

Summary
This review describes the recent advances in the study of food microbial ecology, with a focus on food fermentations. High‐throughput sequencing (HTS) technologies have been widely applied to the study of food microbial consortia and the different applications of HTS technologies were exploited in order to monitor microbial dynamics in food fermentative processes. Phylobiomics was the most explored application in the past decade. Metagenomics and metatranscriptomics, although still underexploited, promise to uncover the functionality of complex microbial consortia. The new knowledge acquired will help to understand how to make a profitable use of microbial genetic resources and modulate key activities of beneficial microbes in order to ensure process efficiency, product quality and safety.
Introduction
Culture‐independent techniques have helped to change the way to study food microbial ecology, leading to consider microbial populations as consortia (Cocolin and Ercolini, 2015). Further evolution was stimulated by the advent of high‐throughput sequencing (HTS) technologies in the mid 2000s, which in the past decade became ubiquitous in microbial ecology studies. HTS entails higher sensitivity compared with traditional culture‐independent methods beyond being considered quantitative. Two different HTS approaches can be used. In the most commonly applied option, marker‐genes are amplified from genomic DNA (or RNA, after a reverse‐transcription step) through PCR and sequenced. Taxonomic relevant genes are usually sequenced through this approach, leading to the description of the phylobiome (van Hijum et al., 2013), that is the taxonomic composition of the microbial community and the relative abundance of its members. In metagenomics or metatranscriptomics studies, no PCR is performed and total DNA or cDNA is sequenced. Besides the taxonomical composition of the community, this approach allows obtaining the abundance of all microbial genes. The output of metagenomics analysis, based on DNA sequencing, are the potential activities of the microbial communities, because a specific gene may not be really expressed in that condition or because DNA may arise from dead or metabolically inactive cells. In order to identify the genes actually expressed in a food sample, RNA sequencing (RNA‐seq) is the most appropriate path to take, which is what happens in metatranscriptomics.
The study of microbial ecology is relevant in biotechnology as it is the basis for not only the development of fermentations but also for the comprehension of the microbial interactions that drive a premium quality process. The availability of such a powerful tool box offers tantalizing opportunities to study food microbes understanding how their potential functions can be changed or modulated with the ultimate scope of improving food quality.
Monitoring microbes in food fermentations
Amplicon‐based HTS targeting genes of taxonomic relevance has become the most widely exploited approach in food microbial ecology. In the past decade, it was widely used to monitor microbial communities during fermentation of different types of foodstuffs and beverages (Fig. 1). Table 1 reports an extensive, although not complete, list of them. Several questions can be addressed by the description of microbial communities during fermentations. An in‐depth characterization of the normal or abnormal microbial consortia at different stages of fermentations is important in order to evaluate lot‐to‐lot consistency, identify biomarkers for product quality or spoilage, and learning how to manipulate fermentation conditions to improve the process control.
Figure 1.
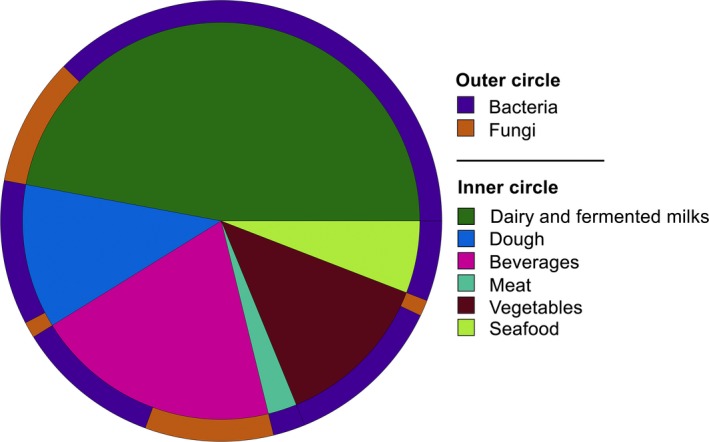
Pie chart showing the abundance of HTS studies of fermented foods and beverages grouped according to the food matrix. For each food environment, the outer circle shows the proportion of studies analysing bacterial or fungal communities.
Table 1.
HTS studies of food fermentations. Studies are grouped according to the type of food and ordered by year of publication
| Target gene | Short description | Year | Food Group | Reference |
|---|---|---|---|---|
| 16S rRNA gene (Bacteria) | Kefir grains and kefir milk | 2011 | Dairy and fermented milks | Dobson et al. (2011) |
| 16S rRNA gene (Bacteria) | Danish raw milk cheese during ripening | 2011 | Dairy and fermented milks | Masoud et al. (2011) |
| 16S rRNA gene (Bacteria) | Kefir grains from different parts of Brazil | 2012 | Dairy and fermented milks | Leite et al. (2012) |
| 16S rRNA gene (Bacteria) | Mozzarella cheese (Italy) and intermediates from two manufactures | 2012 | Dairy and fermented milks | Ercolini et al. (2012) |
| 16S rRNA gene (Bacteria) | Latin style cheese | 2012 | Dairy and fermented milks | Lusk et al. (2012) |
| 16S rRNA gene (Bacteria) | Curd, fresh and smoked Polish cheese (Oscypek) | 2012 | Dairy and fermented milks | Alegria et al. (2012) |
| 16S rRNA gene (Bacteria) | Artisanal soft, semi‐hard and hard cheeses from raw or pasteurized cow, goat, or sheep milk | 2012 | Dairy and fermented milks | Quigley et al. (2012) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Kefir grain and kefir milk from different sources | 2013 | Dairy and fermented milks | Marsh et al. (2013) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Swabs from cheesemaking environment and cheese | 2013 | Dairy and fermented milks | Bokulich et al. (2013a) |
| 16S rRNA gene (Bacteria) | Croatiam raw ewe's milk cheese during ripening | 2013 | Dairy and fermented milks | Fuka et al. (2013) |
| 16S rRNA gene (Bacteria) | Turkish kefir grains | 2014 | Dairy and fermented milks | Nalbantoglu et al. (2014) |
| 16S rRNA gene (Bacteria) | Whey cultures and cheese curds from water‐buffalo Mozzarella, Grana Padano and Parmigiano Reggiano cheese (Italy) manufacturing | 2014 | Dairy and fermented milks | De Filippis et al. (2014) |
| 16S rRNA gene (Bacteria) | Ewe milk, curd and Canestrato cheese (Italy) during ripening | 2014 | Dairy and fermented milks | De Pasquale et al. (2014a) |
| 16S rRNA gene (Bacteria) | Cow milk, curd and Caciocavallo cheese (Italy) during ripening | 2014 | Dairy and fermented milks | De Pasquale et al. (2014b) |
| 16S rRNA gene (Bacteria) | Cow milk (from different lactation stages), curd and Fontina cheese (Italy) from three dairies | 2014 | Dairy and fermented milks | Dolci et al. (2014) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Bloomy, natural and washed cheese rinds | 2014 | Dairy and fermented milks | Wolfe et al. (2014) |
| 16S rRNA gene (Bacteria) | Traditional Pico cheese (Portugal) manufactured in three different dairies, monitored during ripening | 2014 | Dairy and fermented milks | Riquelme et al. (2014) |
| 16S rRNA gene (Bacteria) | Samples of milk, whey, curd and ripened Poro cheese (Mexico) | 2014 | Dairy and fermented milks | Aldrete‐Tapia et al. (2014) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Tarag (fermented dairy product) from China and Mongolia | 2014 | Dairy and fermented milks | Sun et al. (2014) |
