

Abstract
We identify four novel DNA-binding complexes in the nuclear-encoded Lhcb1 promoter of the chlorophyte alga Dunaliella tertiolecta that are regulated by photosynthetic pathways in the plastid. The binding activities of three of the complexes were positively correlated with time-dependent changes in Lhcb1 transcript abundance, implicating their roles as transcriptional enhancers in a retrograde signal transduction pathway. Using a combination of inhibitors, uncouplers, and antimycin A, and by following the kinetic pattern of gene regulation, we infer two different sensors in the signal transduction pathway. On short time scales of 0.5 to about 4 h, the transthylakoid membrane potential appears to be a critical determinant of gene expression, whereas on time scales of 8 h or longer, the redox state of the plastoquinone pool becomes increasingly more important. The differentiation of these two types of signals was observed in parallel effects on gene transcription and on the patterns of DNA-binding activities in the Lhcb1 promoter. These signals appear to be transduced at the nuclear level via a coordinated ensemble of DNA-binding complexes located between −367 and −188 bp from the start codon of the gene. The regulation of these elements allows the cell to up- or down-regulate the expression on Lhcb1 in response to changes in irradiance.
All photosynthetic organisms can adjust the rate of photon absorption to optimize photosynthetic electron transport (PET) in relation to changes in spectral irradiance. Long and persistent shifts in photon flux density (PFD) often involve changes in gene expression that modify the activity, concentration, and structure of the photosynthetic machinery (Falkowski and LaRoche, 1991; Anderson et al., 1995; Durnford and Falkowski, 1997). In comparison with higher plants, photoacclimation is far more pronounced in unicellular algae, where not only the magnitude and rates of the responses are extraordinary, but also the response can easily be distinguished from developmental processes (Falkowski and Chen, 2003). In particular, species in the marine unicellular chlorophyte genus Dunaliella can undergo an almost 10-fold change in the abundance of antenna chlorophyll (Chl)-binding proteins on a per cell basis within a single generation (Sukenik et al., 1988). In eukaryotic algae, these changes involve transcription and translation of both nuclear and chloroplast genes. Consequently, tight interorganellar regulatory networks are required to coordinate gene expression in these two compartments. In this paper, we examine how changes in the abundance of the nuclear-encoded Lhcb gene in response to changes in PFD are sensed by the plastid and how the signals are correlated with binding factors in the promoter region.
At least three signaling pathways involved in plastid regulation of nuclear gene expression have been described for plant cells. The first is mediated by tetrapyrrole biosynthesis, the second requires plastid protein synthesis, and the third involves electron carrier redox poise and potentially other photosynthetic signals (for review, see Rodermel, 2001; Surpin et al., 2002; Gray et al., 2003). The first two pathways play important roles in developmental processes and in response to dark/light cycles, whereas chloroplast redox signaling appears to control the nuclear photosynthetic gene expression during the acclimation to changes in PFD, temperature, and CO2 (Durnford and Falkowski, 1997; Huner et al., 1998; Surpin et al., 2002; Strand et al., 2003).
Among the thylakoid electron carriers, the plastoquinone (PQ) pool is considered to be an ideal candidate for signaling excess or insufficient PSII activity relative to the capacity for carbon fixation (Falkowski et al., 1986; Pfannschmidt et al., 1999). Because there are several PQ molecules per reaction center, and the oxidation of PQH2 is the slowest reaction within the PET chain, the redox state of the PQ pool is sensitive to both excitation pressure on the donor side of PSII and downstream sink capacity on the acceptor side of PSI. Hence, the ratio of PQ/PQH2 is a biological light meter that reflects the ratio between the rate of light absorption by the photosynthetic apparatus and the rate of linear photosynthetic electron transfer (Falkowski et al., 1986; Fujita et al., 1987; Escoubas et al., 1995).
Using a variety of PET and other inhibitors, Escoubas et al. (1995) demonstrated that Lhcb1 expression is dependent on the redox state of the PQ pool in Dunaliella tertiolecta. Subsequently, redox regulation of Lhcb1 and other nuclear-encoded genes were reported in algae and higher plants (Maxwell et al., 1995; Karpinski et al., 1997, 1999; Pfannschmidt et al., 2001). However, it is unclear whether the PQ pool is the only chloroplastic signal initiator and how the signal is transduced. It has been suggested that the site of control involves a stromal-redox component, such as ferredoxin, thioredoxin, and/or glutathione (Danon and Mayfield, 1994; Montané et al., 1998; Oswald et al., 2001). The role of the cytochrome b6f complex-mediated light-harvesting complex (LHC)II kinase as a possible redox sensor independent of the PQ pool has also been proposed (Kovács et al., 2000; Rintamäki et al., 2000; Pursiheimo et al., 2001; Yang et al., 2001; Zer and Ohad, 2003). In addition to controlling the xanthophyll cycle-mediated nonphotochemical quenching (Demmig-Adams et al., 1996), the transthylakoid membrane pH gradient (ΔpH) has been shown to control the phosphorylation of LHCII (Fernyhough et al., 1984) and regulates nuclear-encoded photosynthetic genes (Horton et al., 1996; Wilson and Huner, 2000). Furthermore, chloroplast ATP levels have been suggested to play roles in the signal transduction pathway that regulates LHCII kinase/phosphatase activities and state transitions (Baker et al., 1982; Fernyhough et al., 1984; Bulté et al., 1990; Wollman, 2001), as well as in adjusting the ratio between linear and cyclic electron transfer (Joliot and Joliot, 2002).
The mechanism(s) by which a chloroplast redox signal is relayed to the nucleus remains unknown. Plastid redox signals may be transmitted via one or more plastid factors, the existence of which was postulated decades ago (Bradbeer et al., 1979). The involvement of one or more phosphorylatable protein messenger(s) in the chlorophyte algae, D. tertiolecta (Escoubas et al., 1995) and Chlamydomonas reinhardtii (Depège et al., 2003), as well as the terrestrial plant Arabidopsis (Arabidopsis thaliana; Carlberg et al., 2003), has been suggested. It is also possible that redox signaling occurs via the same phosphorelay system, involving heterotrimetric G proteins and protein kinase C, which regulates the transcription of PsaF in response to light and the functional state of the plastid (Chandok et al., 2001).
In this study, we measured the kinetics of Lhcb1 transcript abundance and DNA-binding activity associated with a 180-bp Lhcb1 promoter region in D. tertiolecta following light transitions, or following the addition of a variety of inhibitors, or combinations of the two types of treatments. Our results reveal, for the first time to our knowledge, that the binding activity of multiple DNA-binding complexes, associated with the Lhcb1 promoter region, are affected by changes in light intensity, redox poise of electron transfer components between PSII and PSI, and the transthylakoid membrane potential. The relationship between the Lhcb1 transcript abundance and the binding activities of these complexes implicate the roles of the latter as transcriptional enhancers in a retrograde signal transduction pathway. The results further suggest that two types of chloroplastic signals, the redox state of PQ pool as potentially sensed by the cytochrome b6f complex and the transthylakoid membrane potential, cooperatively regulate Lhcb1 expression.
RESULTS
DNA-Binding Patterns in the Lhcb1 Promoter Region
To dissect the binding properties of the Lhcb1 promoter, we generated a set of six 30-bp long oligo DNA probes that sequentially represent the 180-bp promoter region (from −367 to −188 bp; Fig. 1). Based on electrophoretic mobility shift assay (EMSA) of whole-cell protein extracts, we identified four different binding complexes that we call HLF, LF1, LF2, and LF3 (Fig. 2). The largest complex, HLF, bound to all six probes, while the second largest complex, LF1, bound to all except Oligo 3. One of the two smaller complexes, LF2, bound strongly to Oligo 3 (−307 to −278 bp) and Oligo 5 (−247 to −218 bp), whereas LF3 bound primarily to Oligo 5. These results were confirmed using more than one dozen PCR-generated probes in the same promoter region (data not shown). Binding of all complexes is specific; they can be outcompeted by their respective unlabeled oligo DNA, but not by nonspecific competitors such as sonicated salmon sperm DNA (Fig. 2).
Figure 1.

