

Abstract
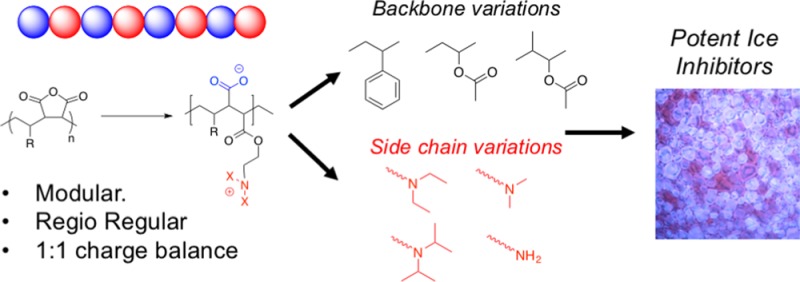
Antifreeze proteins from polar fish species are potent ice recrystallization inhibitors (IRIs) effectively stopping all ice growth. Additives that have IRI activity have been shown to enhance cellular cryopreservation with potential to improve the distribution of donor cells and tissue. Polyampholytes, polymers with both anionic and cationic side chains, are a rapidly emerging class of polymer cryoprotectants, but their mode of action and the structural features essential for activity are not clear. Here regioregular polyampholytes are synthesized from maleic anhydride copolymers to enable stoichiometric installation of the charged groups, ensuring regioregularity, which is not possible using conventional random copolymerization. A modular synthetic strategy is employed to enable the backbone and side chain hydrophobicity to be varied, with side chain hydrophobicity found to have a profound effect on the IRI activity. The activity of the regioregular polymers was found to be superior to those derived from a standard random copolymerization with statistical incorporation of monomers, demonstrating that sequence composition is crucial to the activity of IRI active polyampholytes.
Introduction
Antifreeze (glyco) proteins (AF(G)Ps) have evolved in polar fish species to enable them to survive in subzero environments by specifically interacting with ice crystals.1 AF(G)Ps have three main properties of thermal hysteresis (lowering freezing point, but not melting point), dynamic ice shaping (changing morphology of ice crystals), and also ice recrystallization inhibition (IRI), the inhibition of the growth of already formed ice crystals.1−3 The property of IRI is of particular interest as the growth of ice crystals has been identified as a crucial mechanism of cell death during the thawing of cryopreserved cells and tissues.4 The addition of AF(G)Ps to cryopreservation solutions was found to give some increase in cell recovery but the effect was limited by the onset of dynamic ice shaping, which leads to ice crystals piercing cell membranes.5 Consequently, there has been much interest in developing synthetic mimics which have IRI activity to enable the cryostorage of donor tissues and cells.6,2,7,8 There is an urgent need for new storage mechanisms, with a global shortage of cells such as blood,9−11 which is exacerbated by cells’ limited shelf life. Effective cryoprotectants are also important for applications in frozen food.12−14 Current cryopreservation strategies often involve addition of large quantities of DMSO which can have negative (toxic) effects on both the cells and the recipient.15,16
The most potent polymer-IRI identified to date is poly(vinyl alcohol), which can inhibit all ice growth at sub mg·mL–1 concentrations, although its exact mechanism is unclear,17,18 unlike AFPs, which engage specifically with ice crystal faces.19 Many other polyols have been tested and few have any appreciable activity, suggesting the underlying mechanisms are complex and not just a property of regularly spaced hydroxyl groups.20,21 Addition of PVA to nonvitrifying cryopreservative solutions has been found to enhance the storage of several cell types by reducing thawing induced damage.22−24 It should be noted that this is distinct to PVA’s role in nucleation inhibition.25,26 The role of architecture on PVA’s activity has been studied by Gibson and co-workers with the IRI activity increasing with chain length, with a minimum of 10–20 units being essential.27,28 Voets et al. found that comb-like PVAs had lower activity compared to linear.29
There is increasing evidence that nonhydroxylated polymers (or small molecules) can also display IRI activity, which may give rise to new cryoprotectants. Ben et al. have demonstrated that some surfactants are potent IRIs (but not all) and recently a supramolecular IRI was reported.8 A common feature of these is that hydrophobic domains appear to be crucial to activity, but presented in a manner which does not lead to aggregation. The antimicrobial peptide Nisin A shows potent IRI but only at pH where its histidine residues are protonated, leading to folding into an amphiphilic shape.30 Matsumura et al. have developed the use of polyampholytes (polymers with both cationic and anionic side chains) as new cryoprotectants enabling the DMSO-free storage of stem cells, a key challenge in regenerative medicine.31 Gibson et al. studied a range of polyampholytes and found that they also display significant IRI activity making them a unique class of polymeric AF(G)P mimics.32,33 The rationale for polyampholyte IRI activity is currently unclear as they have no obvious ice binding domains, although they do interact with cell membranes, which may enhance their cryoprotective effect.34 Polyampholytes are an appealing class of polymer to study though, due to their generic nature (e.g any polymer with mixed charges seems to have some activity) and easier synthesis compared to PVA.35
All currently described polyampholytes, however, are comprised of either polydisperse polymers (e.g., carboxylated poly(lysine)31) or random copolymers with nonstoichiometric ratios of the anionic and cationic groups.33,34 We have previously shown that both the molecular weight and the ratio of the two charged components (must be 1:1) are crucial for activity of this emerging class of biomaterials.33 A further issue is that the sequence distribution of these essential units has not been thoroughly studied (other than both are required). Recent advances in controlled radical polymerization by Lutz and others have enabled techniques for control over the site of installation of specific functionality into synthetic polymers by kinetic control36,37 or the synthesis of multiblock copolymers,38 with improved control over sequence; although these methods are still not capable of producing fully sequence controlled materials as found in Nature. The most well-known sequence-defined polymers are those based on maleic anhydride copolymers.39 Due to maleic anhydride’s low propensity to self-propagate, addition of a second monomer enables the formation of perfectly alternating polymers, giving regioregularity. Furthermore, the anhydride ring is an ideal platform for postpolymerization modification40 to insert adjacent carboxy/amine functionalities to give ampholytes.32
Considering the above, the aim of this work was to undertake the first systematic investigation into the role of monomer sequence and location of hydrophobicity on the IRI activity of polyampholytes. Using RAFT polymerization it was possible to obtain well-defined maleic anhydride containing precursors with a range of comonomers, and through ring-opening, varying the side chains, while ensuring a 1:1 balance of charged units. Quantitative IRI activity reveals that side chain hydrophobicity is a powerful tool to enhance activity and that the sequence regulated polymers are more active than random copolymers derived from acrylates, opening a new avenue to potent IRI active materials.
