are crucial for tumor growth by promoting cell proliferation as well as by inhibiting death pathways.40–42 Targeting HSPs has thus been considered for therapeutic intervention in cancers.43
Gene ontology (GO) classification of the protein hits also revealed that a large fraction of the hits are nucleic acid binding proteins (~30%). Given their pivotal role in the translation of genetic expression into functional phenotype, it is not surprising that many ribosomal proteins identified as hits in our study have been previously associated with tumorigenesis. It has been suggested that one of the effects of oncogenic signaling appears to be altered recruitment of mRNAs to ribosomes, which will in turn affect proteins involved in growth regulation and cell-cell interaction.44 The MDM2/MDMX-p53 negative feedback loop can be regulated by a number of ribosomal proteins, such as RPS3, RPS7 and RPS20,45–47 which were identified as hits in our study. Another hit protein, RPS6, has been found to attenuate KRAS-induced DNA damage and p53-mediated tumor suppression during development of pancreatic cancer.48 The 60S RPL32 and 40S RPS16, which were also hits in our study, were reported to have altered expression in a model that recapitulates the progression to an androgen-independent state of prostate tumor cells.49 Studies have also shown knockdown of RPL13, another hit in our study, resulted in drastic attenuation of human gastrointestinal cancer cell growth with significant G1 and G2/M arrest of the cell cycle.50 The differential proteolysis patterns of the ribosomal proteins observed here are consistent with the current understanding of ribosomal proteins in the development of cancer.
Correlating Conformational Changes with Protein Function
Some of the hit proteins were enzymes with known catalytic activities. For example, lactate dehydrogenase A (LDHA) catalyzes the transformation of pyruvate into L-lactate with concomitant inter-conversion of NADH and NAD+. One of the principal biochemical characteristics of cancer cells compared to normal cells is a metabolic switch from oxidative phosphorylation to increased glycolysis (i.e., the Warburg effect). LDHA is the final enzyme in the glycolysis pathway and has been shown to play an important role in the development, invasion and metastasis of malignancies.51
In our study, two peptides, NRVIGSGCNLDSAR (residues 156–169) and RVHPVSTMIK (residues 269–278), from the substrate binding domain of LDHA were identified with decreased protease susceptibility in the MCF-7 cell line compared to the MCF-10A cell line (Figure 5B). Structural studies have revealed that His 193, Asp 166, Arg 169, and Thr 244 all make major contributions to the catalytic geometry in the active site of LDHA.52 The active site loop residues 99–110 enclose the active site and are responsible for the NADH-cofactor binding. A peptide adjacent to this active site loop (IVSGKDYNVTANSKLVIITAGAR, residues 77–99) also displayed decreased sensitivity to protease attack in the MCF-7 cells compared with MCF-10A cells. Considering the close link between protein folding and function, we speculate that this greater protection from protease cleavage in the substrate-binding domain of LDHA may reflect an increased stability that results in increased enzymatic activity in the MCF-7 cells. In agreement with this hypothesis, it has been shown that LDH activity (in pyruvate reducing direction) is higher in breast tumors than normal tissues,53 which is consistent with cancer cells utilizing LDH for anaerobic glycolysis.
Several glycolytic enzymes were also identified as hits based on peptides derived from their substrate and/or cofactor binding sites. For example, two GAPDH peptide hits, RVIISAPSADAPMFVM and LISWYDNEFGYSNRVVDLMAHMASK, are located near the NAD binding site (Figure 5C). Also, two peptide hits in pyruvate kinase, VNFAMNVGK and GDLGIEIPAEK, are located near binding sites of the substrate and an allosteric activator, respectively (Figure 5D). Similarly, three phosphoglycerate kinase 1 peptides (VVMRVDFNVPMKNNQITNNQR, ACANPAAGSVILLENLR and AHSSMVGVNLPQKAGGFLMKK) are located close to the substrate binding site (Figure 5E). The altered proteolytic patterns observed in the above enzymes may suggest that the protein-ligand interactions close to these sites are disease-related.