Schematic presentation of the tentative multiple and repetitive binding sites in D. tertiolecta Lhcb1 180-bp promoter region (from −367 to −188 bp relative to the starting codon). Six oligo DNA were constructed within the promoter and used as unlabeled competitors or labeled probes in EMSA.
Figure 2.

EMSA of DNA-binding activities in the 180-bp Lhcb1 promoter region (−367 to −188 bp) represented sequentially by Oligo 1 to 6, as well as the competition of the binding activities by specific (unlabeled oligo) and nonspecific DNA fragments. Protein samples were extracted from HL- and LL-acclimated culture and fractionated by a single step with 50% (NH4)2SO4. All binding reactions contained 1 μg of poly(dI.dC).poly(dI.dC). SS DNA, Sonicated salmon sperm DNA fragments.
There are significant sequence similarities between two sections in the targeted promoter region: from −367 to −288 bp and from −270 to −227 bp (Fig. 1). We identified three tentative binding sites in these two nonoverlapping promoter regions. The first, a TCTAA box, has a consensus sequence TCTAAHGT and is located between −365 to −357 bp (within the Oligo 1 probe) and −266 to −258 bp (within the Oligo 4 probe). In both cases, this site is 12 to 15 bp upstream of two other putative binding sites. The second contains a CACT box with a consensus sequence CARRCACTSGRA and overlaps the third, a GGAA box, which has a core sequence ASMMYYGGAA. Among the three putative binding motifs, only the TCTAA box is also found in the Lhcb1 promoter in C. reinhardtii and in Lhcb2/Lhcb3 promoters in Arabidopsis.
Effects of DCMU on Lhcb1 Transcript Abundances
The effect of 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) on the time course change of Lhcb1 transcript abundance in cells grown under different light conditions is shown in Figure 3. In both high-light (HL)- and low-light (LL)-acclimated cultures, the addition of 200 nm DCMU (which leads to an approximately 50% reduction in PET through PSII) resulted in a rapid decrease of Lhcb1 transcript abundance in the first 2 to 4 h. In HL-acclimated cells, the Lhcb1 transcript levels recovered and increased nearly 2-fold above the previous steady-state level. The pattern of change in Lhcb1 transcript abundance is consistent with that of Chl a/cell (data not shown). Though the addition of DCMU to HL cultures resulted in increased Lhcb1 transcript abundance, the pattern differed both in the magnitude and kinetics compared to the control HL-to-LL shift. In the latter, the relative Lhcb1 transcript levels increased immediately following the light transition, and the level was tripled within the first 4 h, about two times higher than the peak level reached in DCMU-treated cells after 24 h (Fig. 3).
Figure 3.

Time course changes in Lhcb1 and rrna transcript levels in HL-acclimated cells following a shift to LL and 200-nm DCMU additions to HL- and LL-acclimated cells. Top, Densitometric analysis of the time course changes in Lhcb1 level, normalized to rrna level, as percentage to the zero time point value. Bottom, Northern-blot analysis of total cellular RNA probed sequentially with Lhcb1 and rrna.
We further tested the ability of DCMU to mimic LL conditions by adding the inhibitor prior to a shift from LL to HL. If the reduction of the PQ pool were the sole signal responsible for the up-regulation of Lhcb1 transcription, the HL-induced decline in Lhcb1 transcript abundance should be attenuated. However, DCMU failed to prevent decreases in Lhcb1 transcript abundance (Fig. 4). These results strongly suggest that the redox state of the PQ pool per se is not the only photosynthetic signal that regulates Lhcb1 expression.
Figure 4.

Time course changes in Lhcb1 and rrna transcript levels in LL-acclimated cells following a shift to HL with or without the presence of 200 nm DCMU. Top, Densitometric analysis of the time course changes in Lhcb1 level, normalized to rrna level, as percentage to the zero time point value. Bottom, Northern-blot analysis of total cellular RNA probed sequentially with Lhcb1 and rrna.
Effects of Uncouplers on Lhcb1 Transcript Abundance
The initial decrease in the relative abundance of Lhcb1 following the addition of DCMU raises the possibility that signals other than the PQ pool redox status are involved in the regulation of Lhcb1 transcription. In both HL- and LL-acclimated cultures, Lhcb1 transcript abundance decreased within 1 h following the addition of 400 nm nigericin or 2 μm valinomycin and reached a nadir within 2 h (Fig. 5). Over the next 4 to 6 h, transcript levels rose to approximately 150% of the previous steady-state level. The kinetic patterns of change in Lhcb1 transcript abundance were similar for the two uncouplers.
Figure 5.

Time course changes in Lhcb1 and rrna transcript levels in LL- and HL-acclimated cells following additions of 400 nm nigericin or 2 μm valinomycin. Top, Densitometric analysis of the time course changes in Lhcb1 level, normalized to rrna level, as percentage to the zero time point value. Bottom, Northern-blot analysis of total cellular RNA probed sequentially with Lhcb1 and rrna.
Effects of Antimycin A on Lhcb1 Transcript Abundance
The results from the uncoupler treatments suggest the transthylakoid membrane potential, i.e. the sum of proton gradient and the electric potential, may be involved in the short-term regulation of Lhcb1 transcription. We further examined this hypothesis by analyzing the effects of antimycin A on Lhcb1 transcript abundance. Antimycin A inhibits cyclic electron transport around PSI (for review, see Bendall and Manasse, 1995). When 1 μm antimycin A was added to HL cultures, the relative abundance of the Lhcb1 transcript fluctuated moderately with the overall trend little changed (Fig. 6). However, when 1 μm antimycin A was added to HL cultures together with 200 nm DCMU, Lhcb1 transcript abundance increased immediately and quadrupled within the next 8 h. Though there was a near linear increase in Lhcb1 levels over time, there was a consistently reproducible decline at 2 h (Fig. 6).
Figure 6.

Time course changes in Lhcb1 and rrna transcript levels HL-acclimated cells following additions of 1 μm antimycin A or 1 μm antimycin A plus 200 nm DCMU. Top, Densitometric analysis of the time course changes in Lhcb1 level, normalized to rrna level, as percentage to the zero time point value. Bottom, Northern-blot analysis of total cellular RNA probed sequentially with Lhcb1 and rrna.
Effects of DBMIB on Lhcb1 Transcript Abundance
In both HL- and LL-acclimated cultures, the addition of 1.5 μm 2,5-dibromo-6-methyl-3-isopropyl-1,4-benzochinon (DBMIB) led to declines in Lhcb1 transcript abundance (Fig. 7). The transcripts were virtually undetectable 4 h following the addition of the inhibitor and remained so for the rest of incubation. The pattern of the changes in Lhcb1 transcript abundance was consistent with that of Chl a/cell (data not shown).
Figure 7.

Time course changes in Lhcb1 and rrna transcript levels in LL-acclimated cells following a shift to HL and 1.5 μm DBMIB additions to HL- and LL-acclimated cells. Top, Densitometric analysis of the time course changes in Lhcb1 level, normalized to rrna level, as percentage to the zero time point value. Bottom, Northern-blot analysis of total cellular RNA probed sequentially with Lhcb1 and rrna.
While the short-term changes of Lhcb1 transcript abundance are similar between those induced by DBMIB and a shift from LL to HL, the longer-term changes differ. During the acclimation from LL to HL, Lhcb1 transcript abundance partially recovered after 6 h, reaching a new and lower steady-state level. We further examined the Lhcb1 changes in HL cells transferred to LL with DBMIB added just before the light shift. Kinetic fluorescence measurements with a fast repetition rate (FRR) fluorometer (Kolber et al., 1998) indicated that DBMIB induced an immediate and dramatic reduction of the PQ pool, followed by a moderate recovery within 2 h. DBMIB effectively prevented Lhcb1 transcript abundance from increasing due to the reduced irradiance (Fig. 8). These results suggest that the signal from the reduced PQ pool repressed the up-regulation of Lhcb1 transcription that would have otherwise occurred had the shift been done in the absence of the inhibitor.
Figure 8.

Time course changes in Lhcb1 and rrna transcript levels in HL-acclimated cells following a shift to LL with or without the presence of 1.5 μm DBMIB. Top, Densitometric analysis of the time course changes in Lhcb1 level, normalized to rrna level, as percentage to the zero time point value. Bottom, Northern-blot analysis of total cellular RNA probed sequentially with Lhcb1 and rrna.
