

Abstract
Flowers are icons in developmental studies of complex structures. The vast majority of 250,000 angiosperm plant species have flowers with a conserved organ plan bearing sepals, petals, stamens, and carpels in the center. The combinatorial model for the activity of the so-called ABC homeotic floral genes has guided extensive experimental studies in Arabidopsis thaliana and many other plant species. However, a mechanistic and dynamical explanation for the ABC model and prevalence among flowering plants is lacking. Here, we put forward a simple discrete model that postulates logical rules that formally summarize published ABC and non-ABC gene interaction data for Arabidopsis floral organ cell fate determination and integrates this data into a dynamic network model. This model shows that all possible initial conditions converge to few steady gene activity states that match gene expression profiles observed experimentally in primordial floral organ cells of wild-type and mutant plants. Therefore, the network proposed here provides a dynamical explanation for the ABC model and shows that precise signaling pathways are not required to restrain cell types to those found in Arabidopsis, but these are rather determined by the overall gene network dynamics. Furthermore, we performed robustness analyses that clearly show that the cell types recovered depend on the network architecture rather than on specific values of the model's gene interaction parameters. These results support the hypothesis that such a network constitutes a developmental module, and hence provide a possible explanation for the overall conservation of the ABC model and overall floral plan among angiosperms. In addition, we have been able to predict the effects of differences in network architecture between Arabidopsis and Petunia hybrida.
INTRODUCTION
Monod and Jacob (1961) first proposed that complex networks of gene interactions regulate cell differentiation. The first formal models of genetic regulation of cell differentiation anticipated that real biological genetic networks would be too complex to be analyzed without the use of formal mathematical and/or computational tools (Kauffman, 1969). However, because the early models made some assumptions that were biologically unrealistic (Aldana and Cluzel, 2003), experimentalists largely ignored them. Relatively complete genetic descriptions of developmental programs are now available in several model organisms, providing the necessary inputs for developing biologically realistic dynamic models of gene regulatory networks in cell differentiation (Hasty et al., 2002). Such models should aid at building a formal framework for studies of developmental mechanisms and their evolution.
Dynamical network models lead to formal mechanistic explanations of developmental mechanisms and, thus, of the way genetic interactions translate into phenotypic traits. This will enable novel predictions of the effects of specific perturbations of regulatory networks that in turn may form a basis for investigating how network architecture may constrain morphological evolutionary patterns (Salazar-Ciudad and Jernvall, 2002). Dynamic networks attain steady states that are formally referred to as attractors. In gene networks, each attractor corresponds to a combination of gene expression states that specifies a particular cell type (Kauffman, 1993). Hence, a single network architecture that functions as a dynamic circuit may lead to multiple cell types, each characterized by a different set of steady gene expression states. Network models have now been developed for cell-fate determination in Arabidopsis thaliana (e.g., Mendoza and Alvarez-Buylla, 1998, 2000) and segment-polarity determination in Drosophila melanogaster (e.g., Mjolsness et al., 1991; von Dassow et al., 2000; Albert and Othmer, 2003). Recent studies have also started to show that the dynamics of biological gene networks are robust to quantitative gene function alterations (von Dassow et al., 2000). Such robustness may be responsible in part for morphological pattern conservation.
A conserved floral plan is found among the vast majority of angiosperms with four main types of organs (sepals, petals, stamens, and carpels) and corresponding primordial cell types that appear in a stereotypical spatio-temporal pattern: sepals first in the outside, then petals, stamens, and last carpels in the center of the flower (Rudall, 1987). Studies at the molecular level are also suggesting that there is an overall conservation among genes and most of their interactions in angiosperm flower organ determination (Ferrario et al., 2004). We explore here if the Arabidopsis network of gene interactions of floral organ cell fate determination is a robust developmental module.
Experimental studies in Arabidopsis and Antirrhinum majus led to the ABC combinatorial model of gene expression states that predicts the identity of floral organ primordia (Coen and Meyerowitz, 1991) and has guided extensive experimental studies in many plant species (Ferrario et al., 2004). According to this model, A class genes (APETALA1 [AP1] and AP2) specify sepal fate in the outer (first) floral whorl, A plus B genes (AP3 and PISTILLATA [PI]) determine petal development in the second whorl, B plus C genes determine stamens in the third whorl, and the C gene (AGAMOUS [AG]) alone determines carpel fate in the central (fourth) whorl (Coen and Meyerowitz, 1991). The ABC model, however, does not provide a dynamical explanation of how the steady state pattern of gene expression characteristics of each primordial floral cell type is attained and maintained through interactions among ABC and non-ABC genes.