Comparison with SILAC-SPROX Results
One goal of this work was to compare the protein hits identified using the SILAC-LiP approach described here to those previously identified using the SILAC-SPROX approach.23 The SILAC-SPROX technique is an energetics-based method that relies on hydrogen peroxide-mediated oxidation of globally protected methionine residues to report on the chemical denaturant-induced equilibrium unfolding properties of proteins.19 A total of 243 proteins were assayed in both the SILAC-LiP and SILAC-SPROX studies. In these 243 proteins is a subset of 122 proteins that were identified as hit proteins in either one or both of the studies. These 122 protein hits included 84 proteins shown as hits only in the SILAC-LiP experiment, 14 proteins shown as hits only in the SILAC-SPROX experiment, and 24 proteins being identified as hits in both studies. The overlap between the SILAC-SPROX and SILAC-LiP protein hits was relatively small (~20%). This is not surprising as the two techniques probe different properties of proteins. SPROX is sensitive to changes in the more global unfolding/refolding properties of proteins. More local conformational changes, which do not have a strong denaturant dependence to their energetics,54 typically go undetected in SPROX. However, local conformational changes may be more or less sensitive to protease digestion. Changes in the global stability of a protein (or protein domain) may not always be differentially susceptible to protease. Thus, it is not surprising that the two techniques provide complementary biologically relevant information on the disease states.
The fundamental differences between SILAC-SPROX and SILAC-LiP also make it difficult to correlate the SILAC-SPROX behavior of proteins with their SILAC-LiP behavior. Peptides derived from different domains of protein may display very different behavior (i.e., stabilization or destabilization in SPROX, less or more proteolytic susceptibility in LiP). Even when the same peptide probe is detected as a hit in the two experiments, there may not be a direct correlation between thermodynamic stability and protease susceptibility. Indeed, there are examples in the literature where proteins with increased global stability have been found to be less susceptible to proteolytic digestion under native conditions55–56 and where proteins with increased global stability have been found to be more susceptible to proteolytic digestion.57–59
Among the 24 overlapping hit proteins in the SILAC-LiP and SILAC-SPROX experiments, only 5 were identified as hits based on the same peptide probes. However, a clear trend was not observed between the SILAC-SPROX and SILAC-LiP behavior of these peptides. For example, the peptides SFYPEEVSSMVLTK and YDDMATCMK from HSPA8 and YWHAQ (respectively) were stabilized in SILAC-SPROX and more protected from proteolytic digestion in SILAC-LiP in the MCF-7 cell line as compared to the MCF-10A cell line, and the peptide AAHSEGNTTAGLDMR from CCT2 was destabilized in SILAC-SPROX and less protected from proteolytic digestion in SILAC-LiP in the MCF-7 cell line than in the MCF-10A cell line. However, the peptides NALESYAFNMK and VVDLMAHMASKE from HSPA1A and GAPDH (respectively) were destabilized in SILAC-SPROX and also more protected from proteolytic digestion in SILAC-LiP in the MCF-7 cell line as compared to the MCF-10A cell line.
Altered protease susceptibility in LiP can be detected only at sites in a protein where the conformational changes occur, while altered thermodynamic stability in SPROX can be probed by any global protected methionine-containing peptide from a given protein (or protein domain). On the one hand, this creates a challenge in SILAC-LiP experiments of having to detect a tryptic (or semi-tryptic) peptide from the specific site of the conformational change. On the other hand, if such peptides are successfully detected in the SILAC-LiP experiment they can provide detailed information about the site of the conformational change. Interestingly, a number of the SILAC-LiP peptide hits identified here mapped to regions of protein structure that are at or near known ligand binding sites (see e.g., Figure 5B–E). This suggests that the conformational changes detected here may result in or be the result of altered ligand binding interactions.
Conclusions
A comparative, global proteomic analysis of the protein conformational differences between the MCF-10A and MCF-7 breast cancer cells resulted in the identification of a subset of proteins with differential protease susceptibility. A bioinformatics analysis revealed that nucleic acid binding proteins represented the largest fraction of the hits showing altered conformations in the different disease states. Also of note is the overrepresentation of protein complex biogenesis process. Changes in protein conformation can result from a variety of disease-related phenomena. However, the biological phenomena responsible for the conformational changes identified in this work, cannot be directly determined from the data in this study. Additional biophysical and biochemical studies are needed on the protein hits in this study to better understand the molecular basis of the increased/decreased proteolytic stabilities detected here. The utility of the detected changes in proteolytic susceptibility as biomarkers of disease also requires the analysis of clinical samples. One attractive feature of the global conformational analysis performed here is that a relatively small amount of material (less than 200 μg of total protein) is required for analysis. This greatly facilitates the extension of this methodology to the analysis of clinical samples.
Supplementary Material
Supplementary Text: Summary of the control experiment performed as part of this work.
Supplementary Table S-7. Proteomic coverage obtained in the control experiment using light- and heavy-labeled MCF-7 cells.
Supplementary Figure S-1. Global analysis of the H/L ratios generated for the assayed peptides in the control experiment.