Effects of Various Treatments on the Redox Status of PQ Pools
Transient Chl fluorescence kinetics was measured to assess the oxidation rate of QA− and the redox status of the PQ pool. The changes in the percentages of oxidized PQ to the PQ pool following the additions of DCMU and DBMIB resembled those following light transitions from HL to LL and from LL to HL respectively (Fig. 9). Addition of DCMU increased oxidized PQ by approximately 50% more than that following the HL-to-LL transition. Addition of DBMIB reduced oxidized PQ by approximately 50% less than that by an LL-to-HL transition. Additions of nigericin, valinomycin, and antimycin A increased oxidation of the PQ pool; however, their effects appeared insignificant. Overall, the differences of effects on the redox status of PQ pools by different treatments between the short and the longer term were very small.
Figure 9.

Short- and longer-term effects of various treatments on the PQ pool redox status. Error bars represented 1× sd and n ranges from 3 to 12. Both HL and LL cultures were used in PET inhibitors and uncouplers treatments.
Photoacclimation Involves Changes of DNA-Binding Patterns in the Lhcb1 Promoter
Within 2 h after an LL-acclimated culture was transferred to the HL condition, Lhcb1 transcript levels decreased more than 10-fold (Fig. 4). The change in transcript abundance was concurrent with substantial decreases of LF1, LF2, and LF3 binding (Fig. 10A). Lhcb1 transcript levels reached the lowest level within 6 h following the shift and then quickly recovered to 40% of the initial level. The rapid recovery of Lhcb1 transcript abundance is concurrent with the dramatic increase of DNA-binding activities of LF1, LF2, and LF3 complexes (Figs. 4 and 10A). Lhcb1 transcript levels reached a new, 40% lower, steady-state level 12 h following the shift. EMSA revealed that after 24 h, the binding activities of all four complexes were much lower than those in LL cells. The strong positive correlation between Lhcb1 levels and the binding activities of LF1, LF2, and LF3 in the Lhcb1 promoter region are also closely coupled in time, suggesting that these binding complexes enhance Lhcb1 transcription. On the other hand, binding activities of LF factors (Fig. 10A) did not increase significantly while Lhcb1 transcript abundance nearly tripled following an HL-to-LL shift (Fig. 3). Instead, the increased Lhcb1 transcript abundance is coupled with a decreasing trend of the HLF-binding activity (Fig. 10A), suggesting a complicated role of HLF as a transcriptional repressor in regulating Lhcb1 transcription.
Figure 10.

EMSA of time course changes of DNA-binding activities in −247 to −218-bp region of Lhcb1 promoter following the light transitions (A), additions of PET inhibitors (B), and antimycin A with or without the presence of DCMU (C). All protein samples were subjected to a single-step ammonium sulfate precipitation at 50% saturation level. LL 0 h, LL-acclimated culture prior to the light transition; L-H, LL culture transferred to HL condition; HL 0 h, HL-acclimated culture prior to light transition; H-L, HL culture transferred to LL conditions; LL+DBMIB, LL culture treated with 1.5 μm DBMIB; HL+DCMU, HL culture treated with 200 nm DCMU; HL+antimycin A, HL culture treated with 1 μm antimycin A for 8 h; HL+antimycin A and DCMU, HL culture treated with 1 μm of antimycin A and 200 nm of DCMU for 8 h.
Effects of Photosynthetic Inhibitors and Uncouplers on DNA-Binding Activities in the Lhcb1 Promoter Region
We further investigated the relationship between Lhcb1 transcript abundance and the DNA-binding patterns in the presence of PET inhibitors and uncouplers. The DNA-binding activities of LF1, LF2, and LF3 declined substantially 3 h after the DCMU addition to HL-acclimated cells, followed by dramatic increases 9 h later (Fig. 10B). The Lhcb1 transcript abundance and the binding activities of LF1 and LF2 in the Lhcb1 promoter region are not only positively related but also closely coupled in time, once again suggesting that LF factors may enhance Lhcb1 transcription. The changes of binding activities of LF2 and LF3 following the addition of nigericin or valinomycin to HL cultures were similar but much smaller than those induced by DCMU (data not shown). While there were few changes in both the overall binding pattern and activities of individual complexes in cells treated by antimycin A alone, the binding activities of HLF, LF1, LF2, and LF3 were substantially enhanced in cells treated by both antimycin A and DCMU (Fig. 10C). The addition of 1.5 μm DBMIB to the LL culture led to an overall steady decrease of the binding activities of all complexes throughout the incubation, with HLF and LF1 activities virtually eliminated in the end, except that the binding of LF1 and LF2 increased at the 9-h time point (Fig. 10B). The overall patterns of changes in DNA-binding activities of all complexes are similar between those induced by the LL-to-HL shift and those by the DBMIB, yet their corresponding longer-term changes in Lhcb1 transcript abundance differ (Fig. 7).
Effects of Thiol Group-Modifying Agents on DNA-Binding Complexes
Phosphatases, including calf intestine alkaline phosphatase, λ-protein phosphatase, and protein Tyr phosphatase, did not significantly affect in vitro binding activities of any of the complexes (data not shown).
The effects of the thiol-oxidizing agent, azodicarboxylic acid bis-dimethylamide (diamide), and the thiol-modifying agent, N-ethyl maleimide (NEM), on the activities of the four DNA-binding complexes are shown in Figure 11. In vitro binding activities of HLF, LF1, LF2, and LF3 were all eliminated when whole-cell protein extracts were incubated with NEM before the addition of the oligo probes (Fig. 11). This was accompanied by the appearance of a new DNA-binding complex that migrated between LF1 and LF2. When NEM was added after the binding reaction was completed, however, the binding activity of HLF was enhanced while that of LF1 was reduced. Adding NEM after the completion of the binding reaction also resulted in less inhibition on the activities of other binding complexes than adding it prior to the binding reaction. The differences in sequence in which NEM was added affected DNA-binding activities, suggesting that the thiol group(s) in the complexes are not only directly involved in the binding in vitro, but can also enhance and reduce binding affinity of the HLF complex. While NEM dramatically inhibits in vitro binding activities of LF1, LF2, and LF3, diamide exhibits much less of an effect, especially if it is added after the binding reactions were completed (Fig. 11). However, diamide and NEM had a similar effect on HLF-binding activity. Taken together, the results of thiol group-modifying reagents treatment suggest that the assembly and binding of transcription factors to Lhcb1 promoter is thiol group dependent.
Figure 11.

Effects of thiol group-modifying reagents on DNA-binding activities of various DNA-binding factors in D. tertiolecta Lhcb1 promoter region from −247 to −218 bp. Whole-cell protein samples extracted from HL- and LL-acclimated cultures were subjected to a single-step ammonium sulfate precipitation at 50% saturation level.
DISCUSSION
Chloroplastic Signals Affecting DNA-Binding Activities in the Lhcb1 Promoter Region
Four DNA-binding complexes were detected when the Lhcb1 promoter fragments were incubated with whole-cell protein extracts isolated from D. tertiolecta. The binding activity of the HLF complex is generally stronger in HL cells than in LL cells based on the experimental results conducted on exactly the same culture as the one used by Escoubas et al. (1995); however, from the results presented in this study, which used a different culture (with the same clonal designation), we did not observe the same pattern or a consistently inverse relationship between HLF-binding and Lhcb1 mRNA levels, which suggests that the HLF complex may not be, by itself, a strong transcriptional repressor. In contrast, all three LF complexes are more abundant in cells grown under LL than those under HL. Their binding activities, when affected by light transitions or photosynthesis inhibitors, are not only positively related to the Lhcb1 transcript abundance but also kinetically concordant with the latter (Fig. 10). Such results suggest that LF complexes may be part of a chloroplast-to-nucleus signal transduction pathway and function as transcriptional enhancers of Lhcb1. The dynamic changes of their binding activities, as well as the similarities of the binding patterns between those induced by the light transitions and those induced by various inhibitors, strongly suggest that they respond not only to the redox status of the PQ pool, but also to other chloroplast-derived signals that converge at the transcriptional level.