| 16S rRNA gene (Bacteria) | Samples of core and rind of Herve cheese (Belgium) | 2014 | Dairy and fermented milks | Delcenserie et al. (2014) |
| 16S rRNA gene (Bacteria) | Chinese traditional fermented milk (yond bap) from cow or goat milk | 2015 | Dairy and fermented milks | Liu et al. (2015a) |
| 16S rRNA gene (Bacteria); 18S rRNA gene (Fungi) | Naturally fermented cow milks from Mongolia | 2015 | Dairy and fermented milks | Liu et al. (2015b) |
| 16S rRNA gene (Bacteria); 26S rRNA gene (Fungi) | Milk kefir grains from different Italian regions | 2015 | Dairy and fermented milks | Garofalo et al. (2015) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Matsoni (fermented milk) samples from several geographic areas | 2015 | Dairy and fermented milks | Bokulich et al. (2015a) |
| 16S rRNA gene (Bacteria); 26S rRNA gene (Fungi) | Environmental swabs from a dairy plant and cheeses (Italy) | 2015 | Dairy and fermented milks | Stellato et al. (2015) |
| 16S rRNA gene (Bacteria) | Continental cheese produced early and late in the day, at different ripening times | 2015 | Dairy and fermented milks | O'Sullivan et al. (2015) |
| 16S rRNA gene (Bacteria) | Commercial high‐moisture Mozzarella cheese produced with different acidification methods | 2016 | Dairy and fermented milks | Guidone et al. (2015) |
| 16S rRNA gene (Bacteria) | Undefined strain starters (milk cultures) for high‐moisture Mozzarella cheese | 2016 | Dairy and fermented milks | Parente et al. (2016b) |
| 16S rRNA gene (Bacteria) | Grana‐type cheese (Italy) during ripening | 2016 | Dairy and fermented milks | Alessandria et al. (2016) |
| 16S rRNA gene (Bacteria) | Natural whey culture, milk, curd and Caciocavallo cheese (Italy) during ripening | 2016 | Dairy and fermented milks | De Filippis et al. (2016a) |
| 16S rRNA gene (Bacteria) | Environmental swabs from a dairy plant and cheeses (Italy) | 2016 | Dairy and fermented milks | Calasso et al. (2016) |
| 16S rRNA gene (Bacteria) | Spatial distribution of microbiota in Italian ewes’ milk cheese | 2016 | Dairy and fermented milks | De Pasquale et al. (2016) |
| 16S rRNA gene (Bacteria) | Rye, durum and common wheat sourdough | 2013 | Doughs | Ercolini et al. (2013) |
| 16S rRNA gene (Bacteria) | Traditional sweet leavened doughs | 2013 | Doughs | Lattanzi et al. (2013) |
| 16S rRNA gene (Bacteria) | Rye sourdoughs propagated for two months at 20 and 30°C | 2014 | Doughs | Bessmeltseva et al. (2014) |
| 16S rRNA gene (Bacteria) | Flour and sourdough made of durum wheat grown under organic and conventional farming | 2015 | Doughs | Rizzello et al. (2015) |
| 16S rRNA gene (Bacteria); 18S rRNA gene (Fungi) | Flour, doughs and related food environments | 2015 | Doughs | Minervini et al. (2015) |
| 16S rRNA gene (Bacteria) | Wheat sourdoughs used for traditional breads in different regions of France | 2015 | Doughs | Lhomme et al. (2015a) |
| 16S rRNA gene (Bacteria) | Sourdoughs used for the manufacture of traditional French breads | 2015 | Doughs | Lhomme et al. (2015b) |
| 16S rRNA gene (Bacteria) | Dough samples during manufacture of Chica (a fermented maize product) in Argentina | 2015 | Doughs | Elizaquível et al. (2015) |
| 16S rRNA gene (Bacteria) | Rye sourdoughs from four Estonian bakeries | 2016 | Doughs | Viiard et al. (2016) |
| 16S rRNA gene (Bacteria) | Botrityzed wine during fermentation, three vintages, inoculated and uninoculated bactches | 2012 | Fermented beverages ‐ grapes | Bokulich et al. (2012a) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Grape must samples collected in California over two different vintages | 2014 | Fermented beverages ‐ grapes | Bokulich et al. (2014a) |
| 16S rRNA gene (Bacteria); 26S rRNA gene and ITS1‐2 (Fungi) | Grape must samples collected in different Portuguese regions during fermentation | 2015 | Fermented beverages ‐ grapes | Pinto et al. (2015) |
| 26S rRNA gene (Fungi) | Spanish grape must samples during fermentation | 2015 | Fermented beverages ‐ grapes | Wang et al. (2015) |
| 18S rRNA gene (Fungi) | Italian traditional wine fermentations | 2016 | Fermented beverages ‐ grapes | De Filippis et al. (2016b) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Grape must samples collected in three wineries in Northern Italy during fermentation | 2016 | Fermented beverages ‐ grapes | Stefanini et al. (2016) |
| 16S rRNA gene (Bacteria) | Beer (American Coolship Ale) during fermentation and related environment | 2012 | Fermented beverages ‐ malt | Bokulich et al. (2012b) |
| 16S rRNA gene (Bacteria) | Barley during malting, two different seasons | 2014 | Fermented beverages ‐ malt | Justé et al. (2014) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Traditional Korean alcoholic beverage (Makgeolli), and the starter (Nuruk), during fermentation | 2012 | Fermented beverages ‐ rice | Jung et al. (2012) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Kimoto sake during manufacturing and related environmental samples | 2014 | Fermented beverages ‐ rice | Bokulich et al. (2014b) |
| 16S rRNA gene (Bacteria); ITS1‐2 (Fungi) | Tea fungus (kombucha) samples during fermentation | 2014 | Fermented beverages ‐ tea | Marsh et al. (2014) |
| 16S rRNA gene (Bacteria); ITS1‐4 (Fungi) | Pu‐erh Japanese traditional tea during fermentation | 2015 | Fermented beverages ‐ tea | Zhao et al. (2015) |
| 16S rRNA gene (Bacteria) | Fermented meat (salami) during ripening | 2015 | Meat | Greppi et al. (2015) |
| 16S rRNA gene (Bacteria) | Fermented meat (salami) during ripening | 2015 | Meat | Polka et al. (2015) |
| 16S rRNA gene (Bacteria) | Fermented seafood | 2010 | Seafood | Roh et al. (2010) |
| 16S rRNA gene (Bacteria) | Narezushi (salted and fermented fish, rice, peppers) | 2011 | Seafood | Koyanagi et al. (2011) |
| 16S rRNA gene (Bacteria) | Traditional fermented sushi (kaburazushi),during fermentation | 2013 | Seafood | Koyanagi et al. (2013) |
| 16S rRNA gene (Bacteria) | Traditional fermented shrimp (Saeu‐jeot) during fermentation | 2013 | Seafood | Jung et al. (2013) |
| 16S rRNA gene (Bacteria) | Traditional fermented shrimp (Saeu‐jeot) during fermentation at different temperatures | 2014 | Seafood | Lee et al. (2014) |
| 16S rRNA gene (Bacteria) | Korean fish sauce (Myeolchi‐Aekjeot) | 2015 | Seafood | Lee et al. (2015) |
| 16S rRNA gene (Bacteria) | Pearl millet slurried with or without groundnuts at the beginning and end of fermentation | 2009 | Vegetables | Humblot et al. (2009) |
| 16S rRNA gene (Bacteria) | Vegetable pickle from rice bran (Nukadoko) | 2011 | Vegetables | Sakamoto et al. (2011) |
| 16S rRNA gene (Bacteria) | Baechu (Chinese cabbage) and Chonggak (radish) kimchi prepared with and without starter | 2012 | Vegetables | Jung et al. (2012) |
| 16S rRNA gene (Bacteria) | Fermented Korean soybean paste (Doenjang) | 2012 | Vegetables | Nam et al. (2012a) |
| 16S rRNA gene (Bacteria) | Traditional Korean fermented food (Kochujang) made of rice, pepper, soybeans | 2012 | Vegetables | Nam et al. (2012b) |
| 16S rRNA gene (Bacteria) | Ten different varieties of kimchi, during fermentation | 2012 | Vegetables | Park et al. (2012) |