Here, we integrate data on ABC and non-ABC genes in a simple, dynamical network model for Arabidopsis that extends and improves our previous efforts (Mendoza and Alvarez-Buylla, 1998; Mendoza et al., 1999). The new model exploits a more complete understanding of genetic components and interactions, and in contrast with our previous model, we now obtain steady states that are coherent with experimental data. To achieve this, we developed logical rules grounded on experimental data to capture the qualitative information that appears to form the basis of this regulatory gene network. This new approach avoids the circular criteria used before to estimate the gene interaction parameters (Mendoza and Alvarez-Buylla, 1998).
The logical rules for each gene in the network were derived from molecular genetic experimental data (see references in logical rules section). The global dynamical behavior of the network was analyzed by obtaining all steady states of gene expression for all possible initial conditions. Interestingly, we found that the model is canalized to developmental pathways that converge only to a few documented gene expression patterns rather than reaching a diverse range of gene expression states depending on initial conditions. The model also attained expression profiles coherent with experimental data when loss and gain of function mutants were simulated. Hence, these qualitatively documented interactions are sufficient to recover expression states that have been experimentally observed in inflorescence meristem cells and in sepal, petal, stamen, and carpel primordial cells, suggesting that these result from the gene network architecture rather than from the parameters of the gene interactions. This was further verified directly by simulating perturbed networks.
Thus, the model integrates in a formal manner the available data on gene interactions underlying cell-fate determination during floral organ formation. This model is intended to be useful to test the coherence of experimental data and to hypothesize testable gene interactions that remain to be discovered. Several of such predictions are presented here. Finally, the model provides a tool for exploring the relative conservation of the basic structure of flowers among angiosperms, and we demonstrate that it can successfully predict the effects of evolutionary differences in the network architecture between Arabidopsis and Petunia hybrida.
RESULTS
The Model
We derived a discrete network model of cell-fate determination during floral organ primordia formation in Arabidopsis flower development. All parameters of the model are grounded on available experimental data for this species (see Methods; Figures 1 to 4). The updating of the gene's expression state depends on the tables of logical rules and on the states of the input genes in the previous time step. For each initial condition, the system attains an attractor after a number of iterations. Attractors can either be of type “fixed point” (steady state), in which the expression state of each gene is fixed, or “limit cycles.” In the latter, two or more gene activation combinations alternate periodically. The set of initial conditions that lead to each attractor is referred to as its basin of attraction.
Figure 1.

Logical Rules for FT, LFY, TFL1, EMF1, and SEP.
The state of each network node (rightmost column in each table) depends on the combination of activity states of its input nodes (all other columns in each table). X represents any possible value. An asterisk denotes cases where subjective decisions had to be made and where we tested the alternative. The alternatives produced equivalent results to those obtained with the original values (see text). FT (A), LFY (B), TFL1 (C), EMF1 (D), and SEP (E).
Figure 2.

Logical Rules for AP1, AP2, FUL, AP3, and PI.
The state of each network node (rightmost column in each table) depends on the combination of activity states of its input nodes (all other columns in each table). X represents any possible value. Comparative symbols (< and >) are used when the relative values are important to determine the state of activity of the target node. AP1 (A), AP2 (B), FUL (C), AP3 (D), and PI (E).
Figure 3.

Logical Rules for AG and WUS.
The state of each network node (rightmost column in each table) depends on the combination of activity states of its input nodes (all other columns in each table). X represents any possible value. Comparative symbols (< and >) are used when the relative values are important to determine the state of activity of the target node. Asterisks denote cases where subjective decisions had to be made and where we tested the alternative. The alternatives produced equivalent results to those obtained with the original values (see text). AG (A) and WUS (B).
Figure 4.

Gene Network Architecture for the Arabidopsis Floral Organ Fate Determination.
Network nodes represent active proteins of corresponding genes, and the edges represent the regulatory interactions between node pairs (arrows are positive, and blunt-end lines are negative). Dashed lines are hypothetical interactions for which there is no experimental support (see logical rules). The network includes F-box proteins (UFO), membrane bound signaling molecules (TFL1 and FT), cofactors involved in transcriptional regulation (EMF1 and LUG), chromatin remodeling proteins (CLF), and transcription factors (all others). Interactions have been confirmed to be direct transcriptional regulations in a few cases (LFY on AG, Busch et al., 1999; LFY on AP1, Wagner et al., 1999), and the rest can either be direct or indirect and can be transcriptional or other. See Results and Methods for model details.
Logical Rules for Each Network Node and Experimental Support
For all network nodes (genes) with inputs we provide a table of logical rules in Figures 1 to 3 and summarize the experimental evidence from which these rules were derived. Each table in Figures 1 to 3 lists the numerical values of the node's activity state as a function of the activity states that the nodes from which it receives input had in a previous time step. Eight of the nodes are Boolean with two possible outcomes: 0 (off) and 1 (on); but for the other seven nodes, experimental data enabled us to establish three levels of expression, 0, 1, and 2, with 1 representing an intermediate level of expression. The rationale that we used to codify the available experimental data into the logical rules is explained in Methods, but we summarize here the experimental evidence from which the rules were derived.