Our EMSA results indicate that larger binding complexes, such as HLF and LF1, were formed when the same protein sample was incubated with the nonoverlapping promoter fragments in the 180-bp region (Fig. 2). Escoubas et al. (1995) proposed that a G-box-like element located in the 180-bp region (from −265 to −254 bp) might be the binding site for the repressor-like HLF complex. However, the HLF complex binds to promoter regions lacking this element, clearly indicating that a G-box is not required for binding of this complex. It is possible that the 180-bp promoter region contains a number of motifs that are part of a multipartite cis-regulatory unit with repeating subunits, providing binding sites for several regulatory factors. The larger binding complexes, HLF and LF1, may be functional entities consisting of multiple DNA-binding factors whose protein-to-protein interactions determine their binding sites. It is also likely that the smaller DNA-binding complexes may represent the partial or intermediate versions of larger complexes with different regulatory functions. In addition, the coexistence of binding activities by all complexes in short promoter fragments strongly suggests that the binding sites of these complexes are closely located or even partially overlapped and/or all binding complexes share the same DNA-binding protein factor(s). The binding sites of LF2 and LF3 appear to be located in the −247 to −218-bp promoter region (Oligo 5) that contains both CACT and GGAA boxes (Figs. 1 and 2). The CACT box and GGAA box are uninterrupted only in Oligo 5, and the strongest binding of LF2, as well as the exclusive binding of LF3, was detected only in that region of the promoter.
Light-mediated transcription of Lhc genes in higher plants is regulated by the binding of regulatory proteins to upstream promoter regions (Argüello-Astorga and Herrera-Estrella, 1998). Detailed molecular studies of trans-acting factors and cis-acting elements have focused primarily on phytochrome-mediated signaling, whereas the regulation via chloroplast redox signaling remains poorly understood. While the D. tertiolecta Lhcb1 promoter contains redundant control elements with identical or tightly linked light-responsive motifs (Fig. 1) similar to those in higher plants (Anderson and Kay, 1995; Terzaghi and Cashmore, 1995), the sequences of these tentative cis-elements differ from the GT-1 and G-boxes. In C. reinhardtii, the promoters of many light-responsive genes, including Lhcb1, also lack these cis-acting elements, which appear to be conserved in higher plants (Goldschmidt-Clermont and Rahire, 1986; Imbault et al., 1988; Hahn and Kück, 1999). The differences in promoter architecture suggest that the intermediate messenger, as well as possibly the mechanisms of light-regulation of photosynthetic gene expression, differ between higher plants and chlorophyte algae. However, the presence of a TCTAA site in Lhcb genes of D. tertiolecta, C. reinhardtii, and Arabidopsis suggests that some core chloroplastic signaling pathways may be highly conserved.
Multiple Signals Involved in Transcriptional Regulation Lhcb1
The nuclear-localized Lhcb genes are tightly regulated by light intensity in both algae and higher plants. Escoubas et al. (1995) demonstrated that the transient increase in the transcriptional rate from a shift of HL to LL was the result of enhanced transcription. Similar conclusions have also been reached for Dunaliella salina (Masuda et al., 2002, 2003) and C. reinhardtii (Jasper et al., 1991; Durnford et al., 2003). Although plastid regulation of Lhcb1 expression has been shown to occur at a transcriptional level (for review, see Gray et al., 2003), posttranscriptional regulatory mechanisms also may be involved (Hermsmeier et al., 1994; Durnford et al., 2003). Results of our northern blots revealed substantial and rapid decreases of the Lhcb1 transcript levels in D. tertiolecta cells following the additions of DCMU (Fig. 3), DBMIB (Fig. 7), and nigericin and valinomycin (Fig. 5), as well as the shift to HL conditions (Fig. 4). These precipitous drops cannot be explained simply by a decrease in transcription rate and the subsequent dilution of existing transcripts due to cell division. Although the stability of Lhcb1 transcripts differs little between HL- and LL-acclimated cells (Escoubas et al., 1995; Masuda et al., 2002), our results strongly suggest that there is enhanced degradation of Lhcb1 mRNA immediately following the addition of inhibitors/uncouplers or a shift to the HL conditions. This phenomenon was reported in C. reinhardtii (Durnford et al., 2003) and further supported in a study on D. salina in which a significant decrease in the half-life of Lhcb transcripts was detected immediately following an LL-to-HL shift (Masuda et al., 2003). The PQ pool redox state has also been suggested to regulate mRNA stability of the nuclear-encoded PetE gene in higher plants (Sullivan and Gray, 2002). Hence, mRNA degradation may also contribute to the short-term down-regulation of Lhcb1 expression in D. tertiolecta.
On longer time scales however, the redox state of the PQ pool clearly regulates the transcription of Lhcb1 gene. DBMIB always results in both short-term and longer-term decreases in Lhcb1 transcript levels in both HL and LL culture (Fig. 7) and prevented increases in Lhcb1 transcription during HL-to-LL transitions (Fig. 8). Transient fluorescence measurements indicated that in all DBMIB treatments, the PQ pool was rapidly reduced within the first 1 to 2 h before some recovery (Fig. 9), suggesting that the reduced PQ pool and/or cytochrome b6f are among the chloroplastic signals responsible for down-regulation of Lhcb1 transcription. DCMU, which always led to an oxidized PQ pool, induced increases in both Lhcb1 transcript levels and Chl synthesis 6 to 8 h following the addition of the inhibitor (Fig. 3). Although the short-term decline in Lhcb1 transcript abundance immediately following DCMU addition could be mediated by a different chloroplastic signal and/or changes in transcript stability, the longer-term DCMU effects on Lhcb1 transcription and Chl synthesis phenotypically resemble those by an HL-to-LL shift. These results suggest that a signal initiated from the oxidized PQ pool regulates longer-term photoacclimative responses.
There is, however, strong evidence that the redox state of the PQ pool is not the only photosynthetic signal that regulates Lhcb1 transcription, especially in the short term. Despite the fact that DCMU additions always resulted in a rapidly oxidized PQ pool (Fig. 9), both Lhcb1 transcript levels (Fig. 3) and Chl synthesis decreased substantially within the first 4 to 6 h (data not shown). Furthermore, when added to an LL culture prior to a shift to HL, DCMU failed to prevent decreases in both Lhcb1 transcript level (Fig. 4) and Chl synthesis, although the inhibitor prevented the PQ pool from being reduced by the elevated light intensity. DCMU's inability to prevent Lhcb genes from being down-regulated by HL was also reported in C. reinhardtii (Teramoto et al., 2002). Together, these results unequivocally demonstrate that the PQ pool redox status is not the sole signal involved in regulating Lhcb1 transcription in the short term; either the signal from the PQ redox state is overwritten by the other signal(s) or the PQ pool signal belongs to a longer-term chloroplastic signaling pathway, or both. Changes in ΔpH have been suggested as a rapid and powerful photosynthetic signal initiator (Horton et al., 1996; Wilson and Huner, 2000). It is conceivable that DCMU addition resulted in an immediate decrease in membrane potential, manifested by ΔpH, Δψ, and ATP production, triggering signals to rapidly down-regulate Lhcb1 transcription. While such negative signals may overwrite the up-regulation redox signals from the oxidized PQ pool, the balance is reversed in the longer term as the redox state of the latter electron carrier becomes dominant.
Like DCMU, addition of nigericin and valinomycin to LL and HL cultures resulted in sizable decreases in Lhcb1 transcript levels within the first 2 h, which then recovered and climbed to a new steady-state level at about 50% higher than the previous level (Fig. 5). Since nigericin uncouples photophosphorylation by dissipating ΔpH while valinomycin eliminates Δψ, the similarities of their impact on Lhcb1 transcription clearly indicate that their regulation of Lhcb1 transcription may be at least partially achieved via photophosphorylation. The initial down-regulation of Lhcb1 expression may be resulted from an immediate decrease of ΔpH/ATP production caused by the uncouplers. On the longer time scale, however, the uncouplers facilitate PQH2 oxidation (Rees et al., 1989; Hope et al., 1994), resulting in a more oxidized cytochrome b6f complex, which appears to up-regulate Lhcb1 transcription.
