

Abstract
The cell surface receptor CD44 is a glycoprotein belonging to the hyaluronan-binding proteins, termed hyaladherins. CD44 is expressed in a wide variety of isoforms in many cells, and in particular is present on the surface of malignant cells where it is involved in the onset and progression of cancer. In a first attempt to identify novel CD44 binding agents, we first characterized via nuclear magnetic resonance (NMR) techniques, several agents that were reported to bind to hCD44. To our surprise, however, none of these putative CD44 binding agents including peptides, one of which is in phase 2 clinical trial (A6 peptide), and a recently reported fragment hit, were found to significantly interact with recombinant hCD44(21–178). Nonetheless, we further report that a fragment screening campaign, using solution NMR as detection method, identified a viable fragment hit that bound in a potentially functional pocket on the surface of CD44, opposite to the HA binding site. We hypothesize that this pocket could be indirectly associated with the cellular and in vivo activity of the A6 peptide hence providing a novel framework for a possible development of therapeutically viable CD44 antagonists.
Keywords: CD44, A6 peptide, protein-protein interactions inhibitors, FBLD, FBDD, NMR
Graphical Abstract
With the aim of finding an inhibitor of hCD44 we first evaluated the binding properties of its putative ligands using solution NMR spectroscopy. We observed that apart from its natural ligand HA8 and the antibody DF1485 none of these putative ligands bound significantly to recombinant hCD44(21–178). However a fragment screening campaign identified and validated fragment hit that bound to a possibly ‘druggable’ back pocket of hCD44(21–178) that may have functional implications.
Introduction
The cell surface receptor CD44, a type I transmembrane glycoprotein, was first described in 1983[1]. This hyaluronan-binding protein is expressed on the surface of many cell types[2], where it is involved in leukocyte migration to inflamed sites, T cell activation, and tumor metastasis[3]. Several studies suggest that the interactions between the adhesion of CD44 with its main natural ligand, the hyaluronic acid (HA) are critical for its function [2a, 4]. HA is a linear copolymer of repeating N-acetyl-D-glucosamine and D-glucuronic acid units, and represents one of the most abundant component of the extra-cellular matrix [5]. There are several isoforms of CD44 [6], and this variability is enhanced by the presence of post-translational modifications, most notables is its glycosylation (both N- and O-linked carbohydrate side chains)[7]. Each of the different isoforms, however, is characterized by a fixed N-terminal domain that functions as docking site for HA[8]. In particular, the recognition of HA by CD44 is permitted by the presence in the amino-terminal region of about 90-amino acids containing the highly conserved Link domain (residues 32–124) [2b].
It has been observed that CD44 expression enhances metastatic behavior of cancer cells[9] enabling matrix metalloproteases (MMPs) activation, thus enabling many biological processes[8, 10] that result into the cleavage of CD44, and concomitant cancer cells detachment and migration[2b]. Moreover, in Chronic Lymphocytic Leukemia (CLL), CD44 has been reported to signal B-cell survival by activating both the PI3K/AKT as well as the MAPK/ERK pro-survival pathways, and by inducing the expression of the anti-apoptotic protein Mcl-1[11]. Accordingly, Mcl-1 inhibitors or an anti-CD44 antibody can revert these effects and induce apoptosis in primary CLL cells [12].
Hence, in recent years several laboratories have attempted to identify CD44 binding agents for the development of novel therapeutics. The involvement of CD44 in cells migration and its overexpression on a wide spectrum of tumor cells, make this receptor also a suitable target for delivery of chemotherapeutics in cancer cells. Most approaches targeting CD44 have either used antibodies or small peptides binding to CD44 epitopes. Other targeting strategies used the natural CD44 ligand, hyaluronic acid (HA), as a backbone or scaffold for drug attachment, or as both a targeting moiety and delivery agent for small molecule therapeutics[13].
Of these putative CD44 interacting agents, the peptide A6, an 8-amino acid peptide (acetyl-KPSSPPEE-amino), has been shown to have anti-invasive, anti-migratory, and anti-angiogenic activities in a variety of in vitro and in vivo model systems[14]. Recent studies[15] with human CLL B-cell lymphocytes have shown that A6 down modulates the expression of CD44 and ZAP-70 (a marker for an aggressive form of CLL), and inhibits B-cell receptor (BCR) signaling, resulting in a direct, dose-dependent, cytotoxicity in vitro. Currently the putative CD44 binding peptide A6 is in phase 2 clinical trial in patients with CLL and SLL (Small Lymphocytic Lymphoma)[9] (ClinicalTrials.gov Identifier: NCT02046928).
Analysis of the three-dimensional structure of CD44 and its interaction site with HA reveals a wide and shallow binding site which is likely difficult to target with small organic molecules [16]. The HABD has no obvious well-formed or deep pockets that would serve as attractive binding sites for small molecule inhibitors and is known to undergo small but important conformational changes upon HA binding [17]. Similarly, the stem region is unlikely to possess a well-defined three-dimensioned structure to accommodate a ligand. Nonetheless, the biological relevance of CD44 in cancer metastasis provides further impetus for the identification of suitable and novel CD44 binding agents, either peptides or small molecules. To this end we sought to first characterize the binding of reported putative CD44 binding compounds. Moreover, we used an NMR guided fragment-based screening campaign to identify possibly novel agents.