Experimental Section
General
Phosphate-buffered saline (PBS) solutions were prepared using preformulated tablets (Sigma-Aldrich) in 200 mL of Milli-Q water (>18.2 Ω mean resistivity) to give [NaCl] = 0.138 M, [KCl] = 0.0027 M, and pH 7.4. Vinyl acetate (>99%), styrene (>99%), and isopropenyl acetate (99%) were purchased from Sigma-Aldrich and were filtered through a plug of basic alumina to remove inhibitors prior to use. 4,4′-Azobis(4-cynaovaleric acid) (>98%) was recrystallized from methanol and stored at −18 °C in the dark. Maleic anhydride (99%), benzyl bromide (98%), 2-cyano-2-propyl benzodithioate (>97%), N-Boc-ethanolamine (98%), 2-dimethylaminoethanol (99.5%), 2-(diethylamino)ethanol (>99.5%), 2-(diisopropylamino)ethanol (>99%), and trifluoroacetic acid (99%) were purchased from Sigma-Aldrich. Potassium ethyl xanthate (98%) was purchased from Alfa Aesar. All solvents were purchased from VWR or Sigma-Aldrich and used without further purification. S-benzyl O-ethyl carbondithioate was synthesized by Thomas Congdon.41
Physical and Analytical Methods
1H and 13C NMR spectra were recorded on Bruker Avance III HD 300 MHz, HD 400 MHz, or HD 500 MHz spectrometers using deuterated solvents obtained from Sigma-Aldrich. Chemical shifts are reported relative to residual nondeuterated solvent. All size exclusion chromatography (SEC) data were recorded in DMF or THF on Agilent 390-LC MDS instruments equipped with differential refractive index (DRI) detectors. Both systems were equipped with 2xPLgel Mixed D columns (300 × 7.5 mm) and a PLgel 5 μm guard column. The eluents are DMF with 5 mmol NH4BF4 additive or THF with 2% TEA (triethylamine) and 0.01% BHT (butylated hydroxytoluene) additives (depending on the system used). All samples were run at 1 mL.min–1 at 50 °C. Poly(methyl methacrylate) standards (Agilent EasyVials) were used for calibration. Analyte samples were filtered through a nylon membrane with 0.22 μm pore size before injection.
Respectively, experimental molar mass (Mn,SEC) and dispersity (Đ) values of synthesized polymers were determined by conventional calibration using Agilent GPC/SEC software. Ice wafers were annealed on a Linkam Biological Cryostage BCS196 with T95-Linkpad system controller equipped with a LNP95-Liquid nitrogen cooling pump, using liquid nitrogen as the coolant (Linkam Scientific Instruments UK, Surrey, U.K.). An Olympus CX41 microscope equipped with a UIS-2 20x/0.45/∞/0–2/FN22 lens (Olympus Ltd., Southend on sea, U.K.) and a Canon EOS 500D SLR digital camera was used to obtain all images. Image processing was conducted using ImageJ, which is freely available from http://imagej.nih.gov/ij/.
Ice Recrystallization Inhibition Assay
A 10 μL droplet of polymer in PBS solution is dropped from 1.4 m onto a glass microscope coverslip, which is on top of an aluminum plate cooled to −78 °C using dry ice. The droplet freezes instantly upon impact with the plate, spreading out and forming a thin wafer of ice. This wafer is then placed on a liquid nitrogen cooled cryostage held at −8 °C. The wafer is then left to anneal for 30 min at −8 °C. Three photographs are then taken of the wafer in different locations at 20× zoom under cross polarizers. The number of crystals in the image is counted, again using ImageJ, and the area of the field of view divided by this number of crystals to give the average crystal size per wafer, and reported as a % of area compared to PBS control.
Synthetic Section
Synthesis of Poly(maleic anhydride-alt-styrene)
As a representative example, 2-cyano-2-propyl benzodithioate (0.092 g, 416 μmol), maleic anhydride (1.01 g, 10.3 mmol), styrene (0.280 g, 2.69 mmol), ACVA (4,4′-azobis(4-cyanovaleric acid); 0.020 g, 72.3 μmol), and 1,4-dioxane (3 mL) were added to a sealed vial. The solution was degassed by bubbling N2 through the solution for 30 min, and the reaction was then allowed to polymerize at 80 °C for 5 h. The polymerization reaction was stopped by plunging the resulting solution into liquid nitrogen. Poly(maleic anhydride-alt-styrene) was recovered as a pink solid after precipitation into diethyl ether. The diethyl ether was decanted and the solid dried under vacuum overnight forming a pale pink solid.
1H NMR (DMSO): δ 1.75 (CH2, br, 2H), 2.01 (CH, br, 1H), 3.43 (CHC(O)OC(O)CH, br, 2H), 7.25 (aromatic C-H, br, 5H). 13C NMR (DMSO): δ 31 (CH2), 51 (CHC(O)OC(O)CH), 127 (aromatic C–H), 138 (CH–C6H6), 171 (CHC(O)OC(O)CH). IR anhydride C=O 1775, 1850 cm–1. MnSEC(DMF) = 9200 Da. Mw/Mn = 1.08
Synthesis of Poly(maleic anhydride-alt-vinyl acetate)
As a representative example, S-benzyl O-ethyl carbonodithioate (0.020 g, 0.0948 mmol), maleic anhydride (0.98 g, 10 mmol), vinyl acetate (0.25 g, 2.90 mmol), 4,4′-azobis-4-cyanovaleric acid (0.0013 g, 4.63 μmol), and 1,4-dioxane (1 mL) were added to a sealed vial. The solution was degassed by bubbling N2 through the solution for 30 min, and the reaction was then allowed to polymerize at 80 °C for typically 16 h. The polymerization reaction was stopped by plunging the resulting solution into liquid nitrogen. Poly(maleic anhydride-alt-vinyl acetate) was recovered as a beige solid after precipitation into diethyl ether. The diethyl ether was then decanted and the solid dried under vacuum overnight forming a pale beige solid.
1H NMR (DMSO): δ 1.88 (CH2, br, 2H), 1.94 (CH3COO, br, 3H), 3.38 (CHC(O)OC(O)CH, br, 2H), 5.10 (CHOC(O), br, 1H). 13C NMR (DMSO): δ 29 (CH2), 67 (CH–OC(O)), 131 (CHC(O)OC(O)CH), 139 (CH3COO), 163 (CHC(O)OC(O)CH), 171 (CH3COO). IR anhydride C=O 1704, 1788 cm–1. MnSEC(DMF) = 5600 Da. Mw/Mn = 1.46.