The role of ATP and/or ΔpH as possible chloroplast signal initiators is further supported by the experiments using antimycin A with or without DCMU. Our fluorescence measurements indicate that antimycin A slightly enhanced the oxidation of the PQ pool (Fig. 9); however, by itself, it had little overall effect on Lhcb1 transcript levels in HL cells (Fig. 6). It is clear that either antimycin A did not have any impact on mitochondria at the concentration used in this experiment or such impact did not affect Lhcb1 transcript abundance. On the contrary, when both DCMU and antimycin A were added, Lhcb1 transcript level rose rapidly from the beginning and quadrupled within 8 h (Fig. 6). The strong, positive, short-term impact of the combinational treatment by DCMU and antimycin A on Lhcb1 transcript levels suggests that, when linear electron transfer is inhibited, cyclic electron flow around PSI may play an important role in maintaining redox poise of cytochrome b6f as well as transthylakoid membrane potential. In the presence of DCMU, inhibition by antimycin A on PSI cyclic electron flow further down-regulates the ΔpH, thus leading to an oxidation state of the PQ pool. We hypothesize that the effect may be sensed via the redox state of cytochrome b6f complex, as this protein complex has direct access to both stromal and luminal sides of the thylakoid membrane (Kurisu et al., 2003). Indeed, a phenotype of photosynthetically competent mutants of Chlamydomas, which have impaired binding of PQH2 to cytochrome b6f is a chronic down-regulation of Lhcb expression (Falkowski and Chen, 2003). There is growing evidence that the cytochrome b6f complex, the redox status of which is determined not only by linear electron transport through the PQ pool but also by cyclic electron transfer around PSI, may play a critical role in the regulation of Lhcb1 expression via LHCBII kinase(s) (Pursiheimo et al., 2001; Yang et al., 2001; Kurisu et al., 2003; Zer and Ohad, 2003). Although the molecular mechanism through which the redox state of the PQ pool is transduced to the kinase is unknown, the implication of the quinol binding site, Qo, of the cytochrome b6f complex has been made both in vivo in C. reinhardtii (Zito et al.; 1999, Finazzi et al., 2001) and in thylakoid preparations from spinach (Vener et al., 1995, 1997).
While the combined effects of DCMU and antimycin A resemble those by DBMIB, there is one major difference between the two scenarios: the PQ pool and cytochrome b6f in DBMIB-treated cells are much more reduced than those in cells treated by both DCMU and antimycin A (Fig. 9). Their opposite effects on Lhcb1 transcription clearly point out the importance of the redox status of the PQ pool as a chloroplastic signal, as well as the notion that different signals regulate nuclear Lhcb1 transcription in a cooperative fashion. However, the effect of DCMU plus antimycin A largely resembles the acclimation to LL in terms of the simultaneous reduction of ΔpH/ATP and the oxidation of the PQ pool and potentially cytochrome b6f complex. The similar time course changes of Lhcb1 transcript abundance between the two scenarios suggest that a reduction in both linear intersystem electron flow and cyclic electron flow around PSI stimulates Lhcb1 transcription.
Role of Thiol Groups in DNA-Binding Activities
Our EMSA results revealed that the binding affinity of all four complexes clearly involves thiol group modification in vitro (Fig. 11), a phenomenon that has been reported for several transcription factors in higher plants and animal cells (for review, see Sun and Oberley, 1996). Based on the PET inhibitor and uncoupler results, it is very unlikely that such modifications occur in vivo via the ferredoxin/thioredoxin redox couple on the acceptor side of PSI (e.g. Danon and Mayfield, 1994). Escoubas et al. (1995) suggested, based on the phosphatase inhibitor studies in vivo, that a phosphorylation cascade, potentially coupled to the PQ redox-sensitive kinase/phosphatase system(s), relayed chloroplastic signals to the nucleus. Our results suggest, however, that the DNA-binding activities of the complexes examined are not altered by phosphatases (data not shown), and therefore the signal transduction pathway must involve additional mechanisms other than a phosphorylation cascade (Escoubas et al., 1995; Carlberg et al., 2003). Unless the binding factors can be translocated across the plastid envelope membrane, thiol group modification must occur in the cyotsol. Interestingly, thiol groups are highly sensitive to H2O2. Hydrogen peroxide is produced inside the chloroplasts of cells that are exposed to excessive light intensities, and it is readily diffusible out of chloroplasts. The H2O2 molecule constitutes a dominant chloroplast-derived reactive oxygen species, and it has been implicated in the signaling of excessive light intensity and regulating the expression of nuclear genes (Mullineaux and Karpinski; 2002; Wilson et al., 2003).
CONCLUSION
Although the lack of a transformation system limits our ability to modify the promoter region of Lhcb1 in D. tertiolecta, our results reveal, for the first time to our knowledge, three complexes that bind to the Lhcb1 promoter are closely coupled to the chloroplastic signals and are correlated with enhanced transcription of the gene in vivo. Sequence analysis of the targeted Lhcb1 promoter region revealed multiple and repetitive sequences as possible binding sites for these DNA-binding complexes. Results from our time course study of Lhcb1 transcription affected by light transitions and various photosynthesis inhibitors indicate the nuclear Lhcb1 gene expression may be cooperatively regulated by multiple chloroplastic signals. Specifically, our results suggest that the short-term expression of Lhcb1 is primarily regulated by transthylakoid membrane potential signals triggered by ΔpH/ATP pool, while the longer-term expression is primarily regulated by the redox signals initiated from the PQ pool.
MATERIALS AND METHODS
Materials
Restriction, modifying, and amplifying enzymes were purchased mainly from Promega (Madison, WI) and used according to vendor's instructions. Chemicals for making medium and various buffer solutions and reagents were purchased from Fisher Scientific (Hampton, NH) and Sigma (St. Louis). [α-32P]dATP and [γ-32P]ATP (3,000 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston) and AP Biotech (Piscataway, NJ). Primers and oligo DNA were synthesized and purified by Integrated DNA Technologies (Coralville, IA).
Cell Cultures
Dunaliella tertiolecta (clone DUN, Provasoli-Guillard Culture Collection, West Boothbay Harbor, ME) was grown in 20-L-size batch culture using filtered off-shore North Atlantic seawater amended with f/2 nutrients (Guillard and Ryther, 1962) and bubbled with air. Cultures were grown between 24°C and 28°C in an EGC growth chamber, under continuous light (cool-white) and maintained optically thin (<1 × 106 cells/mL) to avoid self shading. Cultures were routinely grown between 1,000 and 1,600 μmol quanta m−2 s−1 (HL) or approximately 80 to 120 μmol quanta m−2 s−1 (LL). Light intensity was controlled by a combination of the number of fluorescent tubes used as light source and through the use of black insect screening as a neutral-density filter. Cells were typically acclimated to a particular light intensity for 60 to 72 h prior to initiating experiments to ensure the cells were in the expected physiological state. Chl and cell density were analyzed as previously described (Escoubas et al., 1995).
RNA Extraction and Northern Blots
At each sampling point, 90 to 150 mL culture (3−10 × 107 cells) was harvested by centrifugation. The cells were immediately lysed in an RNA extraction buffer (1.2% [w/v] SDS, 30 mm EDTA, 50 mm Tris-HCl, pH 8.0, 220 mm NaCl, and 50 mm β-mercaptoethanol). The subsequent RNA processing was carried out following commonly used protocols with minor modification (Sambrook et al., 1989).