| 16S rRNA gene (Bacteria) | Olive surfaces and brine during fermentation | 2013 | Vegetables | Cocolin et al. (2013) |
| 16S rRNA gene (Bacteria) | Kimchi samples during fermentation (100 days) | 2013 | Vegetables | Jeong et al. (2013) |
| 16S rRNA gene (Bacteria) | Fermented Korean soybean lumps (meju) | 2014 | Vegetables | Jung et al. (2014) |
| 16S rRNA gene (Bacteria) | Started and unstarted Bella di Cerignola table olives | 2015 | Vegetables | De Angelis et al. (2015) |
Dairy is by far the most explored environment (Fig. 1) and a broad variety of cheeses were studied through amplicon‐based HTS, allowing monitoring of curd fermentation (Ercolini et al., 2012; De Filippis et al., 2014) or cheese ripening (Fuka et al., 2013; De Pasquale et al., 2014b; O'Sullivan et al., 2015; Alessandria et al., 2016; De Filippis et al., 2016a) and exploring the spatial distribution of microbes in different parts of the same cheese (O'Sullivan et al., 2015; De Filippis et al., 2016a; De Pasquale et al., 2016).
In many cases, the study of food microbiota highlighted possible relationships between microbial community structure/dynamics and physicochemical parameters, such as pH, water activity (aw), salt concentration and temperature (Bessmeltseva et al., 2014; Wolfe et al., 2014; Lhomme et al., 2015a,b; Minervini et al., 2015; De Filippis et al., 2016a). In other studies, the microbiota was related to raw material origin (Bokulich et al., 2014a; Rizzello et al., 2015) or quality (Dolci et al., 2014; O'Sullivan et al., 2015; Alessandria et al., 2016), as well as to development of flavour‐impact compounds (De Pasquale et al., 2014a, 2016; De Filippis et al., 2016a). Moreover, food‐related environments were found to harbour a resident microbiota, beneficially involved in dairy (Bokulich and Mills, 2013a; Stellato et al., 2015; Calasso et al., 2016), alcoholic (Bokulich et al., 2012a, 2014b) and sourdough (Minervini et al., 2015) fermentations, although the presence of potential spoilers was also emphasized in some cases (Bokulich et al., 2015a; Stellato et al., 2015).
Although fungi can be very important in some kinds of food fermentations, there is a huge difference in the number of published studies describing fungal and bacterial communities through HTS (Fig. 1). While 16S ribosomal RNA (rRNA) is the common choice for bacteria, more variability in the target gene to use was highlighted for fungi (Table 1). The most used target is the internal transcribed spacer (ITS), most of all thanks to the presence of a well‐curated database (https://unite.ut.ee). However, the uneven ITS length among species may promote preferential amplification of shorter fragments during the PCR step and therefore higher number of sequences for those OTUs with shorter ITS fragment. Since the OTU abundance is proportional to the number of reads obtained, this may lead to an incorrect estimation of OTU abundance (Bokulich and Mills, 2013b; Ercolini, 2013; De Filippis and Ercolini, 2016). Therefore, the use of different targets would be also advisable, such as the 26S (Garofalo et al., 2015; Stellato et al., 2015; Wang et al., 2015) or the 18S rRNA (Liu et al., 2015b; Minervini et al., 2015) genes.
Although PCR‐dependent, the amplicon‐based HTS is considered quantitative: the number of reads obtained for each operational taxonomic unit (OTU) is proportional to the abundance of that OTU in the sample and the higher sensitivity allows identifying also sub‐populations previously difficult to detect. However, the data generated need to be interpreted with the awareness of culture‐independent PCR biases that have been reviewed elsewhere (Sipos et al., 2010), such as the possibility of preferential amplification, due to the different efficiency of the primer towards selected species, that may result in the under‐representation of some clades (Pinto and Raskin, 2012).
In several occasions RNA was preferred as target instead of DNA, that may arise from dead or inactive cells (Ercolini et al., 2013; Rizzello et al., 2015; De Filippis et al., 2016a). Since RNA is more easily degraded, it allows obtaining a structure of the microbiota that is potentially ascribable to the viable populations. Nevertheless, the treatment of food samples with propidium monoazide (PMA) was recently proposed as an alternative (Erkus et al., 2016) to the use of RNA templates to describe live microbial populations. PMA selectively binds to DNA freely present in the matrix, arising from dead or compromised cells, but cannot enter intact cell membranes. Therefore, treatment of the sample with PMA before DNA extraction inhibits its amplification by PCR and allows obtaining a profile of the viable microbial community (Erkus et al., 2016).
Notwithstanding the thick body of literature accumulated on food microbial communities assessed by amplicon sequencing, most of the studies are basically descriptive. In addition, well‐known microbial players have been identified and thus limited new information was provided on food fermentative processes.
However, thanks to the extensive amount of 16S rRNA gene data generated by amplicon‐based food microbiota descriptions, there currently is an unprecedented possibility of data sharing. Sequence data can be made available through public databases, e.g. the Sequence Read Archive of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/Traces/sra) or the European Nucleotide Archive of the European Bioinformatics Institute (http://www.ebi.ac.uk/ena). In this way, researchers from all over the world can easily access datasets generated by other laboratories, re‐analyse and use them for meta‐studies. FoodMicrobionet (http://www.foodmicrobionet.org, Parente et al., 2016a) was recently launched with the aim to collect the results obtained in different HTS studies of food microbial ecology and to provide an easy‐to‐use tool for visualization and comparison of the microbiota in diverse foodstuffs. Samples are classified using the FoodEx classification, as suggested by the European Food Safety Authority (http://www.efsa.europa.eu/en/data/data-standardisation). Further metadata include the nature of food (raw, intermediate, finished), the use of thermal treatments and the occurrence of spoilage and/or fermentation. Users can easily extract subsets of samples for the food matrix of interest, recreate abundance tables in long and wide format and visualize them in a network or use in comparative studies. As an example, graphical representations of the network structure of dairy samples extracted from Foodmicrobionet (v. 2.0) are provided here (Fig. 2). Figure 2A, a bipartite network representation, shows high abundance of starter lactic acid bacteria (SLAB) in intermediates and undefined starters coming from the production of different types of cheeses, while non‐starter LAB (NSLAB) display higher abundances in ripened cheeses, with the exception of spoiled products. Such separation is even more evident in the network shown in Fig. 2B, where long‐ripened (> 30 days) clustered apart from fresh/mid‐ripened cheeses and intermediates of production (natural whey cultures, fermented curds).