Synthesis of Poly(maleic anhydride-alt-isopropenyl acetate)
As a representative example, 2-cyano-2-propyl benzodithioate (5.6 mg, 25.3 μmol), maleic anhydride (0.99 g,10.1 mmol), isopropenyl acetate (0.27 g, 2.70 mmol), ACVA (4,4′-azobis(4-cyanovaleric acid); 1 mg, 3.47 μmol), and 1,4-dioxane (1 mL) were added to a sealed vial. The solution was degassed by bubbling N2 through the solution for 30 min, and the reaction was then allowed to polymerize at 80 °C for typically 24 h with the addition of further ACVA (1 mg, 3.47 μmol) after 12 h. The polymerization reaction was stopped by plunging the resulting solution into liquid nitrogen. Poly(maleic anhydride-alt-isopropenyl acetate) was recovered as a brown solid after precipitation into diethyl ether. The diethyl ether was then decanted and the solid dried under vacuum overnight forming a brown/black solid. 1H NMR (DMSO): δ 1.09 (CH3, br, 3H), 1.83 (CH2, br, 2H), 2.03 (CH3COO, br, 3H), 3.38 (CHC(O)OC(O)CH, br, 2H). 13C NMR (DMSO): δ 22 (CH2), 31 (CH3), 54 (CH3COO), 66 (COC(O)), 129 (CHC(O)O), 134 (CHC(O)O), 168 (CHC(O)OC(O)CH), 170 (CH3COO). IR anhydride C=O 1708, 1788 cm–1. MnSEC(DMF) = 18200 Da. Mw/Mn = 1.70.
Postpolymerization Modification of Poly(maleic anhydride-alt-styrene) with N-Boc Ethanolamine
As a representative example, poly(maleic anhydride-alt-styrene) (0.06 g), was dissolved in THF (1 mL). After dissolution, N-boc ethanolamine (0.32 g, 1.99 mmol) was added in excess. The solution was stirred for 24 h at 40 °C. The product was diluted with water and dialyzed (Fisher, 1000–3500 Da MWCO) for 48 h (7 water changes). The water was removed under reduced pressure and the solid dissolved in methanol (2 mL) before addition of excess trifluoroacetic acid (2 g, 17 mmol). The resulting solution was again concentrated in vacuo, dissolved in water, and dialyzed (Fisher, 1000–3500 Da MWCO) for 48 h (7 water changes). The resulting product was then freeze-dried to evolve a white solid. IR ring opened C=O 1700, 1625 cm–1 (broad).
Synthesis of Poly(2-(dimethylamino)ethyl Acrylate-co-tert-butyl acrylate)
As a representative example, 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (0.044 g, 120 μmol), tert-butyl acrylate (0.769 g, 6 mmol), 2-(dimethylamino)ethyl acrylate (0.859 g, 6 mmol), ACVA (4,4′-azobis(4-cyanovaleric acid); 6.7 mg, 24 μmol), and 1,4-dioxane (1 mL) were added to a sealed vial. The solution was degassed by bubbling N2 through the solution for 30 min, and the reaction was then allowed to polymerize at 80 °C for 2 to 4 h (depending on the target length). The polymerization reaction was stopped by plunging the resulting solution into liquid nitrogen. Poly(2-(dimethylamino)ethyl acrylate-co-tert-butyl acrylate) was recovered as a yellow liquid after precipitation into cold petroleum ether. The product was then dried under vacuum to remove any remaining solvent to evolve a viscous yellow liquid.
1H NMR (DMSO): δ 1.38 (CH2, br, 2H), 1.44 (OC(CH3)3, s, 9H), 2.16 (CH, s, 1H), 2.21 (N(CH3)2), s, 6H), 3.57 (NCH2, br, 2H), 4.18 (OCH2, t, 2H). MnSEC (THF) = 8600 Da. Mw/Mn = 1.32.
Synthesis of Poly(2-(dimethylamino)ethyl Acrylate-co-tert-butyl Acrylate-co-methyl acrylate)
As a representative example, 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (0.055 g, 150 μmol), tert-butyl acrylate (0.865 g, 6.75 mmol), methyl acrylate (0.123 g, 1.5 mmol), 2-(dimethylamino)ethyl acrylate (0.966 g, 6.75 mmol), ACVA (4,4′-azobis(4-cyanovaleric acid); 8.4 mg, 30 μmol), and 1,4-dioxane (1 mL) were added to a subasealed vial. The solution was degassed by bubbling N2 through the solution for 30 min, and the reaction was then allowed to polymerize at 80 °C for 2 h (depending on the target length). The polymerization reaction was stopped by plunging the resulting solution into liquid nitrogen. Poly(2-(dimethylamino)ethyl acrylate-co-tert-butyl acrylate-co-methyl acrylate) was recovered as a yellow liquid after precipitation into cold petroleum ether. The product was then dried under vacuum to remove any remaining solvent to evolve a viscous yellow liquid.
1H NMR (DMSO): δ 1.39 (CH2, br, 2H), 1.44 (OC(CH3)3, s, 9H), 2.16 (CH, s, 1H), 2.21 (N(CH3)2), s, 6H), 3.57 (NCH2, br, 2H), 3.68 (OCH3, s, 3H), 4.18 (OCH2, t, 2H). MnSEC (THF) = 7800. Mw/Mn = 1.22.
Results and Discussion
To enable the first detailed study on structure–activity relationships in well-defined and regioregular polyampholytes, copolymers based on maleic anhydride were designed to give a perfectly alternating structure.39 This structure (Figure 1) ensures that the positive and negative charges are in an exact stoichiometric ratio (as previous results have shown this to be crucial33) but also avoids any composition drift or random placement of functionalities associated with postpolymerization modification or statistical copolymerization. By judicious choice of the comonomer polymerized with maleic anhydride, the backbone hydrophobicity, as well as side chain hydrophobicity can be sequentially modified, and is discussed later. Styrene/MA (PSx) and isopropenyl acetate/MA (PIPACx) were polymerized using a dithioester RAFT agent and the vinyl acetate/MA (VACx) using a xanthate. Following polymerization all polymers were isolated by precipitation and characterized by SEC and 1H NMR, Table 1.
Figure 1.