Northern-blot analysis was performed to determine the time course abundance of Lhcb1 (previously known as cab1; LaRoche et al., 1991) transcripts of D. tertiolecta cultures grown under different conditions. Northern-blot analysis was carried out following widely used protocols (Sambrook et al., 1989) with minor modifications. An equal amount of total RNA (approximately 5–10 μg) was fractionated by electrophoresis on a 1% (w/v) agarose gel with 1 m formaldehyde. The intensities of ethidium bromide-stained rRNA bands were visually examined to ensure that equal amounts of total RNA were loaded for all samples. The RNA was then transferred to a charged nylon membrane (Nytran, Schleicher & Schuell, Keene, NH) and crosslinked to the membrane using a Stratalinker (Stratagen, La Jolla, CA). 32P-labeled DNA probes were hybridized to the northern blots overnight at 42°C in the PerfectHyb solution (Ambion, Austin, TX). The blots were then washed twice with 2× SSC (0.3 m NaCl and 0.03 m sodium citrate) and 0.1% (w/v) SDS for 30 min at 42°C, rinsed with 0.2× SSC and 0.1% (w/v) SDS, and exposed to BioMax MR/MS films (Eastman-Kodak, Rochester, NY). A D. tertiolecta Lhcb1-specific fragment was amplified from the 3′-untranslated region of a D. tertiolecta Lhcb1 cDNA clone (cDNA4) with specific primers (forward primer, 5′-TAAATGCTGAAATGCAG-3′ and reverse primer, 5′-TAAGGCCAGTAAGAAT-3′). The fragment was labeled with [α32P]dATP using a random-primed labeling kit (Promega). To account for variations in total RNA loading, Lhcb1 signals were standardized to rRNA levels determined by hybridization of a Chlamydomonas reinhardtii rDNA probe (Clone P92) to the same northern blots whose Lhcb1 probes were stripped by incubating in stripping solution (0.2× SSC, 0.2% [w/v] SDS) at 95°C for 20 min followed at 65°C overnight. For densitometric analysis of signals, original northern autoradiograms were scanned into a computer using an Epson 1680 Pro Scanner (1,600 × 3,200 dpi hardware resolution, 48-bit color with 3.6 Dmax optical density, Epson America, Long Beach, CA). The public domain NIH Image program (developed at the United States National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image) was used to perform densitometric analysis of labeled bands. To minimize artifacts introduced during the northern assay and subsequent quantitative analysis, we compare only samples on the same gel/same batch of gels and plotted them on a relative scale (percentage of zero point value).
Protein Extraction and Measurement
Culture harvest and protein extraction were conducted essentially as previously described (Escoubas et al., 1995). A modified high-salt extraction buffer (25 mm HEPES-KOH, pH 8.0, 0.5 m KCl, 1 mm EDTA, 20 mm β-mercaptoethanol, 0.2 m Suc, and 1 mm magnesium acetate) was used. The cellular homogenate, clarified by ultracentrifugation at 200,000g for 30 min at 4°C, is routinely precipitated with (NH4)2SO4 at 50% (w/v) saturation. The precipitated proteins are collected by ultracentrifugation at 35,000g for 20 min at 4°C. The protein pellet is resuspended in a storage buffer (25 mm HEPES-KOH pH 7.9, 40 mm KCl, 5 mm β-mercaptoethanol, 1 mm EDTA, and 10% [v/v] glycerol), and desalted using Econo-Pac 10DG columns packed with Bio-Gel P-6G gel (Bio-Rad Laboratories, Hercules, CA). The protein concentration is determined in duplicates with the BCA assay kit (Pierce Chemical, Rockford, IL). A protease inhibitor mixture (1 mm phenylmethylsulfonyl fluoride, 5 mm ɛ-amino-n-caproic acid, 1 mm benzamidine chloride, 100 μm sodium metabisulfite, 1 mg/mL aporotinin, and 1× General Protease Inhibitor Cocktails by Sigma) was added to both extraction and storage buffers.
Preparation of Lhcb1 Upstream Fragments for EMSA
Our entire study of DNA-binding activities in D. tertiolecta Lhcb1 promoter was centered on a 180 bp region (−367 to −188 bp). Six 30-bp long double-stranded oligo DNA (Oligo 1–6) were constructed by annealing six pairs of commercially synthesized single-stranded oligos (Integrated DNA Technologies), which sequentially represent both strands of the 180-bp upstream region of Lhcb1. These oligo DNA were isolated via agarose gel electrophoresis and subsequently purified using Centrex MF-0.4 micro-centrifuge filters (Schleicher & Schuell) and Microcon-10 devices (Millipore, Bedford, MA). The purified oligo DNA were quantified and used as either labeled probes or unlabeled competitors in EMSA. The locations and sizes of the oligo DNA are presented in Figure 1.
EMSA
EMSA was used to study DNA-binding activities in the Lhcb1 upstream region of D. tertiolecta cultures grown under different conditions. In all EMSA, the labeled oligo DNA probes were added at over-saturating level so that the DNA protein-binding activities were limited only by the amount of the DNA-binding proteins presented in the binding mixtures. The binding reaction and the EMSA were performed based on previously described protocols with minor modifications (Escoubas et al., 1995). For competition EMSA, the competitors were added together with the probes. Treatment with the thiol-modifying agents was carried out essentially as described in Jiang et al. (1997). Treatment with the thiol-oxidizing agent diamide (Sigma) was conducted by incubating crude protein extract in the binding buffer for 1 h at 37°C in the presence of 1 mm diamide either before or after a 40 min incubation with labeled promoter fragments. The same procedures were followed for treatment with 1 mm thiol group modifying agent NEM (Sigma). To ensure a proper comparison, the same amount of total protein samples extracted from different cultures from the same experiment was used in each binding reaction. For all physiological experiments, EMSA were done using protein samples obtained from a single 50% (w/v) (NH4)2SO4 cut. The results from the incubation of the protein samples with Oligo 5 are presented since the activities of all four complexes could be clearly detected only with this probe.
Light Transition Studies
The light transition studies were conducted to examine the changes of DNA-binding activities in the Lhcb1 upstream region in responding to the change of light intensities. Two one-step transition experiments from LL (70–100 μmol quanta m−2 s−1) to HL (1,200–1,400 μmol quanta m−2 s−1) and from HL-to-LL transitions were carried out. For each transition experiment, 20-L batch culture of D. tertiolecta was grown and acclimated to the initial light intensity. At the beginning of the experiment, the culture was equally divided into two 10-L subcultures, with one remaining under the initial light conditions while the other was transferred to a different light condition. The incubation lasted for 8 h following the light shift. Samples for Chl, cell density, and RNA analysis were taken at the mid and end point of the incubation. The biomass from each subculture was harvested for protein extraction at the end of the experiment.
Inhibitor Studies
Results from preliminary experiments based on measurements of variable Chl fluorescence, Chl, and cell density were used to choose the proper concentrations for each inhibitor, ensuring their effective inhibition on photosynthesis while still allowing cells to grow. Stock solutions of photosynthetic electron transfer inhibitors DCMU and DBMIB were made in dimethyl sulfoxide. Stock solutions of photophosphorylation uncouplers nigericin and valinomycin were made in ethanol, as was the PSI cyclic electron transfer inhibitor antimycin A. Based on the dose response analyses, we used the following concentration for each inhibitor: DCMU, 200 nm; DBMIB, 1 μm initial with repletion of 250 nm every 4 to 5 h to remedy the labile nature of DBMIB; nigericin, 400 nm; valinomycin, 2 μm; and antimycin A, 1 μm. At the zero time of each inhibitor experiment, batch cultures of D. tertiolecta acclimated to HL or LL conditions were divided into the treatment subculture that receives the inhibitor and the control subculture that receives the solvent (in which the inhibitors stock solutions were made) with the level identical to what was added to the treatment subculture. Variable fluorescence of each subculture was monitored frequently throughout the entire incubation using a pulse amplitude-modulated fluorometer to ensure the effectiveness of the inhibitors on photosynthetic electron transfer. Samples for Chl, cell density, RNA, and protein were taken according to predetermined time intervals.
For the combination treatments involving both light transitions and inhibitor addition, the inhibitors were added to the cultures just prior to the light shift.
FRR Fluorometry
The photosynthetic performance and redox status of the PQ pool were assessed using the FRR fluorometer (Kolber et al., 1998) with a modified detection unit (large area avalanche photodiode detector, Advanced Photonix, Camarillo, CA). The instrument generates a train of short (0.6 μs) blue (470 nm) flashlets in the microsecond-to-millisecond timescale. Electron transport elicited by these light pulses induces transient changes in Chl a fluorescence emission reflecting redox and light acclimation status of the photosynthetic machinery. The FRR Chl fluorescence transient was recorded in the dark, and the fluorescence parameters Fv/Fm and σ470nm (functional cross section of photosynthetic units) were determined as described previously (Kolber et al., 1998). The cells in the measuring chamber were then exposed to continuous background irradiance simulating the light conditions used in the experiments for 1 min, and the second FRR fluorescence transient was recorded. The oxidation status of PQ pool was estimated as 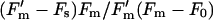 , where Fs is the steady-state fluorescence intensity and
, where Fs is the steady-state fluorescence intensity and 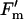 is the maximum fluorescence in the light.
is the maximum fluorescence in the light.