Figure 2.
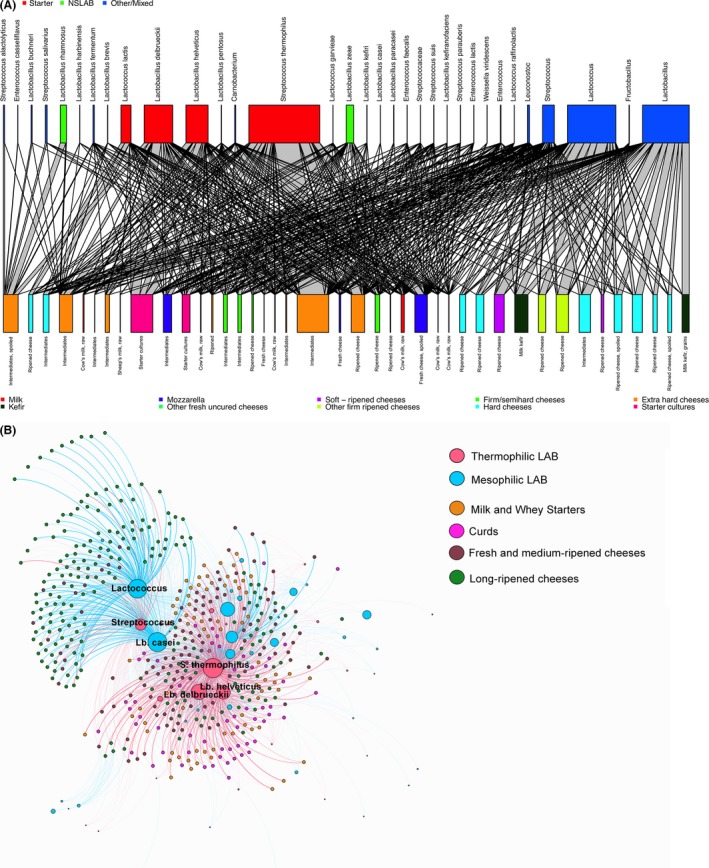
Graphical representations of the network structure of dairy samples extracted from Foodmicrobionet (v. 2.0). In (A), individual dairy samples are coulored according to the type of dairy product and aggregated by the state of the food (finished, intermediate, raw), the occurrence of spoilage, fermentation or ripening.
Interesting and relatively new application in the amplicon‐based HTS is its use for the monitoring of microbial populations beyond the species level. Genes with high polymorphism within a species can be selected for this application, potentially allowing discrimination between different strains within a species (Ercolini et al., 2013). In spite of the limitations due to possible sequencing errors, this application was recently exploited for monitoring Streptococcus thermophilus in whey and milk cultures and in cheeses, using lactose permease (lacS, De Filippis et al., 2014) and phosphoserine phosphatase (serB, Parente et al., 2016b; Ricciardi et al., 2016) as species‐specific and polymorphic target genes.
Exploring microbial functions directly in food
Shotgun metagenome or metatranscriptome sequencing offers several advantages over amplicon‐based approach (Fig. 3). Whole genomes are sequenced directly (often after fragmentation and library preparation), without any prior PCR step, avoiding the possibility of amplification biases. Moreover, a picture of the entire microbial community can be obtained, tracking and comparing the abundance of bacteria and other organisms at the same time, although the methods have to be chosen carefully, in order to avoid preferential nucleic acids extractions from bacterial cells. The exceptional advantage of shotgun sequencing is the possibility to monitor the abundance of microbial activities directly in the food matrix, and hence to collect information on the genetic capacity of the whole microbial community. Moreover, it is possible to recover complete or draft microbial genomes from the metagenomes, achieving in this way a strain‐level resolution. Although metagenome and metatranscriptome sequencing potentially give a much higher amount of information their cost is still substantially higher compared with the amplicon‐based approaches. Additional technical precautions are needed in metatranscriptomics studies. For example, when sequencing messenger RNA (mRNA), all efforts are needed in order to ‘freeze’ the gene expression at sampling and protect mRNA from degradation by using stabilizing solutions, since microbial mRNA has extremely short half‐life (Rauhut and Klug, 1999; Deutscher, 2006). In addition, mRNA is only a small percentage of the total RNA extracted from the sample, which is mainly rRNA. Therefore, a further step is usually necessary, in order to reduce the amount of rRNA by bacterial or eukaryotic rRNA depletion. Finally, the bioinformatics analysis of shotgun data is much more computationally intensive and complex compared with amplicon sequencing, since well‐defined pipelines have not been implemented yet.
Figure 3.
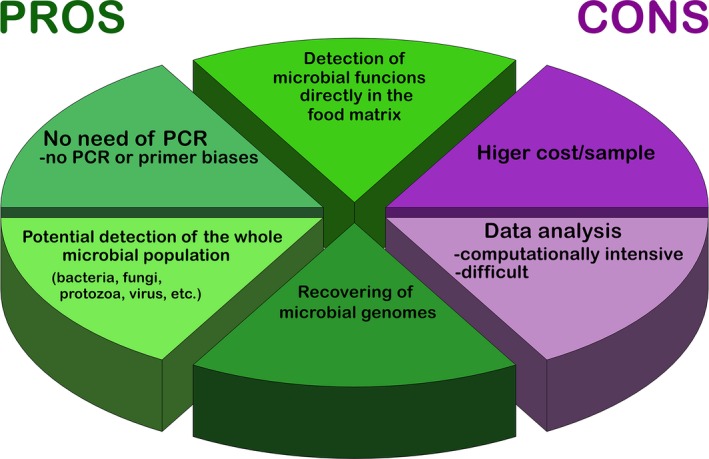
Pie chart showing pros and cons in the use of shotgun metagenomics and metatranscriptomics compared to the amplicon‐based approach.
The above reasons have limited the application of metagenomics and metatranscriptomics to only few pioneer studies, again mainly focused on dairies. Insights about microbial activities involved in cheese ripening process were provided. Monitoring of microbial gene expression during ripening of a Camembert‐type (Lessard et al., 2014) and a surface‐ripened cheese (Dugat‐Bony et al., 2015) by metatranscriptomics revealed the presence of microbial genes involved in flavour production from amino acids and highlighted that these activities were enhanced in the first phase of ripening. Moreover, Monnet et al. (2016) reported differences in amino acid catabolism between the two yeasts inoculated on a Reblochon‐type cheese: Geotrichum candidum reached a maximum expression of amino acids‐related genes in the first phase of ripening, while Debaryomyces hansenii took over after 30 days, highlighting a different role of these yeasts in flavour production. Indeed, Wolfe et al. (2014) suggested that pathways leading to the production of flavouring compounds, such as those involved in branched‐chain and sulfur‐containing amino acid degradation, were enriched in washed‐rind compared with natural‐ and bloomy‐rind cheeses metagenomes. These studies provided an in‐depth analysis of the cheese maturation process and allowed to better understand the metabolic activities of the different community members and their possible interactions. Indeed, cheese microbial communities were proposed as simplified model to study both microbial assembly and metabolism (as affected by abiotic factors), providing a possible interesting model for understanding microbial dynamics in more complex environments (Wolfe and Dutton, 2015). In a recent example, the evolution of bacterial activities during manufacturing and ripening of a traditional cheese made with an undefined starter was monitored by metatranscriptomics highlighting how to manipulate microbial gene expression through modifications of the process parameters (De Filippis et al., 2016a). An increase in ripening temperature was shown to foster NSLAB growth and the expression levels of genes related to proteolysis, lipolysis and to the production of flavours from amino acids, thus promoting cheese ripening. These kinds of studies help to shed light on microbial community dynamics involved in fermentative processes and suggest how microbiome responses can be modulated in order to optimize production efficiency and product quality.