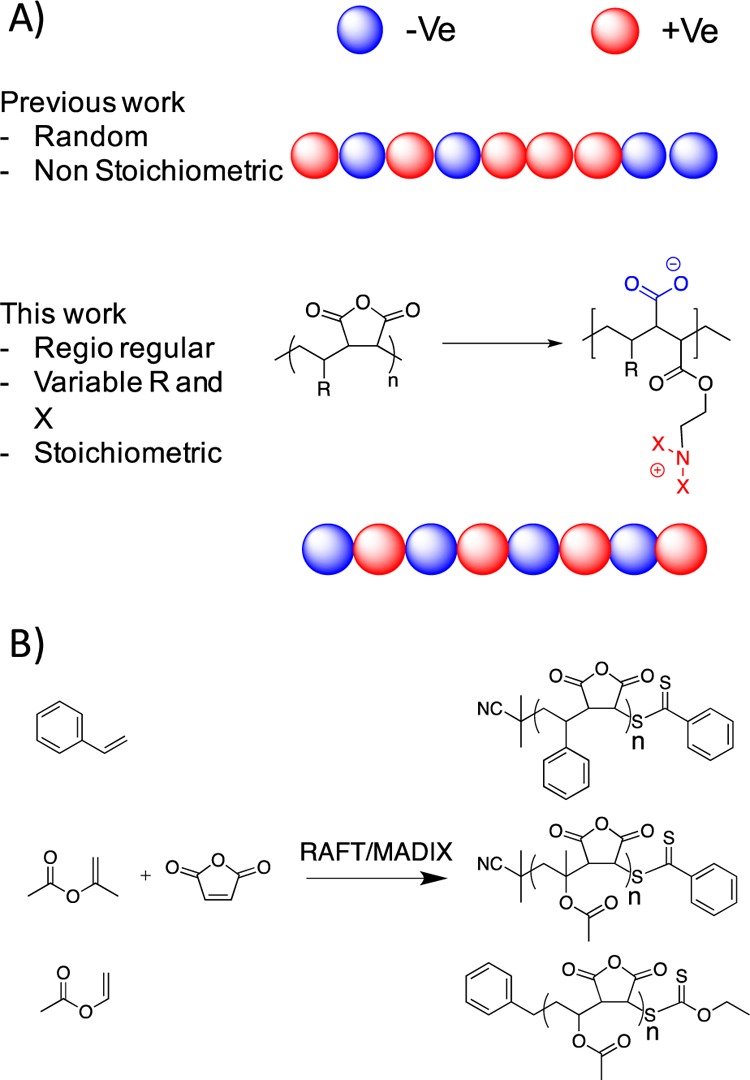
(A) Concept taken here to produce regioregular ampholytes compared to other previous synthetic strategies; (B) Copolymerization of maleic anhydride with styrene, isopropenyl acetate, and vinyl acetate.
Table 1. Alternating Polymers Synthesized.
| MA copolymera | [M]/[CTA]b | Mnc (SEC) (g mol–1) | Đd | DPe |
|---|---|---|---|---|
| PS100 | 310 | 20300 | 1.11 | 100 |
| PS46 | 150 | 9200 | 1.08 | 46 |
| PIPAC36 | 100 | 7200 | 1.11 | 36 |
| PIPAC92 | 420 | 18200 | 1.70 | 92 |
| PVAc30 | 140 | 5600 | 1.46 | 30 |
| PVAc57 | 280 | 10500 | 1.34 | 57 |
PS100 indicates a poly(styrene-alt-maleic anhydride) copolymer with 100 of the alternating repeat units, see Figure 1A.
Total monomer to RAFT agent ratio. Monomers were used in a 4:1 ratio of maleic anhydride to comonomer.
Determined by SEC.
Đ is Mw/Mn from SEC.
Number-average degree of polymerization from SEC.
To ensure an alternating polymer was formed (discussed further below), a 4-fold excess of the maleic anhydride to comonomer was used (reducing homopolymer blocks). The reactions were also stopped at low conversion for the same reason, meaning relatively large [M]:[CTA] ratios were required to achieve the desired molecular weights. A consequence of this was that the total [I] was low and, hence, contributed to the observed dispersities being a bit larger than expected for a controlled radical polymerization, alongside the nonideal SEC solvents for these polymers.35,42 Vinyl ester monomers (VAc and IPAc) are also deactivated so more challenging to polymerize than other common monomers.35
Key to this present investigation is that the polymers produced had not only a 1:1 ratio of each monomer component, but that they are regioregular with a perfectly alternating structure, as would be predicted by their reactivity ratios. To investigate this, quantitative 13C NMR was employed; homopolymers of the non-MA component should show distinct peaks associated with, for example, styrene–styrene units, whereas an alternating copolymer should not show any of these. Figure 2A shows styrene homopolymer compared to copolymer with the cross peak clearly (asterisk) not being present in the copolymers. The same is seen for the VAc polymers in Figure 2B. For PIPAc it was not possible to obtain a homopolymer control (as this monomer does not self-polymerize readily35), but the 13C still showed only a single peak in the region of the PIPAC backbone confirming the alternating structure.
Figure 2.
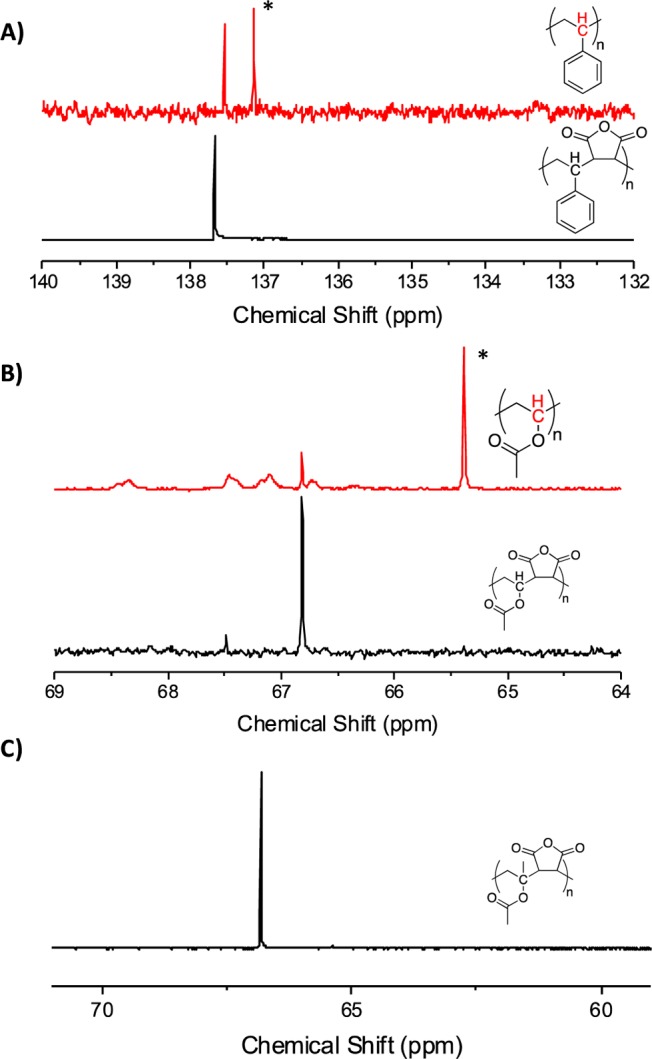
13C NMR sequence analysis of alternating copolymers. *Indicates peak associated with homopolymer.