Acknowledgments
We are very grateful to Ms. Mary Ann Tran, Mr. Carlos Gonzales, and Ms. Katerina Zrotalova for participation in the experiments. We thank Drs. Mario Giordano, Yoram Gerchman, and Yi Sun for helpful discussions. We are indebted to Mr. K. Wyman for technical help, to Dr. Dan Tchernov for graphics preparation, and to Ms. Emmeline Romana and Ivy Jones for administrative support.
This work was supported by the U.S. Department of Energy (contract no. DEAC02–76CH00016).
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.104.038919.
References
- Anderson JM, Chow WS, Goodchild DJ (1995) The grand design of photosynthesis: acclimation of the photosynthetic apparatus to environmental cues. Photosynth Res 46: 129–139 [DOI] [PubMed] [Google Scholar]
- Anderson S, Kay S (1995) Functional dissection of circadian clock- and phytochrome-regulated transcription of the Arabidopsis CAB2 Gene. Proc Natl Acad Sci USA 92: 1500–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argüello-Astorga G, Herrera-Estrella L (1998) Evolution of light-regulated plant promoters. Annu Rev Plant Physiol Plant Mol Biol 49: 525–555 [DOI] [PubMed] [Google Scholar]
- Baker NR, Markwell JP, Thornber JP (1982) Adenine nucleotide inhibition of phosphorylation of the light-harvesting chlorophyll a/b-protein complex. Photobiochem Photobiophys 4: 211–217 [Google Scholar]
- Bendall DS, Manasse RS (1995) Cyclic photophosphorylation and electron transport. Biochim Biophys Acta 1229: 23–38 [Google Scholar]
- Bradbeer J, Atkinson YB, Börner T, Hagemann R (1979) Cytoplasmic synthesis of plastid polypeptides may be controlled by plastid-synthesised RNA. Nature 279: 816–817 [Google Scholar]
- Bulté L, Gans P, Rebéillé F, Wollman FA (1990) ATP control on state transitions in vivo in Chlamydomonas reinhardtii. Biochim Biophys Acta 1016: 72–80 [Google Scholar]
- Carlberg I, Hansson M, Kieselbach T, Schroder WP, Andersson B, Vener AV (2003) A novel plant protein undergoing light-induced phosphorylation and release from the photosynthetic thylakoid membranes. Proc Natl Acad Sci USA 100: 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandok MR, Sopory SK, Oelmüller R (2001) Cytoplasmic kinase and phosphatase activities can induce PsaF gene expression in the absence of functional plastids: evidence that phosphorylation/dephosphorylation events are involved in interorganellar cross talk. Mol Gen Genet 264: 819–826 [DOI] [PubMed] [Google Scholar]
- Danon A, Mayfield SP (1994) Light-regulated translation of chloroplast messenger RNAs through redox potential. Science 266: 1717–1719 [DOI] [PubMed] [Google Scholar]
- Demmig-Adams B, Adams I, William W (1996) The role of xanthophyll cycle carotenoids in the protection of photosynthesis. Trends Plant Sci 1: 21–26 [Google Scholar]
- Depège N, Bellafiore S, Rochaix J-D (2003) Role of chloroplast protein kinase Stt7 in LHCII phosphorylation and state transition in Chlamydomonas. Science 299: 1572–1575 [DOI] [PubMed] [Google Scholar]
- Durnford DG, Falkowski PG (1997) Chloroplast redox regulation of nuclear gene transcription during photoacclimation. Photosynth Res 53: 229–241 [Google Scholar]
- Durnford DG, Price JA, McKim SM, Sarchfield ML (2003) Light-harvesting complex gene expression is controlled by both transcriptional and post-transcriptional mechanisms during photoacclimation in Chlamydomonas reinhardtii. Physiol Plant 118: 193–205 [Google Scholar]
- Escoubas J-M, Lomas M, LaRoche J, Falkowski PG (1995) Light intensity regulation of cab gene transcription is signaled by the redox state of the plastoquinone pool. Proc Natl Acad Sci USA 92: 10237–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowski PG, Chen Y-B (2003). Photoacclimation of light harvesting systems in eucaryotic algae. In BR Green and WW Parson, eds, Light-Harvesting Antennas in Photosynthesis. Kluwer Academic Publishers, Dordrecht, The Netherlands
- Falkowski PG, LaRoche J (1991) Acclimatation to spectral irradiance in algae (minireview). J Phycol 27: 8–14 [Google Scholar]
- Falkowski PG, Wyman K, Ley AC, Mauzerall DC (1986) Relationship of steady-state photosynthesis to fluorescence in eucaryotic algae. Biochim Biophys Acta 849: 183–192 [Google Scholar]
- Fernyhough P, Foyer CH, Horton P (1984) Increase in the level of thylakoid protein phosphorylation in maize mesophyll chloroplasts by decrease in the transthylakoid pH gradient. FEBS Lett 176: 133–138 [Google Scholar]
- Finazzi G, Zito F, Barbagallo RP, Wollman F-A (2001) Contrasted effects of inhibitors of cytochrome b6f complex on state transitions in Chlamydomonas reinhardtii: the role of Qo site occupancy in LHCII kinase activation. J Biol Chem 276: 9770–9774 [DOI] [PubMed] [Google Scholar]
- Fujita Y, Ohki K, Murakami A (1987) Chromatic regulation of photosystem composition in the cyanobacterial photosynthetic system: kinetic relationship between change of photosystem composition and cell proliferation. Plant Cell Physiol 28: 227–234 [Google Scholar]
- Goldschmidt-Clermont M, Rahire M (1986) Sequence, evolution and differential expression of the two genes encoding variant small subunits of ribulose bisphosphate carboxylase/oxygenase in Chlamydomonas reinhardtii. J Mol Biol 191: 421–432 [DOI] [PubMed] [Google Scholar]
- Gray JC, Sullivan JA, Wang J-H, Jerome CA, MacLean D (2003) Coordination of plastid and nuclear gene expression. Philos Trans R Soc Lond B Biol Sci 358: 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillard RRL, Ryther JH (1962) Studies of marine planktonic diatoms: I Cyclotella nana Hustedt, and Detonula confervaces (Cleve) Gran. Can J Microbiol 8: 229–239 [DOI] [PubMed] [Google Scholar]
- Hahn D, Kück U (1999) Identification of DNA sequences controlling light- and chloroplast-dependent expression of the lhcb1 gene from Chlamydomonas reinhardtii. Curr Genet 34: 459–466 [DOI] [PubMed] [Google Scholar]
- Hermsmeier D, Schulz R, Senger H (1994) Formation of light-harvesting complexes of photosystem II in Scenedesmus. Planta 193: 406–412 [Google Scholar]
- Hope AB, Valente P, Matthews DB (1994) Effects of pH on the kinetics of redox reaction in and around the cytochrome bf complex in an isolated system. Photosynth Res 42: 111–120 [DOI] [PubMed] [Google Scholar]
- Horton P, Ruban AV, Walters RG (1996) Regulation of light harvesting in green plants. Annu Rev Plant Physiol Plant Mol Biol 47: 655–684 [DOI] [PubMed] [Google Scholar]
- Huner NPA, Oquist G, Sarhan F (1998) Energy-balance and acclimation to light and cold. Trends Plant Sci 3: 224–230 [Google Scholar]
- Imbault P, Wittemer C, Johanningmeier U, Jacobs JD, Hopewell SH (1988) Structure of the Chlamydomonas reinhardtii cabII-1 gene encoding a chlorophyll-a/b-binding protein. Gene 73: 397–407 [DOI] [PubMed] [Google Scholar]
- Jasper F, Quednau B, Kortenjann M, Johanningmeier U (1991) Control of cab gene expression in synchronized Chlamydomonas reinhardtii cells. J Photochem Photobiol B 11: 139–150 [DOI] [PubMed] [Google Scholar]
- Jiang F, Mannervik B, Bergman B (1997) Evidence for redox regulation of the transcription factor NtcA, acting both as an activator and a repressor, in the cyanobacterium Anabaena PCC 7120. Biochem J 327: 513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joliot P, Joliot A (2002) Cyclic electron transfer in plant leaf. Proc Natl Acad Sci USA 99: 10209–10214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinski S, Escobar C, Karpinska B, Creissen G, Mullineaux PM (1997) Photosynthetic electron transport regulates the expression of cytosolic ascorbate peroxidase genes in Arabidopsis during excess light stress. Plant Cell 9: 627–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinski S, Reynolds H, Karpinska B, Creissen G, Mullineaux PM (1999) Systemic signaling and acclimation in response to excess excitation energy in Arabidopsis. Science 284: 654–657 [DOI] [PubMed] [Google Scholar]