FLOWERING LOCUS T
Double embryonic flower1 (emf1) flowering locus t (ft) mutants do not develop embryonic flowers typical of emf1 single mutants (Haung and Yang, 1998), suggesting that the lack of FT activity suppresses the emf1 phenotype because EMF1 represses FT. 35S:LFY 35S:FT double-transgenic plants flower immediately after germination (Kardailsky et al., 1999; Kobayashi et al., 1999), resembling the emf1 phenotype and also supporting that EMF1 represses FT (and LEAFY [LFY]; see below).
LFY
Double mutants of the weak emf1-1 allele and lfy-1 bear lfy-like flowers (Yang et al., 1995), suggesting that, for this trait, lfy is epistatic. These genes have antagonistic activities; hence, we infer that LFY is repressed by EMF1. The fact that in terminal flower1 (tfl1) mutant plants LFY is ectopically expressed in the shoot apex (Ratcliffe et al., 1999) suggests that TFL1 also represses LFY. We consider that when both TFL1 and EMF1 are inactive, LFY is fully active because LFY levels are highest during flower meristem determination when both former genes are inactive. In ap1 and ap1 cauliflower (cal) double mutants, LFY expression is reduced (Piñeiro and Coupland, 1998), and LFY is turned on earlier in 35S:AP1 plants than in the wild type (Liljegren et al., 1999), suggesting that LFY rapidly achieves its highest activity when AP1 is overexpressed. Finally, even though LFY:β-glucuronidase (GUS) expression is the same in the wild type and in fruitfull (ful) mutant plants, it is reduced in ful ap1 cal triple mutants relative to ap1 cal double mutant plants (Ferrándiz et al., 2000), suggesting that the role of FUL in LFY upregulation is important only when AP1 is inactive.
TFL1
In emf1-2 tfl1 double mutants, the emf1-2 mutation is epistatic (Chen et al., 1997). As these two genes do not have opposite functions, this result suggests that EMF1 protein is needed for TFL1 activity in wild-type Arabidopsis. In transgenic 35S:AP1 plants, the shoot apex is terminated in a flower resembling tfl1 mutants (Mandel and Yanofsky, 1995a), whereas TFL1 is severely diminished (Liljegren et al., 1999), and in ap1 cal double mutants TFL1 is ectopically expressed (Liljegren et al., 1999), suggesting that AP1 represses TFL1. The 35S:LFY plants also resemble the tfl1 mutant, and no TFL1 expression is observed in these plants (Ratcliffe et al., 1999), whereas TFL1 is ectopically expressed in lfy mutants (Liljegren et al., 1999). Finally, the tfl1-1 mutation partially suppresses the ap2-1 ap1-1 mutant phenotype (Schultz and Haughn, 1993; Shannon and Meeks-Wagner, 1993), suggesting that both AP1 and AP2 repress TFL1. Hence, TFL1 is repressed by LFY, AP1, and AP2.
EMF1
In double emf1-2 lfy mutants, emf1 is epistatic to lfy, and a 35S:LFY transgene enhances a weak emf1 allele phenotype (emf1-1) (Chen et al., 1997). Together, these results suggest that LFY is a repressor of EMF1.
SEPALLATA1-3
The SEPALLATA1-3 (SEP1-3) genes are fully expressed after floral induction (Savidge et al., 1995; Mandel and Yanofsky, 1998; Pelaz et al., 2000), but we are not aware of a direct inductor of these genes. Although the activator of these genes has not been discovered, we have considered that TFL1, which is a floral repressor, represses the SEP node (dotted t-bars in Figure 4). One or more undiscovered factors expressed in the inflorescence meristem and absent from the floral one or vice versa could play the role of TFL1 in regulating SEP genes, and the network results would not be affected.
AP1
AP1 mRNA is ectopically expressed in the inner two whorls of ag mutant flowers, where AG is normally expressed in wild-type Arabidopsis (Gustafson-Brown et al., 1994). In tfl1 mutants, AP1 is ectopically expressed in the basal lateral meristems and in N-terminal flowers (Gustafson-Brown et al., 1994), suggesting that TFL1 represses AP1. Also, AP1 is expressed in lfy mutants but not in 35S:TFL1 lfy transgenic plants (Ratcliffe et al., 1999), suggesting that TFL1 repression over AP1 is critical. The phenotypes of tfl1 ap1, tfl1 lfy ap1, and tfl1 lfy mutants further support the repression of TFL1 over AP1 (Shannon and Meeks-Wagner, 1993). It has been shown that LFY directly binds to the AP1 promoter and activates it (Wagner et al., 1999). Furthermore, AP1 expression is delayed in lfy-6 null mutants and is ectopic in 35S:LFY plants (Liljegren et al., 1999). In ft lfy double mutants, AP1 mRNA is absent, whereas in the respective single mutants there is AP1 expression, suggesting that at least one of these two genes needs to be present for AP1 activation (Ruiz-García et al., 1997).