With these alternating polymers to hand, it was necessary to introduce the desired amine functionality postpolymerization. N-Boc ethanolamine was used as a nucleophile to ring-open the anhydride, which following deprotection with TFA (trifluoroacetic acid), generates the ampholyte structure, Scheme 1. An alcohol nucleophile was chosen to prevent the undesired ring-closing reaction associated with amines (to give a malemide) which would stop the formation of the carboxylic acid group, essential for IRI activity (later). To indicate installation of the amine (and for convenience when other side chains are used later) the polymers are appended with −NH2 in our naming convention. (e.g., PS-NH2 is PS/MA copolymer with primary amine installed side chain).
Scheme 1. Ring-Opening of Anhydride and Subsequent Deprotection.

Successful ring-opening was confirmed by IR spectroscopy. The two peaks associated with the anhydride carbonyls (1850 and 1790 cm–1) were quantitatively removed and the installation of an ester and carboxylic acid at lower wavelengths could be seen, Figure 3.
Figure 3.
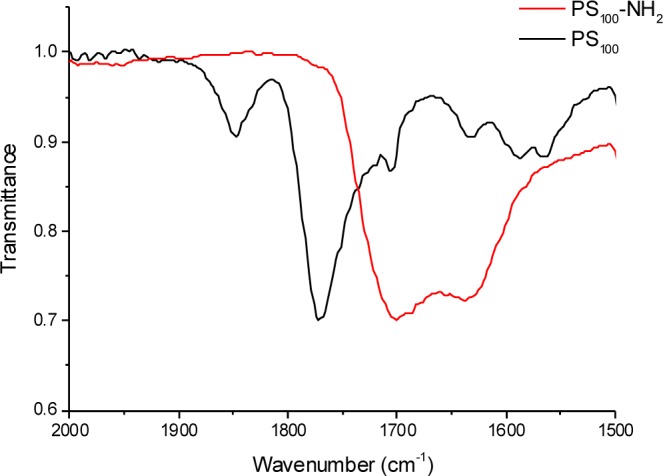
Infrared analysis of PS-100 showing removal of anhydride peaks following ring opening and deprotection.
With this library of polyampholytes containing variable chain lengths and backbone hydrophobicites, the ice recrystallization inhibition (IRI) activity could be evaluated. IRI was measured using the “splat” assay.27 10 μL droplets of the polymers in PBS were dropped onto a glass coverslip sat on a chilled (−80 °C) aluminum plate. This generates a large number of <10 μm ice crystals by rapid nucleation, which were then incubated on a cold stage for 30 min at −8 °C. The average area of the ice crystals (by counting the total number in a fixed area to generate an average, note this is slightly different to our previous reports using the mean largest grain size which did not account for the population21,27,43,44) relative to a PBS control. Smaller numbers indicate more activity. The results of this are shown in Figure 4 as a function of concentration.
Figure 4.
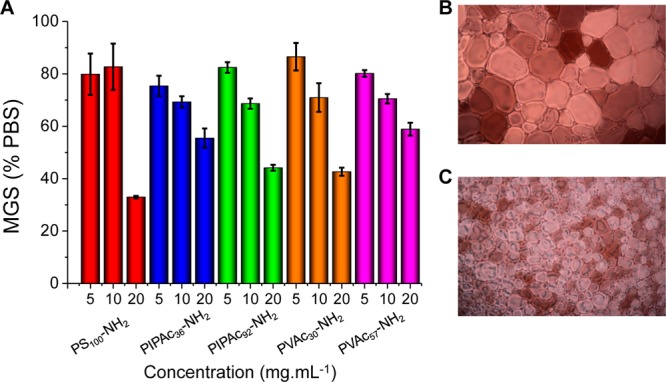
IRI activity of amino side chain polyampholytes with various backbones. (A) IRI activity; (B) Cryo-micrograph of PBS buffer control; (C) Cryo-micrograph of PS100-NH2 at 20 mg·mL–1. MGS = mean grain size reported as an area. Averages are from a minimum of three repeats and error bars are the standard deviation. Images are 660 μm wide.
In line with our previous studies on polyampholytes, these polymers were moderately active, compared to, for example, poly(vinyl alcohol), which is the most potent non-AF(G)P IRI known, but as very few synthetic materials have this property, it is still a remarkable observation.2,6 All the polymers appeared to have similar activity without any strong molecular weight trends, with all of them leading to 35–50% crystal areas at the highest concentration tested (20 mg·mL–1), which is more potent than ampholytes reported by Matsumura and co-workers.34 This does not necessarily rule out molecular weight dependence, just in the range tested. Interestingly, the PS-NH2 copolymers appeared to have higher activity at 20 mg·mL–1 compared to the other polymers, but less at low concentration. Careful analysis of the solutions releveled that the PS-NH2 were actually slightly turbid due to some aggregation. This means that the actual concentration of dissolved chains is less making a critical comparison challenging. The styrene units, when compared to the other comonomers used in this study, are significantly more hydrophobic, and these copolymers are less soluble, demonstrating the delicate balance between hydrophobicity (expected to increase activity) and hydrophilicity (needed to ensure solubility in biological buffers).
The above results showed that modulation of the backbone of polyampholytes, for the range of materials tested, did not have a large impact on their overall activity, or was not suitable due to solubility issues. However, the modular synthetic strategy employed here enables the side-chain to also be varied, by using differently substituted amines. Guided by the above, we wanted to see if instead of backbone hydrophobicity we could vary the alkyl chain on the amine as a route to modulate activity as there is literature evidence that this might enhance activity in other polyampholytes.34 It was decided that the VAc and IPAc based polymers were to be studied further, as these were more soluble than the styrenes to enable comparison. Four different amino alcohols with different hydrophobic chains on the amine were chosen and used to ring open the anhydride unit, Scheme 2. Again, IR spectroscopy was used to confirm successful ring-opening and installation of the desired functional groups.
Scheme 2. Installation of a Range of Different Amine Side Chains into Alternating Copolymers by Ring Opening of Malemide Unit.

This second set of polymers were tested for IRI activity, as a function of concentration using the same methodology as the first set of polymers, Figure 5. For the diethyl, diisopropyl, and primary amine, the activity observed was largely identical across concentrations. However, for the dimethyl, on the longest (PIPAC92) the dimethyl amino polymers were dramatically more active at high concentrations, inhibiting nearly all ice growth. This was a remarkable level of activity, and was only achieved by achieving a fine balance between backbone hydrophobicity, side chain hydrophobicity, chain length, and solubility.
Figure 5.