Results
Expression and characterization of hCD44(21–178) and HA8 binding studies
For all the experiments we used a soluble construct of human CD44 from residue Q21 to residue V178, corresponding to the HA Binding Domain (HABD). Uniformly 15N labeled hCD44(21–178) was expressed in E. coli and purified as reported by Banerji et al.[18]. The yield of the protein was about 14 mg per liter of M9 medium in the monomeric form. The 2D [1H,15N]-sofast-HMQC[19] spectrum of hCD44(21–178) closely resembles the previously reported spectrum[20] and the deposited [1H,15N] resonance assignments (BMRB ID 6093). Furthermore, we tested the ability of our construct to bind its natural ligand, Hyaluronic Acid (HA)[4, 21]. Structural studies have been reported for the complex between HA8 (Figure 1A) and murine CD44 HABD by X-ray crystallography[17] (PDB 2JCR). In addition, the NMR structure of hCD44 HABD in complex with HA8 was also reported[20], although only the coordinates of the protein were deposited (PDB 1POZ). Hence, we prepared a model of the structure of hCD44 HABD in complex with HA8 using SWISS-MODEL[22] and the PDB-ID 2JCR as template (Figure 1A). To test the ability of our hCD44(21–178) to bind HA8, a 20 μM solution of 15N-hCD44(21–178) was titrated with increasing amounts of HA8 (20, 40, 60, 80, 100, 120, 140, 160, 180, 200, and 220 μM, respectively) collecting both 1D 1H and 2D [1H,15N]-sofast-HMQC NMR spectra at each HA8 concentration (Figure 1B and C). Binding of HA8 to hCD44(21–178) could be appreciated by chemical shift perturbations induced in the 1H-aliphatic region of the spectrum of hCD44(21–178) (Figure 1B). In particular, the resonances corresponding to δ-protons of residue L135 seemed most affected by the presence of HA8. Interestingly, L135 is not in direct contact with HA8, hence the observed perturbations are likely a results of conformational changes in hCD44(21–178) induced by ligand binding, in agreement with previously reported structural studies[23]. Similarly, widespread perturbations were observed in the 2D [1H,15N]-sofast-HMQC spectrum of hCD44(21–178) (20 μM) in presence of HA8 (220 μM) (Figure 1C), again suggestive of binding and conformational rearrangements upon complex formation. Finally, we used Isothermal Titration Calorimetry (ITC) to quantify the dissociation constant between HA8 and our construct (Figure 1D). We obtained a Kd value of 24.6 μM, which is in agreement with previously reported values in similar studies by ITC[17], and by SPR[16]. These relatively low binding affinities are expected for the E. coli expressed non-glycosylated form of hCD44(21–178), and the relatively low molecular weight version of HA used in the binding experiments. Nonetheless, our data collectively suggested that our construct was properly folded and retained the ability to bind to small HA derived oligosaccharides. As such, our CD44 construct was suitable for further studies to evaluate the binding properties of several putative CD44 binding agents.
Figure 1. Biophysical validation of HA8.

(A) Semitransparent molecular surface representation of the homology model of hCD44 built with SWISS-MODEL [22a] [22b] [22c] [22d] using the structure of mCD44 in complex with HA8 (PDB 2JCR) as template. HA8 (chemical structure is reported in the upper panel) and the HA binding pocket are colored in orange. The glycosylation sites are highlighted in purple.
(B) 1D 1H-aliphatic spectra of 20 μM hCD44(21–178). The apo spectrum is blue while the spectra recorded in presence of increasing concentrations of HA8 (starting from 20 μM to 220 μM) are in purple to light-blue.
(C) 2D [1H,15N]-sofast-HMQC of 20 μM hCD44(21–178) in the apo form (blue) and with a 11-fold molar excess of HA8 (orange).
(D) Isothermal Titration Calorimetry (ITC) for hCD44(21–178) titrated with HA8. The Kd obtained is 24.6 μM. The relatively low enthalpy of binding (ΔH= −0.7 Kcal/mol) is justified by the weak binding.
Validation of putative binders of hCD44
Over the past several years, a variety studies reported on numerous putative CD44 binding agents, ranging from monoclonal antibodies, to peptides and to small organic molecules (Table 1). Surprisingly, we found that with the notable exception of HA8 and antibodies, none of these previously reported agents bound appreciably to hCD44(21–178) as described below.
Table 1.
| Putative CD44 binders | References and comments | |||
|---|---|---|---|---|
| Natural Ligand | ||||
| HA8 : [GlcNAc-GlcUA]4 | [21] [4a] [4b] [20] | Kd = 24.6 μM by ITCa | ||
| Antibodies | ||||
| DF1485 | [25] | From Santa Cruz Biotechnolgy Kd ≪ 400 nM by NMRb |
||
| Roche | Patent Number: WO 2011095498 A1c | |||
| Peptides | ||||
| A6 Peptide: Ac-KPSSPPEE-NH2 | [25] [9] | In phase 2 for CLL |
|
No direct bind to recombinant hCD44(21–178) by NMRd |
| A5G27: H-RLVSYNGIIFFLK-NH2 | [26] | Laminin α5 peptide | ||
| Ac-SRPQGPFL-NH2 | [28] | From blade I of MMP-9 | ||
| Fragment | ||||
Compound 3:
|
[16] | Reported Kd by SPR is 0.9 mM No appreciable binding by NMR or ITCe |
||
Isothermal Titration Calorimetry (ITC) data as reported in Figure 1. The data are in agreement with similar measurements from ref. [17].
No Kd was reported in literature for this antibody. By NMR spectroscopy with recombinant 15N-hCD44(21–178) we estimated a Kd value ≪ 400 nM.
This antibody is not commercially available. We tested peptides regions cited in the patent as putative CD44 binding elements. However, under the reported experimental condition, none of these peptides bound to hCD44(21–178) significantly.
Using 15N-hCD44(21–178) at 20 μM we could not detect significant binding for these peptides when tested at a concentration up to 500 μM.
In [1H,15N]-sofast-HMQC correlation spectra with 15N-hCD44(21–178) at 20 μM, compound 3 did not show appreciable binding up to 20 mM. In addition, the compound was not able to significantly displace the binding of HA8 to CD44 at 55 mM by ITC (see text and supporting information).
HCAM (DF1485), from Santa Cruz Biotechnology, is a mouse anti-CD44 monoclonal antibody of about 90–95 kDa. The 2D [1H,15N]-sofast-HMQC of hCD44(21–178) at 10 μM is reported in Figure 2A in absence (blue) and in presence (magenta) of the DF1485 antibody at 1:1 stoichiometry. Complex formation was evident by the extensive line broadening beyond detection, typical of a complex of large molecular weight (> 100 kDa). Similar data were obtained at 5 μM hCD44(21–178) and 4 μM DF1485 concentration, indicating an upper limit for the Kd of the complex of ~ 400 nM (Table 1). A second antibody was recently reported (patent number US 20130224108 A1), however it is not commercially available, hence could not be tested.
Figure 2. Biophysical validation of hCD44 putative binders.

(A) CD44 antibody DF1485. 2D [1H,15N]-sofast-HMQC spectra of 10 μM 15N-hCD44(21–178) recorded in absence (blue) and in presence (magenta) of 10 μM DF1485 antibody. Due to the high molecular weight of the antibody-CD44 complex, as a result of the binding, the signals of the protein are broadened beyond detection. After the addition of 500 μM A6 peptide the antibody is not displaced, corroborating the findings of panel B that A6-peptide does not appreciably bind to hCD44(21–178).
A6-peptide. (B) 2D [1H,15N]-sofast-HMQC spectra of 20 μM 15N-hCD44(21–178) in absence (blue) and in presence (green) of 500 μM A6 peptide. No appreciable binding is detected under those experimental conditions. (C) 2D [1H,13C]-HSQC spectra of 5 μM 13C-A6 peptide in absence (green) and in presence (magenta) of 4 μM DF1485 antibody. The signal of the 13C-labeled acetyl methyl of the peptide is not perturbed by the presence of the antibody, indicating the absence of significant interactions between the A6-peptide and the antibody.