Targeting microbial genomic variability for food fermentation processes
Technologically relevant microbial species and strains are extensively used alone or in mixed cultures in industrial processes. While allowing a better control and a standardization of the process, the use of selected cultures can lead to a loss of microbial diversity and flattening of the sensorial properties of the final products. Recent advances in microbial genomics are making it possible to exploit the wealth of genetic and phenotypic variability existing among food‐fermenting microbes (Smid and Hugenholtz, 2010). Thanks to the reduction in sequencing costs, whole genome sequencing of several strains from the same species is now a common practice. Pangenomics (the pool of essentially different genes found within a species) of common food‐borne microbes highlighted important genetic differences, which can be taken into account in order to select the best combination of species/strains for microbial industrial starters. Moreover, recently developed bioinformatics tools consent to recover microbial genomes directly from metagenomes, allowing strain‐level monitoring during the process and genomic comparison (Eren et al., 2015; Scholz et al., 2016). Comparing the genomes of five Lactococcus lactis strains (two wild‐type of non‐dairy origin and three dairy isolates), only about 60% of the genes were found to be shared among them and hundreds of accessory genes were not present in the dairy isolates (Siezen et al., 2008). Some of these coded for industrial‐interesting functions, such as exopolysaccharide (EPS) production, sugar and amino acids metabolism.
Pangenomics may also help to understand microbial evolution and adaptive mechanisms to different food niches. The genome of a Lactobacillus delbrueckii sub. bulgaricus strain industrially used for yoghurt production was compared with other collection strains of the same subspecies and genomic traits conferring higher efficiency in industrial fermentations were highlighted (Hao et al., 2011). The industrial strain showed higher efficiency in EPS production and well‐equipped stress tolerance, explaining its initial selection for yoghurt production. Moreover, its effective proteolytic system clarified the protocooperation mechanism with Strepococcus thermophilus (Hao et al., 2011).
Microbial interactions may be also elucidated. Coupling metagenomics and pangenomics, Erkus et al. (2013) characterized an undefined cheese starter culture with a long history of use. Although only two species were detected, high level of diversity at strain level was found and genome comparison suggested that a kill‐the‐winner mechanism subsisted: the phage sensitivity of the fittest strain was density‐dependent, preventing the eradication of other genetic lineages during back‐slopping regimes.
Comparative genomics of industrial strains is providing a richer and deeper understanding of the genetic composition and variability in these important microbes, promising to rapidly identify genetic loci that shape industrially significant traits. This will enable the development of a genetic catalogue of strains, tailoring starter composition at strain level to meet specific demands.
Conclusions
Food fermentations are often complex phenomena, involving several microbial species and strains. We have learned how to identify and quantify them as well as to study their traits on the bases of state‐of‐the‐art methodologies. As discussed, the most widely used application in food microbiology is the use of amplicon‐based sequencing, leading to an in‐depth description of the ecosystem studied. This can be undoubtedly useful in order to understand microbial dynamics and evolution during food production, as well as to identify the presence of possible spoilers. Nevertheless, the real advance led by HTS is the application of shotgun metagenomics and metatranscriptomics. These approaches are still underexploited in food microbial ecology. Their application to food fermentations may be extremely useful in order to explore microbial functions directly in the food matrix and understand microbial behaviour in response to different process conditions. Moreover, recovering microbial genomes from the metagenomes allows to monitor the evolution of different strains during the process and to compare their genomic potential. These tools promise to be an invaluable help to better understand and possibly tune microbial activities in order to ensure process efficiency, product quality and safety.
Conflicts of interest
None declared.
Microbial Biotechnology (2017) 10(1), 91–102
Funding Information
No specific funding source has to be acknowledged.
References
- Aldrete‐Tapia, A. , Escobar‐Ramírez, M.C. , Tamplin, M.L. , and Hernández‐Iturriaga, M. (2014) High‐throughput sequencing of microbial communities in Poro cheese, an artisanal Mexican cheese. Food Microbiol 44: 136–141. [DOI] [PubMed] [Google Scholar]
- Alegría, A. , Szczesny, P. , Mayo, B. , Bardowski, J. , and Kowalczyk, M. (2012) Biodiversity in Oscypek, a traditional Polish cheese, determined by culture‐dependent and ‐independent approaches. Appl Environ Microbiol 78: 1890–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandria, V. , Ferrocino, I. , De Filippis, F. , Fontana, M. , Rantsiou, K. , Ercolini, D. , and Cocolin, L. (2016) Microbiota of an Italian Grana like cheese during manufacture and ripening unraveled by 16S rRNA‐based approaches. Appl Environ Microbiol 82: 3988–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessmeltseva, M. , Viiard, E. , Simm, J. , Paalme, T. , and Sarand, I. (2014) Evolution of bacterial consortia in spontaneously started rye sourdoughs during two months of daily propagation. PLoS ONE 9: e95449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , and Mills, D.A. (2013a) Facility‐specific “house” microbiome drives microbial landscapes of artisan cheesemaking plants. Appl Environ Microbiol 79: 5214–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , and Mills, D.A. (2013b) Improved selection of internal transcribed spacer‐specific primers enables quantitative, ultra‐high‐throughput profiling of fungal communities. Appl Environ Microbiol 79: 2519–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Bamforth, C.W. , and Mills, D.A. (2012a) Brewhouse‐resident microbiota are responsible for multi‐stage fermentation of American coolship ale. PLoS ONE 7: e35507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Joseph, C.M. , Allen, G. , Benson, A.K. , and Mills, D.A. (2012b) Next‐generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS ONE 7: e36357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Ohta, M. , Richardson, P.M. , and Mill, D.A. (2013) Monitoring seasonal changes in winery‐resident microbiota. PLoS ONE 8: e66437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Thorngate, J.H. , Richardson, P.M. , and Mills, D.A. (2014a) Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc Natl Acad Sci USA 111: E139–E148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Ohta, M. , Lee, M. , and Mills, D.A. (2014b) Indigenous bacteria and fungi drive traditional kimoto sake fermentations. Appl Environ Microbiol 80: 5522–5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Bergsveinson, J. , Ziola, B. , and Mills, D.A. (2015a) Mapping microbial ecosystems and spoilage‐gene flow in breweries highlights patterns of contamination and resistance. Elife 10: 4. doi:10.7554/eLife.04634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A. , Amiranashvili, L. , Chitchyan, K. , Ghazanchyan, N. , Darbinyan, K. , Gagelidze, N. , et al (2015b) Microbial biogeography of the transnational fermented milk matsoni. Food Microbiol 50: 12–19. [DOI] [PubMed] [Google Scholar]