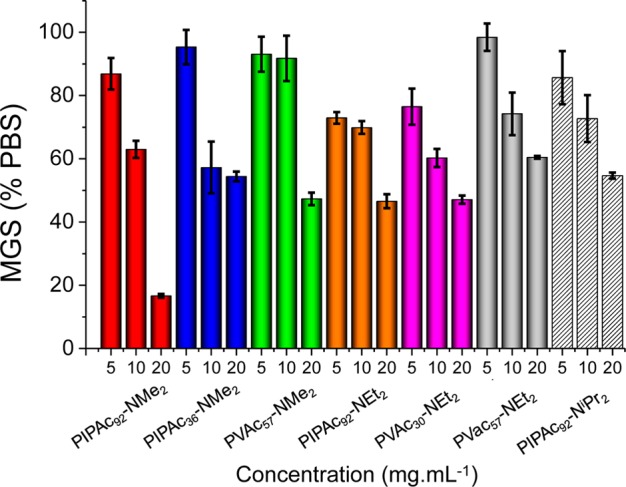
IRI activity of side chain modified polyampholytes. Averages are from a minimum of three repeats and error bars are the standard deviation.
The data in Figure 5 suggests that hydrophobicity plays a role in activity, but the trends are not clear. To obtain a measure of hydrophobicity, the Log P (partition coefficient) for each repeat unit used was calculated and compared to IRI activity (see Supporting Information). This did not reveal any correlation. IRI activity has been linked to localization of hydrophobic/hydrophilic domains in native AFPs, small molecules,45 supramolecular mimics,8 and in other macromolecules,30 and the Log P does not capture this level of detail, but rather just the overall lipophilicity. This observation supports the idea that hydrophobic domains are essential for IRI activity, but that the exact 3-D position of these is crucial, and that simply adding “more hydrophobicity” will not lead to an increase in activity. The results presented also challenge the assumption that to have a potent IRI you must have a good “match” for a specific face of an ice crystal to enable binding (which is essential for AFP function), suggesting multiple potential mechanisms can lead to the macroscopic effect of IRI. The ampholytes used have no obvious binding units for ice such as hydroxyls and the individual homopolymers (e.g., polyamine) have no activity.33
To determine if the sequence and placement of the carboxyl/amine groups promote IRI activity, random sequenced analogues were designed based upon poly(acrylates). This enables investigation into the effect of the sequence and hydrophobicity on IRI under identical testing parameters. Acrylates were chosen as an analogue to the maleic anhydride units which do not have a methyl side chain (as seen in methacrylates). t-Butyl acrylate (BA) was chosen as a precursor to the carboxylic acid group and N,N-dimethyl aminoethyl acrylate (DMAEA) as the dimethyl amino group was identified (above) as a potent cationic group. To probe the role of added hydrophobicity, 10–30% methyl acrylate (MA) was also copolymerized into this while maintaining the charge balance. RAFT polymerization was used to ensure narrow dispersities and control of the molecular weight, Scheme 3. The t-butyl group was removed by TFA treatment post polymerization and confirmed by IR and 1H NMR spectroscopy. Table 2 lists the polymers synthesized.
Scheme 3. Random Copolymers Prepared via RAFT Polymerization.

Table 2. Random Sequence Polyampholytes.
| copolymera | [M]/[CTA]b | Mnc (SEC) (g mol–1) | Đd | conversione (%) | DPf | Mnf (conversion) (g mol–1) |
|---|---|---|---|---|---|---|
| DMAEA-AA22 | 50 | 10000 | 1.22 | 43 | 22 | 2400 |
| DMAEA-AA75 | 100 | 8600 | 1.32 | 75 | 75 | 8100 |
| DMAEA-AA100 | 400 | 10900 | 1.30 | 25 | 100 | 10800 |
| DMAEA-AA128 | 400 | 17300 | 1.30 | 32 | 128 | 13800 |
| DMAEA-AA232 | 400 | 19400 | 1.43 | 58 | 232 | 25000 |
| DMAEA-AA-MA(10%)51 | 100 | 7800 | 1.22 | 51 | 51 | 5400 |
| DMAEA-AA-MA(20%)45 | 100 | 7400 | 1.21 | 45 | 45 | 4600 |
| DMAEA-AA-MA(30%)57 | 100 | 7900 | 1.22 | 57 | 57 | 5800 |
Name of polymer sample, DMAEA-AA22 indicates a poly(dimethylaminoethyl acrylate-co-acrylic acid) copolymer with 22 randomly incorporated units, and DMAEA-AA-MA(10%)51 indicates a poly(dimethylaminoethyl acrylate-co-acrylic acid-co-methyl acrylate) terpolymer with 51 randomly incorporated units of which 10% is methyl acrylate.
Monomer to RAFT agent ratio.
Determined by SEC.
Đ is equal to Mw/Mn.
Determined by 1H NMR.
Estimated from conversion.
The polymer library in Table 2 was screened for IRI activity using the same “splat” method as described earlier. Initial observations revealed these polymers to be significantly less active than the alternating polymers. Even at 50 mg·mL–1 (max concentration used for regioregular polymers was 20 mg·mL–1) only 80% activity relative to PBS was observed which would indicate essentially zero activity, similar to the nonactive negative control poly(ethylene glycol). This observation suggests that both the sequence and the precise architecture of the backbone is essential to the overall function of the polymers. Our previous studies have shown that random poly(methacrylate)-based ampholytes are more potent than these poly(acrylates) suggesting the backbone methyl is important,33 but both are less potent than the alternating polymers from Figure 5. To probe the role of hydrophobicity the polymers containing methyl acrylate to enhance side-chain hydrophobicity were assessed for activity with the chain length for all targeted at 50 repeat units, Figure 6. It was found that addition of 10% MA, gave rise to the most significant enhancement in activity, compared to either 20 or 30% MA. At 50 mg·mL–1, the addition of MA increased activity from ∼80% to ∼40% representing a significant increase. At all these concentrations, the polymers were fully soluble, suggesting that the benefit from additional hydrophobicity is subtle and a fine balance between that and the reduction in density of the ampholyte units. This is in contrast to the alternating polymers where a constant ampholyte unit density was maintained.
Figure 6.