(D) Compound 3[16]. 2D [1H,15N]-sofast-HMQC spectra of 20 μM 15N-hCD44(21–178) recorded in absence (blue) and in presence (red) of 2 mM compound 3 [16]. No binding is detected. The absence of significant interactions is confirmed by Isothermal Titration Calorimetry in a competition assay in which hCD44(21–178) is incubated with a 750-fold molar excess of compound 3 and subsequently the binding of HA8 is probed. The Kd detected for HA8 under this condition was 37.5 μM with a ΔH= −0.83 Kcal/mol, hence not significantly different than the binding of HA8 to hCD44(21–178) in absence of compound 3 (Supplementary Figure S3).
The most advanced putative ligand of hCD44 is the A6-peptide (Table 1), that is currently in phase 2 clinical trial in patients with Chronic Lymphocytic Leukemia (CLL) and Small Lymphocytic Lymphoma (SLL)[9](ClinicalTrials.gov Identifier: NCT02046928). Similar to what we reported for HA8 and DF1485, in Figure 2B are shown the superimposed 2D [1H,15N]-sofast-HMQC spectra of 15N-hCD44(21–178) (20 μM) in absence (blue) and in presence (green) of the A6-peptide (500 μM). The superposition revealed no significant changes in chemical shifts upon titration of the ligand, suggesting the absence of appreciable binding under our experimental conditions. The absence of direct interactions between A6-peptide and recombinant non-glycosylated hCD44(21–178) was also confirmed by ITC (Supplementary Figure S1). These results are in agreement with what was found very recently by Liu et al.[24] using a sensitive Surface Plasmon Resonance (SPR) binding assay. Because the activity of A6 against CD44 was probed in cellular studies using DF1485[25], we also investigated the possibility of a direct interaction between the antibody and A6. For these studies we synthesized a 13C labeled A6 peptide by introducing a 13C methyl in its N-terminal acetyl group (13C-A6 peptide). Subsequently we recorded 2D [1H,13C]-HSQC spectra of 5 μM 13C-A6-peptide in absence and in presence of 4 μM DF1485 antibody (Figure 2C). Unlike CD44, no signal broadening or chemical shift perturbations could be detected for 13C-A6 in presence of DF1485. Hence, it is possible that A6 bound to CD44 only when it is fully glycosylated and/or that it recognized a CD44 region that is outside the HA binding domain.
Because of the possible therapeutic potential of A6, other laboratories pursued studies aimed at identifying other putative CD44 binding peptides. Hibino et al.[26] found that the laminin α5 synthetic peptide A5G27 (Table 1) inhibits cell migration, invasion and angiogenesis by binding the glycosaminoglycans on CD44. However, the peptide presented limited solubility for binding studies, and again it is an unlikely binder for the non-glycosylated version of hCD44. Accordingly from the 2D [1H,15N]-sofast-HMQC spectra of 20 μM 15N-hCD44(21–178) with 200 μM of A5G27 there was no evidence of binding.
Based on evidences related to the interactions between the Matrix Metalloproteinase-9 (MMP-9) and CD44[27], Dufour et al.[28] suggested that MMP-9 could be involved in a heterodimer formation with CD44 and in particular that the interactions could take place between CD44 and an 8-amino acid peptide (SRPQGPFL) constituting the blade I of the PEX domain of MMP-9. This peptide was purchased acetylated at the N-terminus and amidated at the C-terminus (Table 1) and was tested at a concentration of 400 μM against 20 μM 15N-hCD44(21–178). However, also in this case, under these experimental conditions, the 2D [1H,15N]-sofast-HMQC spectra of hCD44(21–178) indicated no significant binding of this peptide to our CD44 construct.
Recently Liu et al.[16] performed a fragment screening of the Maybridge Ro3 Diversity Library with the goal of identifying initial fragment hits. The authors of this work screened the fragments using immobilized CD44 HABD in a SPR based assay. The most potent fragment reported (compound 3 in Table 1) was crystallized (PDB 4MRG). To validate the binding of this fragment, initially we recorded 2D [1H,15N]-sofast-HMQC spectra of 20 μM 15N-hCD44(21–178) (Figure 2D) in absence (blue) and in presence (red) of 2 mM of compound 3. Because under these conditions there was no evidence of binding, we increased the concentration of the fragment up to 20 mM. At this concentration we detected large perturbations in the chemical shift of the 15N-hCD44(21–178) resonances, but after further investigation we noticed that the high concentration of compound 3 had also increased the pH of the solution from the value of 6.7 to about 8. After bringing the pH to the initial value of 6.7, the signals on the 2D [1H,15N]-sofast-HMQC spectrum of 15N-hCD44(21–178) in presence of 20 mM compound 3 matched the spectrum of the apo form, indicating the absence of binding in these experimental conditions (Supplementary Figure S2). In addition, we performed a displacement assay using ITC. The dissociation constant between HA8 and hCD44(21–178) was determined in absence and in presence of a 750-fold molar excess of compound 3. The Kd detected in presence of such large excess of compound 3 was 37.5 μM (Supplementary Figure S3), hence similar to the value obtained in absence of the fragment (Figure 1D), suggesting that the affinity of HA8 for hCD44(21–178) was not significantly affected by the presence of compound 3.
Fragment based ligand discovery (FBLD) by NMR
In the pursue of possible novel agents that bind to CD44 we performed a fragment screening campaign using 1D 1H-aliphatic and 2D [1H,15N]-sofast-HMQC of 15N-hCD44(21–178) as detection methods[29]. Hence we tested a library of 500 fragments from Maybridge (Fisher Scientific). To maximize the sensitivity of the binding assay, we used 15N-hCD44(21–178) at 20 μM while each fragment was tested at 400 μM. To reduce the number of samples, fragments were tested in pools of 10, and mixtures containing hits were further deconvoluted. [30] Hence, of the 50 mixtures tested only 2 mixtures presented significant chemical shift perturbation in both 1D 1H-aliphatic and 2D [1H,15N]-sofast-HMQC spectra. Further deconvolutions of these mixtures were accomplished by testing individual compounds at 400 μM against 20 μM 15N-hCD44(21–178). Of these resulting 20 samples, two structurally related hits produced significant chemical shift perturbations in both 1D and 2D NMR spectra of 15N-hCD44(21–178) (Supplementary Figure S4). Initial fragment evolution studies were performed using commercially available analogues of the two hits (Table S1). These studies resulted in a validated hit, compound 131B6 (Figure 3A). Interestingly, the perturbations induced by 131B6 in the 1D 1H-aliphatic NMR spectrum of hCD44(21–178) (Figure 3A) closely resemble the perturbations induced by HA8 as reported in Figure 1B. However, chemical shift perturbations in 2D [1H,15N]-sofast-HMQC spectrum (Figure 3B), indicated that only a distinct subset of resonances were perturbed by the ligand. Intriguingly, these changes map in a region of the structure of hCD44(21–178) that is opposite to the reported HA8 binding site (Figure 3C). The dissociation constant calculated using the chemical shift perturbation from the 2D [1H,15N]-sofast-HMQC spectra results in a Kd of 7.43 mM (Figure 3D), hence in the same affinity range of the fragments previously reported[16]. Further evaluations of the binding of 131D6 to CD44 where enabled by measurements of intermolecular NOEs (Supplementary Figure S5). Hence, 2D [1H, 1H] NOESY spectra were recorded with 100 μM of hCD44(21–178) in absence (blue) and in presence (red) of 500 μM of 131B6. In the spectrum of the complex, we could clearly observe in the aromatic region of the spectra, the presence of a cross-peak that connected the resonances of the proton of the thiazol (131B6) with the resonances of the δ-protons of the leucine 135 in hCD44(21–178), suggesting a spatial proximity of the compound to this aminoacid (< 5 Å). However, in a subsequent NMR displacement assay the presence of 131B6 did not prevent the binding of HA8, suggesting that under these conditions, these two binding events can exist simultaneously (Supplementary Figure S6A and B). Nonetheless, the screening revealed a possible binding pocket on the surface of CD44 that may be ‘druggable’ and that may have functional implications in cell.