Supplementary Table S-1. Excel spreadsheets summarizing peptides identified in the five biological replicates of MCF-7 versus MCF-10A cell line comparison with double and single digestion.
Supplementary Table S-2. Excel spreadsheet summarizing peptides assayed in the five biological replicates of MCF-7 versus MCF-10A cell line comparison.
Supplementary Table S-3. Excel spreadsheet summarizing peptide and protein hits identified with altered conformations in the MCF-7 vs. MCF-10A cell line comparison.
Supplementary Table S-4. Excel spreadsheets summarizing peptides identified in the three biological replicates of the control experiment for false positive rate determination using heavy- and light-labeled MCF-7 cell lines with double and single digestion.
Supplementary Table S-5. Excel spreadsheet summarizing peptides assayed in the three biological replicates of the control experiment using heavy- and light-labeled MCF-7 cell lines.
Supplementary Table S-6. Excel spreadsheet summarizing peptide and protein hits identified with altered conformations in the control experiment using heavy- and light-labeled MCF-7 cell lines.
Acknowledgments
The authors thank the Duke Proteomics Facility, especially Laura Dubois, for acquiring the LC-MS/MS data. This work was supported by a grant from the National Institute of General Medical Sciences at the National Institutes of Health 2R01GM084174-06 (to M.C.F.).
Footnotes
Supporting Information. The following files are available free of charge.
References
- 1.Karplus M, Kuriyan J. Molecular dynamics and protein function. P Natl Acad Sci USA. 2005;102(19):6679–85. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhalla J, Storchan GB, MacCarthy CM, Uversky VN, Tcherkasskaya O. Local flexibility in molecular function paradigm. Mol Cell Proteomics. 2006;5(7):1212–23. doi: 10.1074/mcp.M500315-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Carrell RW, Lomas DA. Conformational disease. Lancet. 1997;350(9071):134–138. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 4.Sheikh S, Safia, Haque E, Mir SS. Neurodegenerative Diseases: Multifactorial Conformational Diseases and Their Therapeutic Interventions. J Neurodegener Dis. 2013;2013:563481. doi: 10.1155/2013/563481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden MR, Tyagi SC, Kerklo MM, Nicolls MR. Type 2 diabetes mellitus as a conformational disease. JOP. 2005;6(4):287–302. [PubMed] [Google Scholar]
- 6.Brandt-Rauf PW, Monaco R, Pincus MR. Conformational effects of environmentally induced, cancer-related mutations in the p53 protein. P Natl Acad Sci USA. 1994;91(20):9262–6. doi: 10.1073/pnas.91.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siligardi G, Hussain R, Patching SG, Phillips-Jones MK. Ligand- and drug-binding studies of membrane proteins revealed through circular dichroism spectroscopy. Biochimica et biophysica acta. 2014;1838(1 Pt A):34–42. doi: 10.1016/j.bbamem.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 8.Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, Kelly JW. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. P Natl Acad Sci USA. 1998;95(22):12956–12960. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273(3):729–39. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 10.Dearmond PD, Xu Y, Strickland EC, Daniels KG, Fitzgerald MC. Thermodynamic analysis of protein-ligand interactions in complex biological mixtures using a shotgun proteomics approach. J Proteome Res. 2011;10(11):4948–58. doi: 10.1021/pr200403c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu PF, Kihara D, Park C. Energetics-based discovery of protein-ligand interactions on a proteomic scale. J Mol Biol. 2011;408(1):147–62. doi: 10.1016/j.jmb.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adhikari J, Fitzgerald MC. SILAC-Pulse Proteolysis: A Mass Spectrometry-Based Method for Discovery and Cross-Validation in Proteome-Wide Studies of Ligand Binding. J Am Soc Mass Spectrom. 2014;25(12):2073–83. doi: 10.1007/s13361-014-0992-y. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, De Franceschi G, Kahraman A, Soste M, Melnik A, Boersema PJ, de Laureto PP, Nikolaev Y, Oliveira AP, Picotti P. Global analysis of protein structural changes in complex proteomes. Nat Biotechnol. 2014;32(10):1036–44. doi: 10.1038/nbt.2999. [DOI] [PubMed] [Google Scholar]
- 14.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J. Target identification using drug affinity responsive target stability (DARTS) P Natl Acad Sci USA. 2009;106(51):21984–9. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trindade RV, Pinto AF, Santos DS, Bizarro CV. Pulse Proteolysis and Precipitation for Target Identification. J Proteome Res. 2016;15(7):2236–45. doi: 10.1021/acs.jproteome.6b00214. [DOI] [PubMed] [Google Scholar]