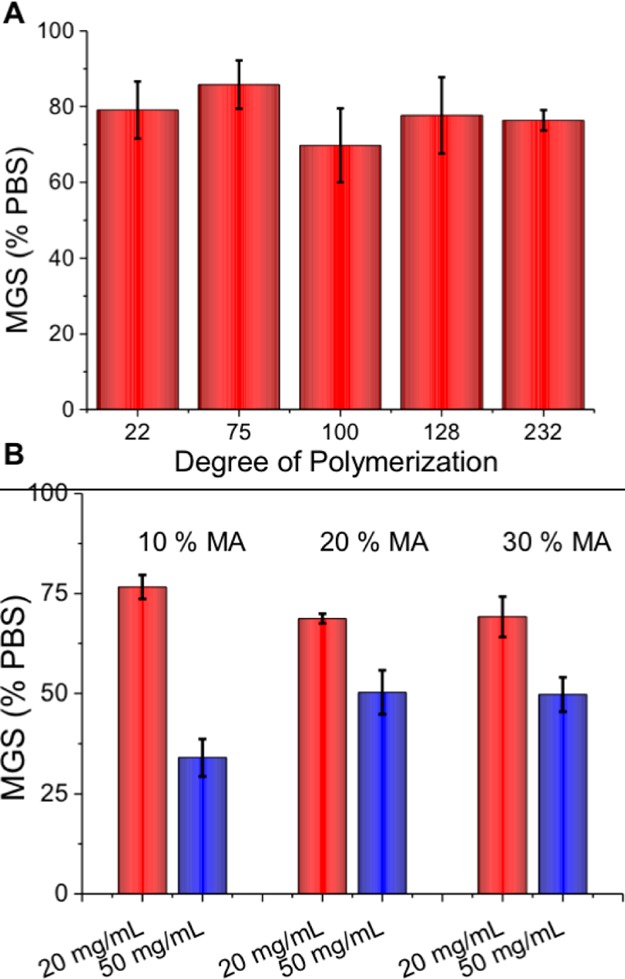
IRI activity of the random poly(acrylate) copolymers. Student t test comparing DMAEA-AA75 and DMAEA-AA-MA(10%)51 at 50 mg·mL–1 showed a p-value of 0.0164 indicating significance.
The above data shows that consideration of the distribution of charged units in polyampholytes is crucial in the rational design of new IRI active macromolecules. The random polymers had less activity than the regioregular, which was shown to be a delicate balance between position of the hydrophobicity, but most importantly maintaining solubility. None of the polymers had noticeable ice shaping effects, but this property (DIS) is normally seen at high concentrations so might be limited by solubility. The polymers used here may also not be directly applicable for cryopreservation and will require a detailed analysis of toxicological profile but also cell uptake/exclusion.
Conclusions
This study reports the first investigation into the effect of sequence/regiochemistry on the ice recrystallization inhibition (IRI) activity of polyampholytes, with the intention of mimicking the function of antifreeze (glyco)proteins. The mode of interaction of polyampholytes with ice is still unknown and is one of the few classes of synthetic polymer known to display this activity. Alternating copolymers were obtained by exploiting the tendency of maleic anhydride to cross-polymerize with a range of (hydrophobic) comonomers. The anhydride ring could then be opened to introduce a range of amino-functionalities as well as a carboxylic acid, with an exact 1:1 ratio of functional groups, positioned adjacent to each other. Such control is not possible by a normal radical copolymerization. Quantitative IRI (ice recrystallization) assays revealed that the hydrophobic comonomer (styrene) had little impact on the IRI activity which may be due to its dramatic effect on aqueous solubility. Conversely, alkylation of the amines (side chain hydrophobicity) lead to changes in activity. Dimethylation lead to more activity than longer alkyl chains, or a primary amine demonstrating that although hydrophobicity can increase IRI activity the nature and placement of this must be carefully considered to prevent aggregation/precipitation and to maximize activity. These observations support the concept that spatially segregated hydrophilic/hydrophobic domains are essential to ensure potent IRI in synthetic materials. A comparison was made against a nonsequenced ampholyte obtained from acrylates. These polymers had significant lower activity compared to the regioregular ones. Addition of side chain hydrophobicity by inclusion of methyl acrylate, however, did enhance the activity but this effect decreased above 10 mol % highlighting that simply adding more hydrophobicity does not increase activity. These results are significant, in that they provide evidence that control of a polymer’s microstructure can be used to enhance IRI activity and provide insight into the design rules needed to synthesize antifreeze protein mimics, particularly for cellular cryopreservation.
Acknowledgments
M.I.G. holds an ERC starting grant (CRYOMAT 638661). The Royal Society are also thanked for funding the cryo-microscopes used in this study. The Midlands Integrative Biosciences Doctoral Training Partnership (MIBTP) are thanked for a studentship for J.L. (BB/M01116X/1).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.6b01691.
Full details of the synthesis of the postpolymerization modifications, along with calculation and comparison of partition coefficients (Log P; PDF).
The authors declare no competing financial interest.
Supplementary Material
References
- Harding M. M.; Anderberg P. I.; Haymet A. D. J. Eur. J. Biochem. 2003, 270 (7), 1381–1392. 10.1046/j.1432-1033.2003.03488.x. [DOI] [PubMed] [Google Scholar]
- Balcerzak A. K.; Capicciotti C. J.; Briard J. G.; Ben R. N. RSC Adv. 2014, 4 (80), 42682–42696. 10.1039/C4RA06893A. [DOI] [Google Scholar]
- Venketesh S.; Dayananda C. Crit. Rev. Biotechnol. 2008, 28 (1), 57–82. 10.1080/07388550801891152. [DOI] [PubMed] [Google Scholar]
- Capicciotti C. J.; Kurach J. D. R.; Turner T. R.; Mancini R. S.; Acker J. P.; Ben R. N. Sci. Rep. 2015, 5, 9692. 10.1038/srep09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H.; Davies P. L.; Carpenter J. F. J. Exp. Biol. 1996, 199 (9), 2071–2076. [DOI] [PubMed] [Google Scholar]
- Gibson M. I. Polym. Chem. 2010, 1 (8), 1141–1152. 10.1039/c0py00089b. [DOI] [Google Scholar]
- Leclère M.; Kwok B. K.; Wu L. K.; Allan D. S.; Ben R. N. Bioconjugate Chem. 2011, 22 (9), 1804–1810. 10.1021/bc2001837. [DOI] [PubMed] [Google Scholar]
- Drori R.; Li C.; Hu C.; Raiteri P.; Rohl A.; Ward M. D.; Kahr B. J. Am. Chem. Soc. 2016, 138 (40), 13396–13401. 10.1021/jacs.6b08267. [DOI] [PubMed] [Google Scholar]
- Currie L. M.; Livesey S. A.; Harper J. R.; Connor J. Transfusion 1998, 38 (2), 160–167. 10.1046/j.1537-2995.1998.38298193098.x. [DOI] [PubMed] [Google Scholar]
- Polge C.; Smith A. U.; Parkes A. S. Nature 1949, 164 (4172), 666. 10.1038/164666a0. [DOI] [PubMed] [Google Scholar]
- Baust J. G.; Gao D.; Baust J. M. Organogenesis 2009, 5 (3), 90–96. 10.4161/org.5.3.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustun N. S.; Turhan S. J. Food Process. Preserv. 2015, 39 (6), 3189–3197. 10.1111/jfpp.12476. [DOI] [Google Scholar]
- Lindner N. M.; Oldroyd J. R.; Sztehlo A.; Towell D. J.. Frozen confectionery product comprising ice structuring proteins. WO2005058058 A1, 2004.