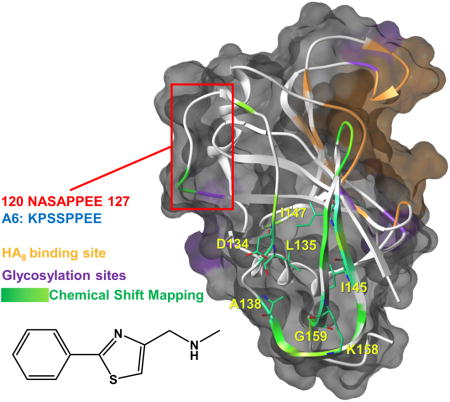
Figure 3. Binding validation of compound 2.

(A) NMR titration of 131B6 (structure on the upper panel) against 20 μM of 15N-hCD44(21–178) using 1D 1H-aliphatic and 2D 15N-sofastHMQC spectra (B). In both experiments, the spectra of the apo protein are depicted in blue, while the spectra in purple to light-blue were collected in presence of the fragment in increasing concentration starting from 1 mM to 15 mM respectively. The resonances corresponding to the most perturbed residues are labeled.
(C) Semitransparent molecular surface representation of the homology model of hCD44 built with SWISS-MODEL [22a] [22b] [22c] [22d] using the structure of mCD44 in complex with HA8 (PDB 2JCR) as template. The HA8 binding pocket is depicted in orange while in purple are highlighted the putative glycosylation sites. The Chemical Shift Perturbations induced by 131B6 are mapped on the secondary structure (ribbon): Δδ > 0.14 ppm in green; Δδ < 0.14 ppm in light green. The most perturbed residues are labeled and localized in a putative back pocket opposite from the HA8 binding pocket. The red square indicates the portion of hCD44 with sequence homology with A6 peptide.
(D) Determination of Kd of 131B6 using the chemical shift perturbation titrations from the 2D [1H,15N]-sofasHMQC experiments; Kd = 7.43 mM)
HTS by NMR
As additional attempt to derive novel CD44 binding agents, we also carried out a screening campaign using the HTS by NMR approach [29, 31]. Hence, we prepared and tested 76 mixtures of a tetra-peptide combinatorial library composed by 19 natural amino acids (the 20 natural amino acids except cysteine) as building blocks [31a]. In addition, we also prepared and tested a tri-peptide combinatorial library (see Methods).[31c] Each mixture was tested at 2 mM total concentration using 2D [1H,15N]-sofast-HMQC spectra with 15N-labaled hCD44(21–178). However, none of the mixtures tested produced a significant chemical shift perturbation in the 2D [1H,15N]-sofast-HMQC spectra of the target under these experimental conditions.
Discussion
CD44, via its interactions with its ligand, the hyaluronic acid, [2a, 4] (Figure 1A), plays an important role in tumor cell migration and tumor metastasis[3c] and as such it is expressed on the surface of several types of tumors [2a]. Despite its demonstrated relevance as viable drug target in oncology, further assessments on the biological role of CD44 in the onset and progression of cancer is hampered by the availability of suitable pharmacological tools. CD44 indeed is known to be not an easily ‘druggable’ target [16], due to the absence of a clear well-defined ligand binding pocket on its surface (Figure 1A). In addition, there are several post-translational modifications that occur in cell, mostly glycosylation, that are critical for the activity of CD44 [7]. However, the exact glycosylation state of the protein cannot be easily determined and reproduced in a test tube. Nonetheless, the recombinant form of the HA binding domain of CD44, retaining some of its binding affinity for HA, may provide a valid platform onto which to attempt to derive CD44 binding agents. Therefore we expressed the hCD44(21–178) as it was reported[4a, 18] by using that such construct which possesses a proper folding for binding HA. Accordingly, we demonstrated the construct’s ability to bind HA8 using both solution nuclear magnetic resonance spectrometry (NMR) (Figure 1B and C) and isothermal titration calorimetry (ITC) techniques (Figure 1D). However and surprisingly, when subsequently we tried to validate the binding of the other reported putative ligands of CD44 (Table 1), these resulted inactive under our experimental conditions, with the sole exception of the antibody DF1485 (Figure 2A). The results for these activities are summarized in Table 1. Several possible causes may be attributable to the lack of binding for these agents to CD44. For example, the peptide A5G27 has a fairly low solubility in our conditions it is also known to bind the glycosaminoglycans on CD44[26]. We could not test the antibody patented by Hoffman-La Roche and the University of Miami (Patent number US 20130224108 A1) because it is not commercially available, but based on the information disclosed in the patent, we could select and test six peptides of which three belongs at the heavy chain of the antibody, and the other three to the light chain. Unfortunately, also all these peptides tested in isolation didn’t seem to bind significantly the protein (see Methods and Materials). Another interesting peptide was reported in literature [28] based on the interactions between CD44 and the PEX domain of MMP-9 (Table 1). Nonetheless, as mentioned, also this peptide under our experimental conditions didn’t show significant binding to CD44. Next, we focused our studies on the binding properties of the A6 peptide given that it is in phase 2 clinical trials for CLL and SLL patients[9](ClinicalTrials.gov Identifier: NCT02046928). The detailed mechanism of action of A6 has not been defined, but studies on metastatic disease suggested that it functions through a CD44-mediated pathway[25]. While cellular studies indicated a possible direct interaction between CD44 and A6, we could not observe any significant binding of A6 to 15N-labeled sample of hCD44(21–178) in 2D [1H,15N]-sofast-HMQC (Figure 2B) and ITC (supplementary Figure S1) experiments. Similarly, we did not observe significant binding of A6 to the hCD44 in complex with HA8, using 15N-labeled hCD44(21–178) (supplementary Figure S7). However and intriguingly, the amino acid sequence of A6 (Table 1) exhibits marked homology with a linear sequence within CD44 (120-NASAPPEE-127) (Figure 3C)[25]. This has prompted us to experimentally verify for a possible recognition of the A6 peptide by the CD44 antibody DF1485, given that such antibody was used in cellular assays to study the mechanism of action of A6. To facilitate these binding studies, we synthesized a 13C-labeled version of A6 and collected 2D [13C,1H]-HSQC spectra of such agent in absence and in presence of the antibody DF1485. However, under these experimental conditions, no appreciable binding was detected (Figure 2C) between A6 and the antibody.