- Calasso, M. , Ercolini, D. , Mancini, L. , Stellato, G. , Minervini, F. , Di Cagno, R. , et al (2016) Relationships among house, rind and core microbiotas during manufacture of traditional Italian cheeses at the same dairy plant. Food Microbiol 54: 115–126. [Google Scholar]
- Cocolin, L. , and Ercolini, D. (2015) Zooming into food‐associated microbial consortia: a “cultural” evolution. Curr Opin Food Sci 2: 43–50. [Google Scholar]
- Cocolin, L. , Alessandria, V. , Botta, C. , Gorra, R. , De Filippis, F. , Ercolini, D. , and Rantsiou, K. (2013) NaOH‐debittering induces changes in bacterial ecology during table olives fermentation. PLoS ONE 8: e69074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis, M. , Campanella, D. , Cosmai, L. , Summo, C. , Rizzello, C.G. , and Caponio, F. (2015) Microbiota and metabolome of un‐started and started Greek‐type fermentation of Bella di Cerignola table olives. Food Microbiol 52: 18–30. [DOI] [PubMed] [Google Scholar]
- De Filippis, F. and Ercolini, D. (2016) Food microbial ecology in the “omics” era In Reference Module in Food Sciences. Smithers G.W. (ed.). USA, ISBN 9780081005965: Elsevier, pp. 1–7. [Google Scholar]
- De Filippis, F. , La Storia, A. , Stellato, G. , Gatti, M. , and Ercolini, D. (2014) A selected core microbiome drives the early stages of three popular Italian cheese manufactures. PLoS ONE 9: e89680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis, F. , Genovese, A. , Ferranti, P. , Gilbert, J.A. , and Ercolini, D. (2016a) Metatranscriptomics revels temperature‐driven functional changes in microbiome impacting cheese maturation rate. Sci Rep 6: 21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pasquale, I. , Calasso, M. , Mancini, L. , Ercolini, D. , La Storia, A. , De Angelis, M. , et al (2014a) Causal relationship between microbial ecology dynamics and proteolysis during manufacture and ripening of protected designation of origin (PDO) cheese Canestrato Pugliese. Appl Environ Microbiol 80: 4085–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pasquale, I. , Di Cagno, R. , Buchin, S. , De Angelis, M. , and Gobbetti, M. (2014b) Microbial ecology dynamics reveal a succession in the core microbiota involved in the ripening of pasta filata caciocavallo pugliese cheese. Appl Environ Microbiol 80: 6243–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pasquale, I. , Di Cagno, R. , Buchin, S. , De Angelis, M. , and Gobbetti, M. (2016) Spatial distribution of the metabolically active microbiota within Italian PDO ewes’ milk cheeses. PLoS ONE 11: e0153213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcenserie, V. , Taminiau, B. , Delhalle, L. , Nezer, C. , Doyen, P. , Crevecoeur, S. , et al (2014) Microbiota characterization of a Belgian protected designation of origin cheese, Herve cheese, using metagenomic analysis. J Dairy Sci 97: 6046–6056. [DOI] [PubMed] [Google Scholar]
- Deutscher, M.P. (2006) Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res 34: 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson, A. , O'Sullivan, O. , Cotter, P.D. , Ross, P. , and Hill, C. (2011) High‐throughput sequence‐based analysis of the bacterial composition of kefir and an associated kefir grain. FEMS Microbiol Lett 320: 56–62. [DOI] [PubMed] [Google Scholar]
- Dolci, P. , De Filippis, F. , La Storia, A. , Ercolini, D. , and Cocolin, L. (2014) rRNA‐based monitoring of the microbiota involved in Fontina PDO cheese production in relation to different stages of cow lactation. Int J Food Microbiol 185: 127–135. [DOI] [PubMed] [Google Scholar]
- Dugat‐Bony, E. , Straub, C. , Teissandier, A. , Onésime, D. , Loux, V. , Monnet, C. , et al (2015) Overview of a surface‐ripened cheese community functioning by meta‐omics analyses. PLoS ONE 10: e0124360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizaquível, P. , Pérez‐Cataluña, A. , Yépez, A. , Aristimuño, C. , Jiménez, E. , Cocconcelli, P.S. , et al (2015) Pyrosequencing vs. culture‐dependent approaches to analyze lactic acid bacteria associated to chicha, a traditional maize‐based fermented beverage from Northwestern Argentina. Int J Food Microbiol 198: 9–18. [DOI] [PubMed] [Google Scholar]
- Ercolini, D. , De Filippis, F. , La Storia, A. , and Iacono, M. (2012) “Remake” by high‐throughput sequencing of the microbiota involved in the production of water buffalo Mozzarella cheese. Appl Environ Microbiol 78: 8142–8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercolini D. (2013) High‐throughput sequencing and metagenomics: moving forward in the culture‐independent analysis of food microbial ecology. Appl Environ Microbiol 79: 3148‐3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercolini, D. , Pontonio, E. , De Filippis, F. , Minervini, F. , La Storia, A. , Gobbetti, M. , and Di Cagno, R. (2013) Microbial ecology dynamics during rye and wheat sourdough preparation. Appl Environ Microbiol 79: 7827–7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren, A.M. , Esen, Ö.C. , Quince, C. , Vineis, J.H. , Morrison, H.G. , Sogin, M.L. , and Delmont, T.O. (2015) Anvi'o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3: e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkus, O. , de Jager, V.C. , Spus, M. , van Alen‐Boerrigter, I.J. , van Rijswijck, I.M. , Hazelwood, L. , et al (2013) Multifactorial diversity sustains microbial community stability. ISME J 7: 2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkus, O. , de Jager, V.C.L. , Geene, R.T.C.M. , van Alen‐Boerrigter, I.J. , Hazelwood, L. , van Hijum, S.A.F.T. , et al (2016) Use of propidium monoazide for selective profiling of viable microbial cells during Gouda cheese ripening. Int J Food Microbiol 228: 1–9. [DOI] [PubMed] [Google Scholar]
- Fuka, M.M. , Wallisch, S. , Engel, M. , Welzl, G. , Havranek, J. , and Schloter, M. (2013) Dynamics of bacterial communities during the ripening process of different Croatian cheese types derived from raw ewe's milk cheeses. PLoS ONE 8: e80734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo, C. , Osimani, A. , Milanović, V. , Aquilanti, L. , De Filippis, F. , Stellato, G. , et al (2015) Bacteria and yeast microbiota in milk kefir grains from different Italian regions. Food Microbiol 49: 123–133. [DOI] [PubMed] [Google Scholar]