- Sidebottom C.; Buckley S.; Pudney P.; Twigg S.; Jarman C.; Holt C.; Telford J.; McArthur A.; Worrall D.; Hubbard R.; Lillford P. Nature 2000, 406 (6793), 256. 10.1038/35018639. [DOI] [PubMed] [Google Scholar]
- Iwatani M.; Ikegami K.; Kremenska Y.; Hattori N.; Tanaka S.; Yagi S.; Shiota K. Stem Cells 2006, 24 (11), 2549–2556. 10.1634/stemcells.2005-0427. [DOI] [PubMed] [Google Scholar]
- Shu Z.; Heimfeld S.; Gao D. Bone Marrow Transplant. 2014, 49 (4), 469–476. 10.1038/bmt.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T.; Lu S. S. Cryst. Growth Des. 2003, 3 (5), 747–752. 10.1021/cg0340300. [DOI] [Google Scholar]
- Budke C.; Koop T. ChemPhysChem 2006, 7 (12), 2601–2606. 10.1002/cphc.200600533. [DOI] [PubMed] [Google Scholar]
- Olijve L. L. C.; Meister K.; DeVries A. L.; Duman J. G.; Guo S.; Bakker H. J.; Voets I. K. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (14), 3740–3745. 10.1073/pnas.1524109113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson M. I.; Barker C. A.; Spain S. G.; Albertin L.; Cameron N. R. Biomacromolecules 2009, 10 (2), 328–333. 10.1021/bm801069x. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Congdon T.; Sahid M. A.; Morgan M.; Vatish M.; Mitchell D. A.; Notman R.; Gibson M. I. Biomater. Sci. 2013, 1 (5), 478–485. 10.1039/c3bm00194f. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. ACS Biomater. Sci. Eng. 2015, 1 (9), 789–794. 10.1021/acsbiomaterials.5b00162. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. Nat. Commun. 2014, 5, 3244. 10.1038/ncomms4244. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Pessin J. E.; Vatish M.; Mitchell D. A.; Gibson M. I. Biomater. Sci. 2016, 47, 935–945. 10.1039/C6BM00129G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wowk B.; Leitl E.; Rasch C. M.; Mesbah-Karimi N.; Harris S. B.; Fahy G. M. Cryobiology 2000, 40 (3), 228–236. 10.1006/cryo.2000.2243. [DOI] [PubMed] [Google Scholar]
- Congdon T.; Dean B. T.; Kasperczak-Wright J.; Biggs C. I.; Notman R.; Gibson M. I. Biomacromolecules 2015, 16 (9), 2820–2826. 10.1021/acs.biomac.5b00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon T.; Notman R.; Gibson M. I. Biomacromolecules 2013, 14 (5), 1578–1586. 10.1021/bm400217j. [DOI] [PubMed] [Google Scholar]
- Phillips D. J.; Congdon T. R.; Gibson M. I. Polym. Chem. 2016, 7 (9), 1701–1704. 10.1039/C5PY01948F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olijve L. L. C.; Hendrix M. M. R. M.; Voets I. K. Macromol. Chem. Phys. 2016, 217 (8), 951–958. 10.1002/macp.201500497. [DOI] [Google Scholar]
- Mitchell D. E.; Gibson M. I. Biomacromolecules 2015, 16 (10), 3411–3416. 10.1021/acs.biomac.5b01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K.; Hyon S. H. Biomaterials 2009, 30 (27), 4842–4849. 10.1016/j.biomaterials.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Cameron N. R.; Gibson M. I. Chem. Commun. 2015, 51 (65), 12977–12980. 10.1039/C5CC04647E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. E. D. E.; Lilliman M.; Spain S. G.; Gibson M. I. Biomater. Sci. 2014, 2 (12), 1787–1795. 10.1039/C4BM00153B. [DOI] [PubMed] [Google Scholar]
- Rajan R.; Hayashi F.; Nagashima T.; Matsumura K. Biomacromolecules 2016, 17 (5), 1882–1893. 10.1021/acs.biomac.6b00343. [DOI] [PubMed] [Google Scholar]
- Harrisson S.; Liu X.; Ollagnier J.-N.; Coutelier O.; Marty J.-D.; Destarac M. Polymers (Basel, Switz.) 2014, 6 (5), 1437–1488. 10.3390/polym6051437. [DOI] [Google Scholar]
- Schmidt B. V. K. J.; Fechler N.; Falkenhagen J.; Lutz J.-F. Nat. Chem. 2011, 3 (3), 234–238. 10.1038/nchem.964. [DOI] [PubMed] [Google Scholar]
- Moatsou D.; Hansell C. F.; O’Reilly R. K. Chem. Sci. 2014, 5 (6), 2246–2250. 10.1039/c4sc00752b. [DOI] [Google Scholar]
- Gody G.; Barbey R.; Danial M.; Perrier S. Polym. Chem. 2015, 6 (9), 1502–1511. 10.1039/C4PY01251H. [DOI] [Google Scholar]
- Ratzsch M. Prog. Polym. Sci. 1988, 13, 277–337. 10.1016/0079-6700(88)90001-9. [DOI] [Google Scholar]
- Gauthier M. A.; Gibson M. I.; Klok H.-A. Angew. Chem., Int. Ed. 2009, 48 (1), 48–58. 10.1002/anie.200801951. [DOI] [PubMed] [Google Scholar]
- Congdon T.; Shaw P.; Gibson M. I. Polym. Chem. 2015, 6 (26), 4749–4757. 10.1039/C5PY00775E. [DOI] [Google Scholar]
- Stenzel M. H.; Cummins L.; Roberts G. E.; Davis T. P.; Vana P.; Barner-Kowollik C. Macromol. Chem. Phys. 2003, 204 (9), 1160–1168. 10.1002/macp.200390089. [DOI] [Google Scholar]
- Phillips D. J.; Congdon T. R.; Gibson M. I. Polym. Chem. 2016, 7 (9), 1–13. 10.1039/C5PY01948F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. E.; Congdon T.; Rodger A.; Gibson M. I. Sci. Rep. 2015, 5, 15716. 10.1038/srep15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capicciotti C. J.; Leclere M.; Perras F. A.; Bryce D. L.; Paulin H.; Harden J.; Liu Y.; Ben R. N. Chem. Sci. 2012, 3 (5), 1408–1416. 10.1039/c2sc00885h. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.