Subsequently, we also tested a recently discovered small organic molecule that was reported to bind the recombinant CD44 HA binding domain with millimoar affinity[16]. We obtained such compound 3 (Matrix Scientific, Columbia, SC) and tested it at 2 mM (Figure 2D) and 20 mM (Supplementary Figure S2) against 15N-hCD44(21–178) by 2D [1H,15N]-sofast-HMQC. Several significant chemical shift perturbations in the 2D [1H,15N]-sofast-HMQC spectrum of hCD44(21–178) were observed initially in presence of 20 mM of compound 3. However, the perturbations were suspiciously widespread for such small ligand. Upon further investigations we noticed that such high concentration of compound 3 led to a sizable increase of the pH, likely due to the compound itself (having a primary ammine) or most likely an impurity (Supplementary Figure S8). Indeed, recording again the spectrum after correcting the pH we observed that there was no significant evidence of binding (Supplementary Figure S2). Similar experiments were conducted also at pH =7.4 leading to the same conclusions (Supplementary Figure S9). The 2D [1H,15N]-sofast-HMQC of 15N-hCD44(21–178) at 20 μM measured in absence and presence of 2 mM compound 3 are reported in Figure 2D. Furthermore we monitored the ability of compound 3 to displace HA8 by ITC. In this experiment, ITC curves for the binding of HA8 to CD44 were collected in absence (Figure 1D) and in presence of up to 55 mM of compound 3. These curves are very similar to these reported previously for the same system. [17] Also by this displacement assay, the presence of compound 3, even at such high concentration, did not significantly displace the binding between CD44 and HA8 (supplementary Figure S3). Furthermore, we checked whether the binding of HA8 could facilitate the binding of compound 3 by NMR or if compound 3 could displace HA8 binding to CD44. Also in this experiment, we could not observe any significant perturbations induced by compound 3 to the hCD44 spectrum in complex with HA8 (supplementary Figure S10). One could conclude, unfortunately that the putative binding and HA8 displacement reported by Liu et al.[16] using a less sensitive SPR assay, may have been a result of pH changes induced by the compound itself or on impurity. However, and intriguingly, Liu et al. also reported the crystal structure of compound 3 bound to mouse CD44 obtained by soaking the compound (PDB entry 4MRG). Of note that authors reported that compound 3 had similar affinity for mouse CD44 versus human CD44. While a definitive explanation for these discrepancies remains unclear, these data may suggest that the use of high concentration screening by SPR or biochemical assays followed by X-ray crystallography, as a way to validate weakly interacting fragments, may still require further validation by NMR or ITC, for example.
In a follow-up study Liu et al.[24] reported that in resolving the crystal structure of hCD44(20–178), electron density of an unidentified peptide docked into a novel hydrophobic binding pocket situated on the opposite side of the HA binding site. Intriguingly, the expression of this construct requires a denaturation/refolding procedure, hence the unidentified peptide could not have remained tightly bound during sample preparation.[24] From the electron density it was possible only to identify a valine residue as the first amino acid of this unknown peptide located into the small binding pocket. Based on these observations we decided to perform a further screening campaign using the HTS by NMR approach[31a]. Aimed at identifying a possible CD44 binding peptide, we decided to screen a four-position combinatorial library composed by 19 natural amino acids (the 20 natural amino acids except cysteine) as building blocks. In this way we tested all the possible combinations of tetra-peptides, including peptides with the valine at the N- and C- terminal. However, no viable hits were detected using 2D [1H,15N]-sofast-HMQC among the tested 76 mixtures (see Methods). At this point we tested with the same approach a tri-peptide combinatorial library assembled using 46, natural and non-natural, amino acids as building blocks. Based on the evidence that only a first amino acid is visible inside the new binding pocket, we decided to screen only the peptides with the first position fixed. Again we didn’t find any peptide interacting with the target protein using 2D [1H,15N]-sofast-HMQC.
Finally we decided to conduct a fragment screening using NMR as detection technique aimed at finding possibly new hits able to bind hCD44. By screening of a 500 small molecule fragment library (Maybridge) we obtained two structurally related initial hits (Supplementary Figure S4). Starting from these initial hits, we tested other analogs (Supplementary Table S1) that resulted in compound 131B6 (Figure 3A). Analyses of data resulting from both the 1D 1H aliphatic (Figure 3A) and 2D [1H,15N]-sofast-HMQC NMR experiments (Figure 3B), concluded that the residues that were most perturbed were situated in a region of hCD44(21–178) forming a possible back pocket in the opposite site from where HA8 binds and close but different from the pocket observed by Liu et al.[24]. From the chemical shift perturbations we could obtain an average dissociation constant of Kd of 7.43 mM (Figure 3D). For both, 131B6 and compound 3[16], we also collected Saturation Transfer Difference (STD) spectra. First we tested as positive control HA8 that as expected gave a significant STD effect (Supplementary Figure S11A). However, when we ran the same experiment with 131B6 (Supplementary Figure S11B), or compound 3 (Supplementary Figure S11C), there was no significant STD effect in either compound. These data further confirm that for fragments with fairly low affinity ranging from 1 to 10 mM a protein based experiment (as the 2D [1H,15N]-sofast-HMQC or the 1D 1H-aliphatic NMR experiments) is more sensitive and robust than a ligand based approach (as the STD)[29]. To further confirm the binding mode of this fragment hit and to obtain further structural information about its possible binding pose we recorded 2D [1H,1H]-NOESY NMR spectra (Supplementary figure S5) for hCD44 (21–178) in absence and in presence of compound 131B6. In the presence of this compound, we observed a cross-peak between the proton of the thiazole of 131B6 and the β-protons of leucine 135 that, from both 1D and 2D NMR experiments, was the most perturbed residue by the compound. These data further confirmed that 131B6 is close in space (< 5 Å) to the leucine 135. Binding of 131B6 to CD44 did not affect the binding of HA8 or A6 as determined by NMR titration with 2D [1H,15N]-sofast-HMQC (Supplementary Figure S6A, S6B and S12). A comparative analysis of binding of compound 3, 131B6, A6 and HA8 to hCD44(21–178) is also reported as supplementary Figure S13 using 1D 1H aliph. NMR spectra.