- Greppi, A. , Ferrocino, I. , La Storia, A. , Rantsiou, K. , Ercolini, D. , and Cocolin, L. (2015) Monitoring of the microbiota of fermented sausages by culture independent rRNA‐based approaches. Int J Food Microbiol 212: 67–75. [DOI] [PubMed] [Google Scholar]
- Guidone, A. , Matera, A. , Ricciardi, A. , Zotta, T. , De Filippis, F. , Ercolini, D. , and Parente, E. (2016) The microbiota of high‐moisture Mozzarella cheese produced with different acidification methods. Int J Food Microbiol 216: 9–17. [DOI] [PubMed] [Google Scholar]
- Hao, P. , Zheng, H. , Yu, Y. , Ding, G. , Gu, W. , Chen, S. , et al (2011) Complete sequencing and pan‐genomic analysis of Lactobacillus delbrueckii subsp. bulgaricus reveal its genetic basis for industrial yogurt production. PLoS ONE 6: e15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hijum, S.A. , Vaughan, E.E. , and Vogel, R.F. (2013) Application of state‐of‐art sequencing technologies to indigenous food fermentations. Curr Opin Biotechnol 24: 178–186. [DOI] [PubMed] [Google Scholar]
- Humblot, C. , and Guyot, J.P. (2009) Pyrosequencing of tagged 16S rRNA gene amplicons for rapid deciphering of the microbiomes of fermented foods such as pearl millet slurries. Appl Environ Microbiol 75: 4354–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, S.H. , Lee, S.H. , Jung, J.Y. , Choi, E.J. , and Jeon, C.O. (2013) Microbial succession and metabolite changes during long‐term storage of Kimchi. J Food Sci 78: M763–M769. [DOI] [PubMed] [Google Scholar]
- Jung, M.J. , Nam, Y.D. , Roh, S.W. , and Bae, J.W. (2012a) Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol 30: 112–123. [DOI] [PubMed] [Google Scholar]
- Jung, J.Y. , Lee, S.H. , Lee, H.J. , Seo, H.Y. , Park, W.S. , and Jeon, C.O. (2012b) Effects of Leuconostoc mesenteroides starter cultures on microbial communities and metabolites during kimchi fermentation. Int J Food Microbiol 153: 378–387. [DOI] [PubMed] [Google Scholar]
- Jung, J.Y. , Lee, S.H. , Lee, H.J. , and Jeon, C.O. (2013) Microbial succession and metabolite changes during fermentation of saeu‐jeot: traditional Korean salted seafood. Food Microbiol 34: 360–368. [DOI] [PubMed] [Google Scholar]
- Justé, A. , Malfliet, S. , Waud, M. , Crauwels, S. , De Cooman, L. , Aerts, G. , et al (2014) Bacterial community dynamics during industrial malting, with an emphasis on lactic acid bacteria. Food Microbiol 39: 39–46. [DOI] [PubMed] [Google Scholar]
- Koyanagi, T. , Kiyohara, M. , Matsui, H. , Yamamoto, K. , Kondo, T. , Katayama, T. , and Kumagai, H. (2011) Pyrosequencing survey of the microbial diversity of “narezushi”, an archetype of modern Japanese sushi. Lett Appl Microbiol 53: 635–640. [DOI] [PubMed] [Google Scholar]
- Koyanagi, T. , Nakagawa, A. , Kiyohara, M. , Matsui, H. , Yamamoto, K. , Barla, F. , et al (2013) Pyrosequencing analysis of microbiota in Kaburazushi, a traditional medieval sushi in Japan. Biosci Biotechnol Biochem 77: 2125–2130. [DOI] [PubMed] [Google Scholar]
- Lattanzi, A. , Minervini, F. , Di Cagno, R. , Diviccaro, A. , Antonielli, L. , Cardinali, G. , et al (2013) The lactic acid bacteria and yeast microbiota of eighteen sourdoughs used for the manufacture of traditional Italian sweet leavened baked goods. Int J Food Microbiol 163: 71–79. [DOI] [PubMed] [Google Scholar]
- Lee, S.H. , Jung, J.Y. , and Jeon, C.O. (2014) Effects of temperature on microbial succession and metabolite change during saeu‐jeot fermentation. Food Microbiol 38: 16–25. [DOI] [PubMed] [Google Scholar]
- Lee, S.H. , Jung, J.Y. , and Jeon, C.O. (2015) Bacterial community dynamics and metabolite changes in myeolchi‐aekjeot, a Korean traditional fermented fish sauce, during fermentation. Int J Food Microbiol 203: 15–22. [DOI] [PubMed] [Google Scholar]
- Leite, A.M. , Mayo, B. , Rachid, C.T. , Peixoto, R.S. , Silva, J.T. , Paschoalin, V.M. , and Delgado, S. (2012) Assessment of the microbial diversity of Brazilian kefir grains by PCR‐DGGE and pyrosequencing analysis. Food Microbiol 31: 215–221. [DOI] [PubMed] [Google Scholar]
- Lessard, M.H. , Viel, C. , Boyle, B. , St‐Gelais, D. , and Labrie, S. (2014) Metatranscriptome analysis of fungal strains Penicillium camemberti and Geotrichum candidum reveal cheese matrix breakdown and potential development of sensory properties of ripened Camembert‐type cheese. BMC Genom 15: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhomme, E. , Lattanzi, A. , Dousset, X. , Minervini, F. , De Angelis, M. , Lacaze, G. , et al (2015a) Lactic acid bacterium and yeast microbiotas of sixteen French traditional sourdoughs. Int J Food Microbiol 215: 161–170. [DOI] [PubMed] [Google Scholar]
- Lhomme, E. , Orain, S. , Courcoux, P. , Onno, B. , and Dousset, X. (2015b) The predominance of Lactobacillus sanfranciscensis in French organic sourdoughs and its impact on related bread characteristics. Int J Food Microbiol 213: 40–48. [DOI] [PubMed] [Google Scholar]
- Liu, X.‐F. , Liu, C.‐J. , Zhang, H.‐Y. , Gong, F.‐M. , Luo, Y.‐Y. , and Li, X.‐R. (2015a) The bacterial community structure of yond bap, a traditional fermented goat milk product, from distinct Chinese regions. Dairy Sci & Technol 95: 369–380. [Google Scholar]
- Liu, W. , Zheng, Y. , Kwok, L.Y. , Sun, Z. , Zhang, J. , Guo, Z. , et al (2015b) High‐throughput sequencing for the detection of the bacterial and fungal diversity in Mongolian naturally fermented cow's milk in Russia. BMC Microbiol 15: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusk, T.S. , Ottesen, A.R. , White, J.R. , Allard, M.W. , Brown, E.W. , and Kase, J.A. (2012) Characterization of microflora in Latin‐style cheeses by next‐generation sequencing technology. BMC Microbiol 12: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, A.J. , O'Sullivan, O. , Hill, C. , Ross, R.P. , and Cotter, P.D. (2013) Sequencing‐based analysis of the bacterial and fungal composition of kefir grains and milks from multiple sources. PLoS ONE 8: e69371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, A.J. , O'Sullivan, O. , Hill, C. , Ross, R.P. , and Cotter, P.D. (2014) Sequence‐based analysis of the bacterial and fungal compositions of multiple kombucha (tea fungus) samples. Food Microbiol 38: 171–178. [DOI] [PubMed] [Google Scholar]
- Masoud, W. , Takamiya, M. , Vogensen, F.K. , Lillevang, S. , Al‐Soud, W.A. , Sørensen, S.J. , and Jakobsen, M. (2011) Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturating gradient gel electrophoresis and pyrosequencing. Int Dairy J 21: 142–148. [Google Scholar]
- Minervini, F. , Lattanzi, A. , De Angelis, M. , Celano, G. , and Gobbetti, M. (2015) House microbiotas as sources of lactic acid bacteria and yeasts in traditional Italian sourdoughs. Food Microbiol 52: 66–76. [DOI] [PubMed] [Google Scholar]