- 16.Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, Kuster B, Nordlund P, Bantscheff M, Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346(6205):1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 17.West GM, Tucker CL, Xu T, Park SK, Han X, Yates JR, 3rd, Fitzgerald MC. Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. P Natl Acad Sci USA. 2010;107(20):9078–82. doi: 10.1073/pnas.1000148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–7. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 19.Tran DT, Adhikari J, Fitzgerald MC. Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)-Based Strategy for Proteome-Wide Thermodynamic Analysis of Protein-Ligand Binding Interactions. Mol Cell Proteomics. 2014;13(7):1800–1813. doi: 10.1074/mcp.M113.034702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2(3):207–12. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 21.Geer MA, Fitzgerald MC. Characterization of the Saccharomyces cerevisiae ATP-Interactome using the iTRAQ-SPROX Technique. J Am Soc Mass Spectrom. 2016;27(2):233–243. doi: 10.1007/s13361-015-1290-z. [DOI] [PubMed] [Google Scholar]
- 22.Kim MS, Song J, Park C. Determining protein stability in cell lysates by pulse proteolysis and Western blotting. Protein Sci. 2009;18(5):1051–9. doi: 10.1002/pro.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adhikari J, West GM, Fitzgerald MC. Global analysis of protein folding thermodynamics for disease state characterization. J Proteome Res. 2015;14(5):2287–97. doi: 10.1021/acs.jproteome.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 25.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1(6):2650–60. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 26.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 27.Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O’Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41(Database issue):D1063–9. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geiger T, Madden SF, Gallagher WM, Cox J, Mann M. Proteomic portrait of human breast cancer progression identifies novel prognostic markers. Cancer Res. 2012;72(9):2428–39. doi: 10.1158/0008-5472.CAN-11-3711. [DOI] [PubMed] [Google Scholar]
- 29.Urano T, Saito T, Tsukui T, Fujita M, Hosoi T, Muramatsu M, Ouchi Y, Inoue S. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature. 2002;417(6891):871–5. doi: 10.1038/nature00826. [DOI] [PubMed] [Google Scholar]
- 30.Vercoutter-Edouart AS, Lemoine J, Le Bourhis X, Louis H, Boilly B, Nurcombe V, Revillion F, Peyrat JP, Hondermarck H. Proteomic analysis reveals that 14-3-3 sigma is down-regulated in human breast cancer cells. Cancer Res. 2001;61(1):76–80. [PubMed] [Google Scholar]
- 31.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51(2):299–321. [PubMed] [Google Scholar]
- 32.Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundstrom M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. P Natl Acad Sci USA. 2006;103(46):17237–42. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benzinger A, Popowicz GM, Joy JK, Majumdar S, Holak TA, Hermeking H. The crystal structure of the non-liganded 14-3-3sigma protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res. 2005;15(4):219–27. doi: 10.1038/sj.cr.7290290. [DOI] [PubMed] [Google Scholar]
- 34.Wilker EW, Grant RA, Artim SC, Yaffe MB. A structural basis for 14-3-3sigma functional specificity. J Biol Chem. 2005;280(19):18891–8. doi: 10.1074/jbc.M500982200. [DOI] [PubMed] [Google Scholar]
- 35.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43(Database issue):D512–20. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13(9):2129–41. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calderwood SK, Gong J. Molecular chaperones in mammary cancer growth and breast tumor therapy. J Cell Biochem. 2012;113(4):1096–103. doi: 10.1002/jcb.23461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guest ST, Kratche ZR, Bollig-Fischer A, Haddad R, Ethier SP. Two members of the TRiC chaperonin complex, CCT2 and TCP1 are essential for survival of breast cancer cells and are linked to driving oncogenes. Exp Cell Res. 2015;332(2):223–35. doi: 10.1016/j.yexcr.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 39.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32(7):805–18. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31(3):164–72. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, Kluger HM. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67(7):2932–7. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 42.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature reviews Cancer. 2005;5(10):761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 43.Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med. 2004;10(2):47–51. doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 44.Holland EC. Regulation of translation and cancer. Cell Cycle. 2004;3(4):452–5. [PubMed] [Google Scholar]
