

Recent publications have reported a previously unrecognized role for the MUC1-C oncoprotein in regulating DNA methyltransferase (DNMT) expression and thereby DNA methylation in human cancer cells [1, 2]. The MUC1-C transmembrane protein is aberrantly overexpressed in diverse human cancers and in certain hematologic malignancies, including acute myelo-genous leukemia (AML) [3]. Dysregulation of DNMTs and disruption of DNA methylation patterns are established hallmarks of the cancer cell [4]. MUC1-C has been linked to other hallmarks, such as (i) induction of the epithelial-mesenchymal transition (EMT), (ii) repression of tumor suppressor genes, (iii) activation of the MYC gene, and (iv) promotion of self-renewal capacity [3, 5]. However, there had been no known relationship between MUC1-C-induced signaling and the regulation of DNMTs and DNA methylation in cancer.
DNMTs catalyze the transfer of a methyl group to cytosine in CpG dinucleotides [4]. DNMT1, which localizes at foci of DNA replication, is largely responsible for maintaining global and gene-specific CpG methylation. In addition, DNMT3a and DNMT3b typically contribute to de novo postreplicative DNA methylation patterns. DNA methylation in promoter regions is associated with transcriptional repression; whereas, DNA methylation in gene bodies can induce increases in transcription and alternative splicing [4]. In this respect, dysregulation of DNMTs in cancer cells can affect global programs of gene repression and activation. Despite this complexity, a notable finding is that both DNMT1 and DNMT3b are required for silencing genes in cancer cells [6].
The MUC1-C cytoplasmic domain is a 72-amino acid intrinsically disordered protein, a finding consistent with other oncogenic molecules that have the plasticity to direct the activation of multiple signaling pathways. Along these lines, the MUC1-C cytoplasmic domain interacts with diverse kinases and effectors that are linked to transformation [3, 5]. For instance, the MUC1-C cytoplasmic activates the WNT pathway by binding directly to β-catenin and promoting the activation of WNT target genes, such as CCND1 and MYC. MUC1-C also activates the inflammatory IKK→NF-κB pathway and drives the expression of NF-κB p65 target genes, including MUC1 itself in an auto-inductive loop. In addition, the MUC1-C→NF-κB p65 pathway induces the ZEB1 tumor suppressor gene and promotes ZEB1-mediated repression of genes encoding (i) factors, such as Crumbs 3 (CRB3), required for maintaining apical-basal polarity, (ii) miR-200c, an inducer of epithelial differentiation, and (iii) E-cadherin, a keystone of the adherens junction complex; all of which contribute to the EMT phenotype (Fig. 1) [7].
Figure 1. MUC1-C→NF-κB signaling integrates EMT and DNA methylation-induced changes in gene expression.
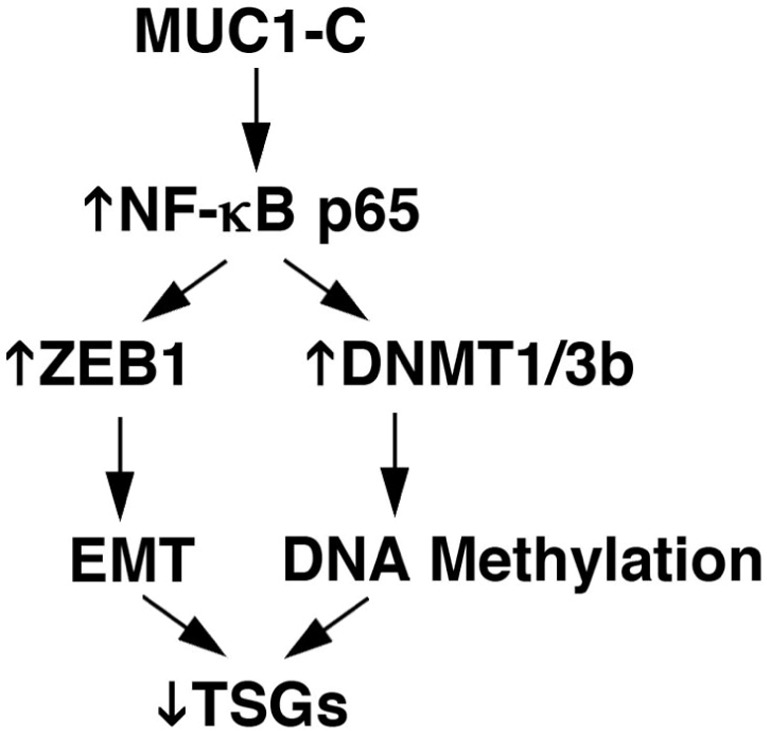
MUC1-C activates the inflammatory TAK1→IKK→NF-κB p65 pathway. MUC1-C also binds directly to NF-κB p65, increases occupancy of MUC1-C/NF-κB p65 complexes on the ZEB1 promoter, induces ZEB1 expression, and drives EMT by ZEB1-mediated mechanisms. Additionally, MUC1-C increases MUC1-C/NF-κB p65 occupancy on the DNMT1/3b promoters, induces DNMT1/3b expression, and alters global and gene-specific DNA methylation patterns. In this way, MUC1-C→NF-κB p65 signaling plays a role in integrating EMT with epigenetic changes in gene expression and the repression of TSGs.