- Monnet, C. , Dugat‐Bony, E. , Swennen, D. , Beckerich, J.‐M. , Irlinger, F. , Fraud, S. , and Bonnarme, P. (2016) Investigation of the activity of the microorganisms in a Reblochon‐style cheese by metatranscriptomic analysis. Front Microbiol 7: 536. doi:10.3389/fmicb.2016.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbantoglu, U. , Cakar, A. , Dogan, H. , Abaci, N. , Ustek, D. , Sayood, K. , and Can, H. (2014) Metagenomic analysis of the microbial community in kefir grains. Food Microbiol 41: 42–51. [DOI] [PubMed] [Google Scholar]
- Nam, Y.D. , Lee, S.Y. , and Lim, S.I. (2012a) Microbial community analysis of Korean soybean pastes by next‐generation sequencing. Int J Food Microbiol 155: 36–42. [DOI] [PubMed] [Google Scholar]
- Nam, Y.D. , Park, S.L. , and Lim, S.I. (2012b) Microbial composition of the Korean traditional food “kochujang” analyzed by a massive sequencing technique. J Food Sci 77: M250–M256. [DOI] [PubMed] [Google Scholar]
- O'Sullivan, D.J. , Cotter, P.D. , O'Sullivan, O. , Giblin, L. , McSweeney, P.L. , and Sheehan, J.J. (2015) Temporal and spatial differences in microbial composition during the manufacture of a continental‐type cheese. Appl Environ Microbiol 81: 2525–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parente, E. , Cocolin, L. , De Filippis, F. , Zotta, T. , Ferrocino, I. , O'Sullivan, O. , et al (2016a) FoodMicrobionet: a database for the visualization and exploration of food bacterial communities based on network analysis. Int J Food Microbiol 219: 28–37. [DOI] [PubMed] [Google Scholar]
- Parente, E. , Guidone, A. , Matera, A. , De Filippis, F. , Mauriello, G. , and Ricciardi, A. (2016b) Microbial community dynamics in thermophilic undefined milk starter cultures. Int J Food Microbiol 217: 59–67. [DOI] [PubMed] [Google Scholar]
- Park, E.J. , Chun, J. , Cha, C.J. , Park, W.S. , Jeon, C.O. , and Bae, J.W. (2012) Bacterial community analysis during fermentation of ten representative kinds of kimchi with barcoded pyrosequencing. Food Microbiol 30: 197–204. [DOI] [PubMed] [Google Scholar]
- Pinto, A.J. , and Raskin, L. (2012) PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE 7: e43093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, C. , Pinho, D. , Cardoso, R. , Custódio, V. , Fernandes, J. , Sousa, S. , et al (2015) Wine fermentation microbiome: a landscape from different Portuguese wine appellations. Front Microbiol 6: 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Połka, J. , Rebecchi, A. , Pisacane, V. , Morelli, L. , and Puglisi, E. (2015) Bacterial diversity in typical Italian salami at different ripening stages as revealed by high‐throughput sequencing of 16S rRNA amplicons. Food Microbiol 46: 342–356. [DOI] [PubMed] [Google Scholar]
- Quigley, L. , O'Sullivan, O. , Beresford, T.P. , Ross, R.P. , Fitzgerald, G.F. , and Cotter, P.D. (2012a) High‐throughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses. Appl Environ Microbiol 78: 5717–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauhut, R. , and Klug, G. (1999) mRNA degradation in bacteria. FEMS Microbiol Rev 23: 353–370. [DOI] [PubMed] [Google Scholar]
- Ricciardi, A. , De Filippis, F. , Zotta, T. , Facchiano, A. , Ercolini, D. , and Parente, E. (2016) Polymorphism of the phosphoserine phosphatase gene in Streptococcus thermophilus and its potential use for typing and monitoring of population structure. Int J Food Microbiol 236: 138–147. [DOI] [PubMed] [Google Scholar]
- Riquelme, C. , Câmara, S. , Dapkevicius Mde, L. , Vinuesa, P. , da Silva, C.C. , Malcata, F.X. , and Rego, O.A. (2015) Characterization of the bacterial biodiversity in Pico cheese (an artisanal Azorean food). Int J Food Microbiol 192: 86–94. [DOI] [PubMed] [Google Scholar]
- Rizzello, C.G. , Cavoski, I. , Turk, J. , Ercolini, D. , Nionelli, L. , Pontonio, E. , et al (2015) The organic cultivation of Triticum turgidum spp. durum reflects on the axis flour, sourdough fermentation and bread. Appl Environ Microbiol 81: 3192–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh, S.W. , Kim, K.H. , Nam, Y.D. , Chang, H.W. , Park, E.J. , and Bae, J.W. (2010) Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing. ISME J 4: 1–16. [DOI] [PubMed] [Google Scholar]
- Sakamoto, N. , Tanaka, S. , Sonomoto, K. , and Nakayama, J. (2011) 16S rRNA pyrosequencing‐based investigation of the bacterial community in nukadoko, a pickling bed of fermented rice bran. Int J Food Microbiol 144: 352–359. [DOI] [PubMed] [Google Scholar]
- Scholz, M. , Ward, D.V. , Pasolli, E. , Tolio, T. , Zolfo, M. , Asnicar, F. , et al (2016) Strain‐level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods 13: 435–438. [DOI] [PubMed] [Google Scholar]
- Siezen, R.J. , Starrenburg, M.J. , Boekhorst, J. , Renckens, B. , Molenaar, D. , and van Hylckama Vlieg, J.E. (2008) Genome‐scale genotype‐phenotype matching of two Lactococcus lactis isolates from plants identifies mechanisms of adaptation to the plant niche. Appl Environ Microbiol 74: 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos, R. , Székely, A. , Révész, S. , and Márialigeti, K. (2010) Addressing PCR biases in environmental microbiology studies. Methods Mol Biol 599: 37–58. [DOI] [PubMed] [Google Scholar]
- Smid, E.J. , and Hugenholtz, J. (2010) Food fermentation processes. Annu Rev Food Sci Technol 1: 497–519. [DOI] [PubMed] [Google Scholar]
- Stefanini, I. , Albanese, D. , Cavazza, A. , Franciosi, E. , De Filippo, C. , Donati, C. , and Cavalieri, D. (2016) Dynamic changes in microbiota and mycobiota during spontaneous “Vino Santo Trentino” fermentation. Microb Biotechnol 9: 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellato, G. , De Filippis, F. , La Storia, A. , and Ercolini, D. (2015) Coexistence of lactic acid bacteria and potential spoilage microbiota in a dairy‐processing environment. Appl Environ Microbiol 81: 7893–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z. , Liu, W. , Bao, Q. , Zhang, J. , Hou, Q. , Kwok, L. , et al (2014) Investigation of bacterial and fungal diversity in tarag using high‐throughput sequencing. J Dairy Sci 97: 6085–6096. [DOI] [PubMed] [Google Scholar]
- Wang, C. , García‐Fernández, D. , Mas, A. , and Esteve‐Zarzoso, B. (2015) Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE. Front Microbiol 6: 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, B.E. , and Dutton, R.J. (2015) Fermented foods as experimentally tractable microbial ecosystems. Cell 161: 49–55. [DOI] [PubMed] [Google Scholar]
- Wolfe, B.E. , Button, J.E. , Santarelli, M. , and Dutton, R.J. (2014) Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, M. , Zhang, D.L. , Su, X.Q. , Duan, S.M. , Wan, J.Q. , Yuan, W.X. , et al (2015) An integrated metagenomics/metaproteomics investigation of the microbial communities and enzymes in solid‐state fermentation of Pu‐erh tea. Sci Rep 5: 10117. [DOI] [PMC free article] [PubMed] [Google Scholar]