An important and interesting observation is that the residues most perturbed upon the binding of 131B6 to hCD44(21–178) spatially overlap to a region that encompasses the sequence of CD44 with high homology to the A6 peptide sequence (Figure 3C). In particular, the resonances of several residues within this sequence were perturbed from the 2D [1H,15N]-sofast-HMQC spectrum collected in presence of 131B6. As previously hypothesized, this homology between the A6 peptide and a linear sequence within CD44 may provide an alternative explanation for the cellular and in vivo activity of A6 whereby A6 may function as a decoy for a yet unknown binding partner of CD44[25]. If this hypothesis is correct, ligands binding in proximity of this loop of CD44 may exert a biological activity, rendering 131B6 a potentially interesting fragment for further hit maturations[32] and subsequent cellular assays. Hence our studies have potentially significant implications on the possible mechanism of A6, and may provide a new rationale for targeting a possible binding pocket on CD44.
Conclusions
CD44 has revealed to be a potentially very interesting receptor for pharmacological intervention targeting tumor metastasis. However, further research in the validation of CD44 as possible drug target is hampered by the lack of suitable pharmacological agents. In this article we characterized a variety of putative CD44 binding agents. With the exception of an antibody and HA8, none of these agents, including the clinical peptide A6, showed appreciable binding to a recombinant HA binding domain construct of CD44. However, an NMR guided fragment screening revealed potentially interesting fragment hit 131B6. The compound bound to CD44 in an area that is opposite to the HA binding site and is composed by a loop region of CD44 (loop β6-β7) that presents a high homology with the anti-CD44 peptide A6. Based on these data, it is tempting to speculate that A6 may function as a decoy for an unidentified CD44 binding protein that uses loop β6-β7 for such interactions. Therefore, further optimizations of 131B6 may result in small molecule agents that similar to A6 may impair the function of CD44.
Methods and Materials
Protein expression and purification
The human CD44 HA Binding Domain (HABD) residues 21–178 was expressed in Escherichia Coli and purified from insoluble inclusion body as described previously [18]. The cDNA of hCD44(21–178) was cloned into the vector pET19b and transformed into E. coli strain BL21(DE3) gold pLysS (Novagen). The overexpression of the protein was obtained growing the transformed cells in LB medium at 37°C with 100 μg/L of ampicillin until reaching an OD600 of 0.6 followed by induction with 0.4 mM IPTG. After OVT incubation at 20°C, cells were harvested by centrifugation. The inclusion body was isolated from the pellet and solubilized in 8 M urea in presence of 1mM dithiothreitol (DTT) at 4°C OVT. The denatured recombinant hCD44(21–178) was subsequently refolded dropping the supernatant solution at 600 μL/h using a syringe pump by 200-fold dilution into buffer containing 250 mM L-Arginine, 100 mM tris-HCl pH = 8, 2 mM reduced glutathione and 1 mM oxidized glutathione. After 48h at 4°C stirring, the solution was ultrafiltrated through Amicon YM10 membranes. The monomeric protein was purified through size exclusion chromatography using a HiLoad 26/60 Superdex 75 prep grade column. For the expression of the 15N-labeled protein used for all heteronuclear NMR experiments, the procedure was the same, but the bacteria were grown in M9 minimal medium supplemented with trace elements, 0.09%v/v glycerol and 0.5 g/ L 15NH4Cl for the labeling.
Reagents
HA8 was purchased from Iduron (UK). The antibody DF1485 was from Santa Cruz Biotechnology (Dallas, TX). Compound 3 [16] was obtained from Matrix Scientific (Columbia, SC, USA). The peptides A6 (Ac-KPSSPPEE-NH2), A5G27 (H-RLVSYNGIIFFLK-NH2), the peptide from the blade I of the PEX domain of MMP-9 (Ac-SRPQGPFL-NH2), the three peptides from the light chain of the Hoffmann-La Roche antibody (Ac-SRYWMS-NH2, Ac-EVNPDSTSINYTPSLKD-NH2, and Ac-PNYYGSRYHYYAMDY-NH2), and the three from the heavy chain (Ac-RASQDINNYL-NH2, Ac-YTSRLHS-NH2, and Ac-QQGSTLPFT-NH2) were purchased from Innopep (San Diego, CA). 13C-A6 peptide was synthesized in our laboratory. 131B6 and all the compounds in Supplementary Table S1 were purchased from Hit2Lead ChemBridge Corporation (San Diego, CA) except for compound 131C1 from Maybridge (Fisher Scientific) and compound 131B12 from Acros Organics.
13C-A6 peptide synthesis
13C-A6 peptide was synthesized using a fmoc solid-phase synthesis using the Rink amide resin. For the coupling reaction each amino acid was dissolved into 5 mL of dry DMF containing 6 equivalents of Oxima pure, DIC and DIAE. Each coupling reaction was carried shaking the resin for 3 hours. After the reaction the resin has been washed 3 times with 8 mL DMF followed by 3 washings with DCM and 3 washings with DMF. The deprotection of the terminal amino acid was achieved by adding to the resin-bound peptide a 20% piperidine solution in DMF (1 mL and 4 mL respectively) for 40 minutes twice. After the last coupling step, the N-terminus was acetylated with 5 mL of dry DMF solution containing 0.05 equivalents of DMAP, 3 equivalents of DIPEA and 2 equivalents of 13CH3COCl (Cambridge Isotope Laboratories, Inc.). The cleavage from the resin and the removal of all the side chains protecting groups was performed by shaking at RT with a cleavage cocktail composed by 94% TFA, 2% phenol, 2% TIPS, and 2% of water for 5.5 hours. TFA was subsequently removed under reduced pressure using a rota-vapor and the peptide was subjected to few cycles of precipitation in diethyl ether and centrifugation before OVT drying in high vacuum. The peptide was purified by a HPLC Breeze system from Waters Co. using preparative reverse phase column to get a purity level > 95%. The final peptide was characterized by NMR and MALDI-TOF spectrometry.
Isothermal Titration Calorimetry (ITC)
The ITC measurements were performed with a ITC200 calorimeter from Microcal (Northampton, MA, USA) at 23°C. For the direct experiments, hCD44 (73 μM) was titrated with a solution containing 1.6 mM HA8 (Figure 1D) or 1 mM A6 peptide (Supplementary Figure S1). For the competition assay (Supplementary Figure S3) the protein was incubated with a 55 mM solution of compound 3 and then titrated with a solution of 1.6 mM HA8. All experiments were conducted in buffer containing 50 mM phosphate, 150 mM NaCl at pH = 6.7 in presence of 5% DMSO. The data were analyzed using Origin software provided by Microcal.