In concert with driving EMT, recent work has linked the MUC1-C→NF-κB pathway to the regulation of DNA methylation in carcinoma cells (Fig. 1) [1]. Interestingly, the findings demonstrate that MUC1-C induces the expression of DNMT1 and DNMT3b, but not DNMT3a [1], in line with the requirement for both DNMT1 and DNMT3b to silence genes in cancer cells [6]. MUC1-C drives NF-κB p65 occupancy on the DNMT1 and DNMT3b promoters and promotes their activation [1]. In this way, MUC1-C regulates global DNA methylation and induces CDH1 promoter methylation with suppression of E-cadherin expression. These findings and the role of MUC1-C in inducing EMT have invoked the likelihood that MUC1-C epigenetically regulates other genes in the EMT program (Fig. 1) [7]. In addition and as raised below, these findings support the notion that MUC1-C is a potential target for derepressing tumor suppressor genes (TSGs), such as CDH1 and others.
DNMT1 is necessary for the self-renewal of leukemia stem cells. Moreover, the anti-leukemic agent decita-bine downregulates DNMT1, but has no effect on DNMT3a/b expression, supporting the potential importance of DNMT1 as a target for the treatment of AML [2]. In this regard and based on the results obtained in carcinoma cells, MUC1-C was found to induce DNMT1 expression by an NF-κB p65-dependent mechanism in AML cells [2]. Targeting MUC1-C with silencing or pharmacologically with the inhibitor GO-203 in AML cell lines and primary blasts was thus associated with (i) the suppression of DNMT1, (ii) decreases in methylation of CpG islands in the CDH1 promoter, and (iii) upregulation of E-cadherin [2]. Targeting MUC1-C in AML cells was also associated with derepression of the PTEN and BRCA1 TSGs [2].
The available evidence thus supports an important role for MUC1-C in the dysregulation of DNA methylation in human cancer cells [1, 2]. Therefore, targeting MUC1-C could represent a therapeutic approach alone and in combination with agents, such as decitabine, for reprogramming the cancer epigenome. Indeed, the combination of GO-203 and decitabine has been shown to be highly effective in downregulating DNMT1 and decreasing AML cell survival [2]. With regard to the potential for clinical translation, GO-203 has completed Phase I evaluation in patients with advanced carcinomas. Moreover, based on the above findings, a Phase Ib/IIa trial of GO-203 in combination with decitabine is underway for the treatment of patients with relapsed/refractory AML.
REFERENCES
- 1.Rajabi H, et al. Oncogene. 2016;35:6439–45. doi: 10.1038/onc.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tagde A, et al. Oncotarget. 2016;7:38974–87. doi: 10.18632/oncotarget.9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kufe DW. Nat Rev Cancer. 2009;9:874–85. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baylin SB, Jones PA. Cold Spring Harb Perspect Biol. 2016;8:a019505. doi: 10.1101/cshperspect.a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kufe DW. Oncogene. 2013;32:1073–81. doi: 10.1038/onc.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhee I, et al. Nature. 2002;416:552–56. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 7.Rajabi H, et al. Oncogene. 2014;33:1680–89. doi: 10.1038/onc.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]