- Kolber ZS, Prasil O, Falkowski PG (1998) Measurements of variable chlorophyll fluorescence using fast repetition rate techniques: defining methodology and experimental protocols. Biochim Biophys Acta 1367: 88–106 [DOI] [PubMed] [Google Scholar]
- Kovács L, Wiessner W, Kis M, Nagy F, Mende D, Demeter S (2000) Short- and long-term redox regulation of photosynthetic light energy distribution and photosystem stoichiometry by acetate metabolism in the green alga, Chlamydobotrys stellata. Photosynth Res 65: 231–247 [DOI] [PubMed] [Google Scholar]
- Kurisu G, Zhang H-M, Smith JL, Cramer WA (2003) Structure of the cytochromeb6f complex of oxygenic photosynthesis: tuning the cavity. Science 302: 1009–1014 [DOI] [PubMed] [Google Scholar]
- LaRoche J, Mortain-Bertrand A, Falkowski PG (1991) Light intensity-induced changes in cab mRNA and light harvesting complex II apoprotein levels in the unicellular chlorophyte Dunaliella tertiolecta. Plant Physiol 97: 147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Polle JEW, Melis A (2002) Biosynthesis and distribution of chlorophyll among the photosystems during recovery of the green alga Dunaliella salina from irradiance stress. Plant Physiol 128: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Tanaka A, Melis A (2003) Chlorophyll antenna size adjustments by irradiance in Dunaliella salina involve coordinate regulation of chlorophyll a oxygenase (CAO) and Lhcb gene expression. Plant Mol Biol 51: 757–771 [DOI] [PubMed] [Google Scholar]
- Maxwell DP, Laudenbach DE, Huner NPA (1995) Redox regulation of light-harvesting complex II and cab mRNA abundance in Dunaliella salina. Plant Physiol 109: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montané M-H, Tardy F, Kloppstech K, Havaux M (1998) Differential control of xanthophylls and light-induced stress proteins, as opposed to light-harvesting chlorophyll a/b proteins, during photosynthetic acclimation of barley leaves to light irradiance. Plant Physiol 118: 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullineaux P, Karpinski S (2002) Signal transduction in response to excess light: getting out of the chloroplast. Curr Opin Plant Biol 5: 43–48 [DOI] [PubMed] [Google Scholar]
- Oswald O, Martin T, Dominy PJ, Graham IA (2001) Plastid redox state and sugars: interactive regulators of nuclear-encoded photosynthetic gene expression. Proc Natl Acad Sci USA 98: 2047–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannschmidt T, Nilsson A, Allen JF (1999) Photosynthetic control of chloroplast gene expression. Nature 397: 625–628 [Google Scholar]
- Pfannschmidt T, Schutze K, Brost M, Oelmüller R (2001) A novel mechanism of nuclear photosynthesis gene regulation by redox signals from the chloroplast during photosystem stoichiometry adjustment. J Biol Chem 276: 36125–36130 [DOI] [PubMed] [Google Scholar]
- Pursiheimo S, Mulo P, Rintamäki E, Aro E-M (2001) Coregulation of light-harvesting complex II phosphorylation and Lhcb accumulation in winter rye. Plant J 26: 317–327 [DOI] [PubMed] [Google Scholar]
- Rees D, Young A, Noctor G, Britton G, Horton P (1989) Enhancement of the [Delta]pH-dependent dissipation of excitation energy in spinach chloroplasts by light-activation: correlation with the synthesis of zeaxanthin. FEBS Lett 256: 85–90 [Google Scholar]
- Rintamäki E, Martinsuo P, Pursiheimo S, Aro E-M (2000) Cooperative regulation of light-harvesting complex II phosphorylation via the plastoquinol and ferredoxin-thioredoxin system in chloroplasts. Proc Natl Acad Sci USA 97: 11644–11649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodermel S (2001) Pathways of plastid-to-nucleus signaling. Trends Plant Sci 6: 471–478 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- Strand A, Asami T, Alonso J, Ecker JR, Chory J (2003) Chloroplast to nucleus communication triggered by accumulation of Mg-protoporphyrinIX. Nature 421: 79–83 [DOI] [PubMed] [Google Scholar]
- Sukenik A, Bennett J, Falkowski PG (1988) Changes in the abundance of individual apoproteins of light-harvesting chlorophyll a/b-protein complexes of photosystem I and II with growth irradiance in the marine chlorophyte Dunaliella tertiolecta. Biochim Biophys Acta 932: 206–215 [Google Scholar]
- Sullivan JA, Gray JC (2002) Multiple plastid signals regulate the expression of the pea plastocyanin gene in pea and transgenic tobacco plants. Plant J 32: 763–774 [DOI] [PubMed] [Google Scholar]
- Sun Y, Oberley LW (1996) Redox regulation of transcriptional activators. Free Radic Biol Med 21: 335–348 [DOI] [PubMed] [Google Scholar]
- Surpin M, Larkin RM, Chory J (2002) Signal transduction between the chloroplast and the nucleus. Plant Cell (Suppl) 14: s327–s338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto H, Nakamori A, Minagawa J, Ono T-a (2002) Light-intensity-dependent expression of Lhc gene family encoding light-harvesting Chlorophyll-a/b proteins of photosystem II in Chlamydomonas reinhardtii. Plant Physiol 130: 323–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzaghi WB, Cashmore AR (1995) Light-regulated transcription. Annu Rev Plant Physiol Plant Mol Biol 46: 445–474 [Google Scholar]
- Vener AV, van Kan PJM, Gal A, Andersson B, Ohad I (1995) Activation/deactivation cycle of redox-controlled thylakoid protein phosphorylation. J Biol Chem 270: 25225–25232 [DOI] [PubMed] [Google Scholar]
- Vener AV, van Kan PJM, Rich PR, Ohad I, Andersson B (1997) Plastoquinol at the quinol oxidation site of reduced cytochrome bf mediates signal transduction between light and protein phosphorylation: thylakoid protein kinase deactivation by a single-turnover flash. Proc Natl Acad Sci USA 94: 1585–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KE, Huner NPA (2000) The role of growth rate, redox state of the plastoquinone pool and the trans-thylakoid pH in photoacclimation of Chlorella vulgris to growth irradiance and temperature. Planta 212: 93–102 [DOI] [PubMed] [Google Scholar]
- Wilson KE, Sieger SM, Huner NPA (2003) The temperature-dependent accumulation of Mg-protoporphyrin IX and reactive oxygen species in Chlorella vulgaris. Physiol Plant 119: 126–136 [Google Scholar]
- Wollman F-A (2001) New EMBO member's review: state transitions reveal the dynamics and flexibility of the photosynthetic apparatus. EMBO J 20: 3623–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D-H, Andersson B, Aro E-M, Ohad I (2001) The redox state of the plastoquinone pool controls the level of the light-harvesting chlorophyll a/b binding protein complex II (LHC II) during photoacclimation. Photosynth Res 68: 163–174 [DOI] [PubMed] [Google Scholar]
- Zer H, Ohad I (2003) Light, redox state, thylakoid-protein phosphorylation and signaling gene expression. Trends Biochem Sci 28: 467–470 [DOI] [PubMed] [Google Scholar]
- Zito F, Finazzi G, Delosme R, Nitschke W, Picot D, Wollman F-A (1999) The Qo site of cytochrome b6f complexes controls the activation of the LHCII kinase. EMBO J 18: 2961–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]