NMR Spectroscopy
All the NMR spectra were acquired on a 600 or a 700 MHz Bruker Avance spectrometers equipped each with a TCI cryoprobe and z-shielded gradient coil. The data were processed and analyzed using TOPSPIN 2.1 (Bruker Biospin, MA) and Sparky 3.1 (University of California, San Francisco, CA). In general, 1D 1H experiments were acquired using 128 scans with 2048 complex data points, 2D [1H,15N]-sofast-HMQC experiments using 48 scans with 2048 and 128 complex data points in the 1H and 15N dimensions, respectively, and the 2D [1H,13C]-HSQC using 64 scans with 4096 and 128 complex points in the 1H and 13C dimensions, respectively. All samples were acquired at 300 K in buffer containing 50 mM phosphate, 150 mM NaCl, and 0.02% NaN3, pH = 6.7.
The 2D [1H,1H]-NOESY NMR spectra were recorded using 72 scans with 2048 and 256 complex data points in the direct and indirect dimensions, respectively, with a NOE mixing time of 0.2 seconds. The Saturation Transfer Difference (STD) NMR spectra were recorded using 128 scans and saturating for 2 seconds at −0.28 ppm or at −10 ppm for the reference spectra.
The dissociation constant (Kd) was calculated monitoring the chemical shift perturbations in the 2D [1H,15N]-sofast-HMQC spectra of hCD44 (20 μM) due to the presence of increasing concentrations of compound 131B6 (1, 2, 3, 5, 7, 9, 11, 13, and 15mM). The weight average perturbations induced on backbone amide resonances corresponding to residues D134, I145, I147 and G159 were determined using the following equation: [33]
Subsequently, Kd values were obtained by fitting these averaged chemical shift perturbation data into the following equation: [34] [35]
where Δδobs is the chemical shift perturbation value observed at each point of the titration, Δδmax is the maximum chemical shift change of the fully complexed protein, and [L]0 and [P]0 are the total concentrations of compound and protein.
Screening by NMR
The Maybridge library of 500 fragments was assembled in 50 mixtures containing 10 fragments each in d6-DMSO. 1D 1H and 2D [1H,15N]-sofast-HMQC experiments were recorded for each mixture at the final concentration of 4 mM (400 μM individual fragment concentration) against 20 μM 15N-hCD44(21–178). The spectra were recorded in 50 mM phosphate buffer pH 6.7, 150 mM NaCl and 0.02% NaN3 with a 2% of d6-DMSO. The hit mixtures were subsequently deconvoluted testing each fragment individually in the same experimental conditions. For the HTS by NMR of combinatorial libraries approach [31a] was tested a four-position combinatorial library (Ac-XXXX-NH2) using 19 amino acids (all the natural amino acid except for cysteine) as building blocks. In this way, 76 (19×4) mixtures each containing 6,859 (19×19×19) individual peptides with one amino acid fixed at one position were synthesized and tested. Hence, we were able to test 130,321 combinations of natural amino acid (19×19×19×19) just by synthesizing and testing 76 mixtures. With the same approach, we tested a tri-peptide library assembled using 46 natural and non-natural amino acid as building blocks (Pepscan Presto BV, Lelystad, The Netherlands) maintaining one amino acid fixed at the first position. In this way we tested 2,116 peptides (1×46×46) just testing 1 mixture.
Supplementary Material
Acknowledgments
Financial support was obtained in part by the NIH, with NCI grants CA081534 (to MP and TJK), grant CA168517 (to MP). CB thanks the University of Padua, Department of Chemical Sciences, for a fellowship. We also thank the Protein Analysis Facility at the Sanford-Burnham-Prebys Medical Discovery Institute supported by NIH Cancer Center grant CA030199. MP holds the Daniel Hays Chair in Cancer Research at the School of Medicine at UCR.
References
- 1.Gallatin WM, Weissman IL, Butcher EC. Nature. 1983;304(5921):30–34. doi: 10.1038/304030a0. [DOI] [PubMed] [Google Scholar]
- 2.a Lesley J, Hyman R, Kincade PW. Adv Immunol. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]; b Naor D, Sionov RV, Ish-Shalom D. Adv Cancer Res. 1997;71:241–319. doi: 10.1016/s0065-230x(08)60101-3. [DOI] [PubMed] [Google Scholar]
- 3.a DeGrendele HC, Estess P, Picker LJ, Siegelman MH. J Exp Med. 1996;183(3):1119–1130. doi: 10.1084/jem.183.3.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shimizu Y, Van Seventer GA, Siraganian R, Wahl L, Shaw S. J Immunol. 1989;143(8):2457–2463. [PubMed] [Google Scholar]; c Gunthert U, Hofmann M, Rudy W, Reber S, Zoller M, Haussmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. Cell. 1991;65(1):13–24. doi: 10.1016/0092-8674(91)90403-l. [DOI] [PubMed] [Google Scholar]
- 4.a Peach RJ, Hollenbaugh D, Stamenkovic I, Aruffo A. J Cell Biol. 1993;122(1):257–264. doi: 10.1083/jcb.122.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liao HX, Lee DM, Levesque MC, Haynes BF. J Immunol. 1995;155(8):3938–3945. [PubMed] [Google Scholar]
- 5.Almond A. Cell Mol Life Sci. 2007;64(13):1591–1596. doi: 10.1007/s00018-007-7032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoller M. Nat Rev Cancer. 2011;11(4):254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 7.Brown TA, Bouchard T, St John T, Wayner E, Carter WG. J Cell Biol. 1991;113(1):207–221. doi: 10.1083/jcb.113.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponta H, Sherman L, Herrlich PA. Nat Rev Mol Cell Biol. 2003;4(1):33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 9.Finlayson M. Front Immunol. 2015;6:135. doi: 10.3389/fimmu.2015.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borland G, Ross JA, Guy K. Immunology. 1998;93(2):139–148. doi: 10.1046/j.1365-2567.1998.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Wu CC, Fecteau JF, Cui B, Chen L, Zhang L, Wu R, Rassenti L, Lao F, Weigand S, Kipps TJ. Proc Natl Acad Sci U S A. 2013;110(15):6127–6132. doi: 10.1073/pnas.1221841110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Herishanu Y, Gibellini F, Njuguna N, Hazan-Halevy I, Farooqui M, Bern S, Keyvanfar K, Lee E, Wilson W, Wiestner A. Leuk Lymphoma. 2011;52(9):1758–1769. doi: 10.3109/10428194.2011.569962. [DOI] [PMC free article] [PubMed] [Google Scholar]; aub Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, Kipps TJ, Choi YS, Bennett F, Reed JC. Blood. 2002;100(5):1795–1801. [PubMed] [Google Scholar]