- 45.Yadavilli S, Mayo LD, Higgins M, Lain S, Hegde V, Deutsch WA. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair (Amst) 2009;8(10):1215–24. doi: 10.1016/j.dnarep.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen D, Zhang Z, Li M, Wang W, Li Y, Rayburn ER, Hill DL, Wang H, Zhang R. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26(35):5029–37. doi: 10.1038/sj.onc.1210327. [DOI] [PubMed] [Google Scholar]
- 47.Daftuar L, Zhu Y, Jacq X, Prives C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS One. 2013;8(7):e68667. doi: 10.1371/journal.pone.0068667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khalaileh A, Dreazen A, Khatib A, Apel R, Swisa A, Kidess-Bassir N, Maitra A, Meyuhas O, Dor Y, Zamir G. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 2013;73(6):1811–20. doi: 10.1158/0008-5472.CAN-12-2014. [DOI] [PubMed] [Google Scholar]
- 49.Karan D, Kelly DL, Rizzino A, Lin MF, Batra SK. Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis. 2002;23(6):967–75. doi: 10.1093/carcin/23.6.967. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi T, Sasaki Y, Oshima Y, Yamamoto H, Mita H, Suzuki H, Toyota M, Tokino T, Itoh F, Imai K, Shinomura Y. Activation of the ribosomal protein L13 gene in human gastrointestinal cancer. Int J Mol Med. 2006;18(1):161–70. [PubMed] [Google Scholar]
- 51.Miao P, Sheng S, Sun X, Liu J, Huang G. Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. IUBMB Life. 2013;65(11):904–10. doi: 10.1002/iub.1216. [DOI] [PubMed] [Google Scholar]
- 52.Read JA, Winter VJ, Eszes CM, Sessions RB, Brady RL. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins. 2001;43(2):175–85. doi: 10.1002/1097-0134(20010501)43:2<175::aid-prot1029>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 53.Talaiezadeh A, Shahriari A, Tabandeh MR, Fathizadeh P, Mansouri S. Kinetic characterization of lactate dehydrogenase in normal and malignant human breast tissues. Cancer Cell Int. 2015;15:19. doi: 10.1186/s12935-015-0171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myers JK, Pace CN, Scholtz JM. Denaturant M-Values and Heat-Capacity Changes - Relation to Changes in Accessible Surface-Areas of Protein Unfolding. Protein Sci. 1995;4(10):2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parsell DA, Sauer RT. The Structural Stability of a Protein Is an Important Determinant of Its Proteolytic Susceptibility in Escherichia-Coli. J Biol Chem. 1989;264(13):7590–7595. [PubMed] [Google Scholar]
- 56.Ahmad S, Kumar V, Ramanand KB, Rao NM. Probing protein stability and proteolytic resistance by loop scanning: a comprehensive mutational analysis. Protein Sci. 2012;21(3):433–46. doi: 10.1002/pro.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park C, Marqusee S. Probing the high energy states in proteins by proteolysis. J Mol Biol. 2004;343(5):1467–76. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 58.Cunningham EL, Jaswal SS, Sohl JL, Agard DA. Kinetic stability as a mechanism for protease longevity. P Natl Acad Sci USA. 1999;96(20):11008–11014. doi: 10.1073/pnas.96.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pedersen JS, Otzen DE, Kristensen P. Directed Evolution of Barnase Stability Using Proteolytic Selection. J Mol Biol. 2002;323(1):115–123. doi: 10.1016/s0022-2836(02)00891-4. [DOI] [PubMed] [Google Scholar]
- 60.Chen VB, Davis IW, Richardson DC. KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci. 2009;18(11):2403–9. doi: 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text: Summary of the control experiment performed as part of this work.
Supplementary Table S-7. Proteomic coverage obtained in the control experiment using light- and heavy-labeled MCF-7 cells.
Supplementary Figure S-1. Global analysis of the H/L ratios generated for the assayed peptides in the control experiment.
Supplementary Table S-1. Excel spreadsheets summarizing peptides identified in the five biological replicates of MCF-7 versus MCF-10A cell line comparison with double and single digestion.
Supplementary Table S-2. Excel spreadsheet summarizing peptides assayed in the five biological replicates of MCF-7 versus MCF-10A cell line comparison.
Supplementary Table S-3. Excel spreadsheet summarizing peptide and protein hits identified with altered conformations in the MCF-7 vs. MCF-10A cell line comparison.
Supplementary Table S-4. Excel spreadsheets summarizing peptides identified in the three biological replicates of the control experiment for false positive rate determination using heavy- and light-labeled MCF-7 cell lines with double and single digestion.
Supplementary Table S-5. Excel spreadsheet summarizing peptides assayed in the three biological replicates of the control experiment using heavy- and light-labeled MCF-7 cell lines.
Supplementary Table S-6. Excel spreadsheet summarizing peptide and protein hits identified with altered conformations in the control experiment using heavy- and light-labeled MCF-7 cell lines.