- 13.Ghosh SC, Neslihan Alpay S, Klostergaard J. Expert Opin Ther Targets. 2012;16(7):635–650. doi: 10.1517/14728222.2012.687374. [DOI] [PubMed] [Google Scholar]
- 14.a Guo Y, Mazar AP, Lebrun JJ, Rabbani SA. Cancer Res. 2002;62(16):4678–4684. [PubMed] [Google Scholar]; b Boyd DD, Kim SJ, Wang H, Jones TR, Gallick GE. Am J Pathol. 2003;162(2):619–626. doi: 10.1016/S0002-9440(10)63855-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mishima K, Mazar AP, Gown A, Skelly M, Ji XD, Wang XD, Jones TR, Cavenee WK, Huang HJ. Proc Natl Acad Sci U S A. 2000;97(15):8484–8489. doi: 10.1073/pnas.150239497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai H, Liu G, Rassenti L, Choi MY, Howell SB, Finlayson M, Kipps TJ. Blood. 2013;122(21):5303–5303. [Google Scholar]
- 16.Liu LK, Finzel BC. J Med Chem. 2014;57(6):2714–2725. doi: 10.1021/jm5000276. [DOI] [PubMed] [Google Scholar]
- 17.Banerji S, Wright AJ, Noble M, Mahoney DJ, Campbell ID, Day AJ, Jackson DG. Nat Struct Mol Biol. 2007;14(3):234–239. doi: 10.1038/nsmb1201. [DOI] [PubMed] [Google Scholar]
- 18.Banerji S, Day AJ, Kahmann JD, Jackson DG. Protein Expr Purif. 1998;14(3):371–381. doi: 10.1006/prep.1998.0971. [DOI] [PubMed] [Google Scholar]
- 19.Schanda P, Kupce E, Brutscher B. J Biomol NMR. 2005;33(4):199–211. doi: 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- 20.Teriete P, Banerji S, Noble M, Blundell CD, Wright AJ, Pickford AR, Lowe E, Mahoney DJ, Tammi MI, Kahmann JD, Campbell ID, Day AJ, Jackson DG. Mol Cell. 2004;13(4):483–496. doi: 10.1016/s1097-2765(04)00080-2. [DOI] [PubMed] [Google Scholar]
- 21.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. Cell. 1990;61(7):1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 22.a Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]; b Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. Nucleic Acids Res. 2009;37(Database issue):D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Guex N, Peitsch MC, Schwede T. Electrophoresis. 2009;30(Suppl 1):S162–173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]; d Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. Nucleic Acids Res. 2014;42(Web Server issue):W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda M, Ogino S, Umemoto R, Sakakura M, Kajiwara M, Sugahara KN, Hayasaka H, Miyasaka M, Terasawa H, Shimada I. J Biol Chem. 2006;281(52):40089–40095. doi: 10.1074/jbc.M608425200. [DOI] [PubMed] [Google Scholar]
- 24.Liu LK, Finzel B. Acta Crystallogr F Struct Biol Commun. 2014;70(Pt 9):1155–1161. doi: 10.1107/S2053230X14015532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piotrowicz RS, Damaj BB, Hachicha M, Incardona F, Howell SB, Finlayson M. Mol Cancer Ther. 2011;10(11):2072–2082. doi: 10.1158/1535-7163.MCT-11-0351. [DOI] [PubMed] [Google Scholar]
- 26.Hibino S, Shibuya M, Hoffman MP, Engbring JA, Hossain R, Mochizuki M, Kudoh S, Nomizu M, Kleinman HK. Cancer Res. 2005;65(22):10494–10501. doi: 10.1158/0008-5472.CAN-05-0314. [DOI] [PubMed] [Google Scholar]
- 27.a Yu Q, Stamenkovic I. Genes Dev. 1999;13(1):35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yu Q, Stamenkovic I. Genes Dev. 2000;14(2):163–176. [PMC free article] [PubMed] [Google Scholar]; c Redondo-Munoz J, Ugarte-Berzal E, Garcia-Marco JA, del Cerro MH, Van den Steen PE, Opdenakker G, Terol MJ, Garcia-Pardo A. Blood. 2008;112(1):169–178. doi: 10.1182/blood-2007-08-109249. [DOI] [PubMed] [Google Scholar]
- 28.Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J. J Biol Chem. 2010;285(46):35944–35956. doi: 10.1074/jbc.M109.091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barile E, Pellecchia M. Chem Rev. 2014;114(9):4749–4763. doi: 10.1021/cr500043b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a Chen J, Zhang Z, Stebbins JL, Zhang X, Hoffman R, Moore A, Pellecchia M. ACS Chem Biol. 2007;2(5):329–336. doi: 10.1021/cb700025j. [DOI] [PubMed] [Google Scholar]; b Huang JW, Zhang Z, Wu B, Cellitti JF, Zhang X, Dahl R, Shiau CW, Welsh K, Emdadi A, Stebbins JL, Reed JC, Pellecchia M. J Med Chem. 2008;51(22):7111–7118. doi: 10.1021/jm8006992. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Leone M, Crowell KJ, Chen J, Jung D, Chiang GG, Sareth S, Abraham RT, Pellecchia M. Biochemistry. 2006;45(34):10294–10302. doi: 10.1021/bi060976+. [DOI] [PubMed] [Google Scholar]
- 31.a Wu B, Zhang Z, Noberini R, Barile E, Giulianotti M, Pinilla C, Houghten RA, Pasquale EB, Pellecchia M. Chem Biol. 2013;20(1):19–33. doi: 10.1016/j.chembiol.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu B, Barile E, De SK, Wei J, Purves A, Pellecchia M. Curr Top Med Chem. 2015;15(20):2032–2042. doi: 10.2174/1568026615666150519102459. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bottini A, Wu B, Barile E, De SK, Leone M, Pellecchia M. ChemMedChem. 2015 doi: 10.1002/cmdc.201500441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fattorusso R, Jung D, Crowell KJ, Forino M, Pellecchia M. J Med Chem. 2005;48(5):1649–1656. doi: 10.1021/jm0493212. [DOI] [PubMed] [Google Scholar]
- 33.Farmer BT, 2nd, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr, Robertson JG, Mueller L. Nat Struct Biol. 1996;3(12):995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 34.Pellecchia M. Chem Biol. 2005;12(9):961–971. doi: 10.1016/j.chembiol.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Smet C, Duckert JF, Wieruszeski JM, Landrieu I, Buee L, Lippens G, Deprez B. J Med Chem. 2005;48(15):4815–4823. doi: 10.1021/jm0500119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.