AP2
The absence of petals in tfl1 ap2 double mutant flowers (Shannon and Meeks-Wagner, 1993) and the presence of these in tfl1 single mutants suggest that there is ectopic AP2 activity in the terminal flowers of tfl1 single mutants and that AP2 is repressed by TFL1.
FRUITFULL
FRUITFULL (FUL) is ectopically expressed in ap1 mutants (Ferrándiz et al., 2000), suggesting that FUL expression is repressed by AP1. Given that FUL has been shown to activate LFY (Ferrándiz et al., 2000), we would expect to find LFY in the tissues where FUL is expressed. However, whereas FUL is strongly expressed in inflorescence meristems, LFY is almost absent from there (Mandel and Yanofsky, 1995b; Ferrándiz et al., 2000). This suggests that there is an unidentified factor that impedes the activation of LFY by FUL in the inflorescence meristem and that the latter is nonfunctional in its capacity to activate LFY in this spatial domain. This interaction has to be at the posttranscriptional level because there is FUL mRNA in the inflorescence meristem. Here, we postulate that TFL1 could be such an inhibitor. It also is possible that other factors play this posttranscriptional inhibitory role. In any case, such missing factors seem to be key for determining a steady state of gene expression characteristic of inflorescence meristematic cells because when the negative posttranscriptional regulation of FUL by TFL1 is not considered, the nonfloral steady gene states disappear (see Supplemental Table 1 online). An alternative to postulating the negative regulation of TFL1 over FUL would be to modify the logical rule of LFY by nullifying the activation of LFY by FUL when TFL1 is on. Both approaches yield equivalent results (data not shown).
AP3
AP3 expression is reduced in lfy mutants (Weigel and Meyerowitz, 1993). In lfy ap1 double mutants, AP3 mRNA is not detected, but in ap1 mutants, AP3 is expressed as in wild-type plants (Hill et al., 1998). In lfy-6 mutants, no stamens or petals are formed, but they develop if an inducible form of LFY is introduced and induced (Wagner et al., 1999). All these data suggest that LFY activates AP3. Both LFY and UNUSUAL FLORAL ORGANS (UFO) have to be overexpressed to cause ectopic expression of AP3 (Parcy et al., 1998), also suggesting that UFO expression is needed for AP3 activation by LFY. LFY and UFO are important for the onset of AP3 expression, but AP3, as well as PI, expression is maintained through self-activation (Hill et al., 1998; Honma and Goto, 2000). In plants bearing a 35S:AP3:GR construct, AP3 is upregulated when induced with dexamethasone, supporting the notion that AP3 self-activates (Honma and Goto, 2000). However, additional cofactors are needed for AP3 self-activation.
The full activation of the B-function genes requires a tetramer with AP1 or AG, as well as SEP, PI, and AP3 proteins as shown by GUS activity in AP3:GUS 35S:PI 35S:AP3 35S:AP1 plants (Honma and Goto, 2001) and because ectopic PI, AP3, SEP, and AP1 expression is sufficient to transform leaves into petal-like organs (Pelaz et al., 2001). AG may substitute for AP1 in maintaining AP3 expression because cauline leaves of 35S:PI 35S:AP3 35S:SEP3 35S:AG are converted into stamen-like organs (Honma and Goto, 2001). Moreover, there is weaker GUS expression in the third whorl of ag-1 AP3:GUS flowers than in wild-type plants (Hill et al., 1998), further supporting that AG activates AP3. The inclusion of other factors (such as SEP, AP1, and AG) required for the self-activation of B genes is sufficient to avoid the unrealistic steady state with B genes and floral repressors turned on at the same time that was found in an earlier study (Mendoza and Alvarez-Buylla, 1998; Mendoza et al., 1999).
PI
We assume that only when AG or AP3 are active, PI expression is maintained because this gene is not active in sepals that are the only floral organs where both AG and AP3 are inactive. However, direct experimental evidence is still lacking to support this interaction. The level of PI mRNA and its domain of expression are severely reduced in lfy-6 null mutants, and there is no GUS expression in lfy PI:GUS early flowers (Honma and Goto, 2000). Moreover, lfy-6 null mutants lack petals and stamens, but when lfy-6 35S:LFY:GR is induced to express LFY ectopically, the wild-type phenotype is rescued (Wagner et al., 1999), supporting PI activation by LFY. Finally, PI self-activation requires the same conditions needed for AP3 self-activation (activity of AP3, PI, the SEP genes and either AP1 or AG; Honma and Goto, 2000, 2001).
AG
In strong ap2 mutant alleles, AG RNA accumulates in the four floral whorls, and in ap2-2 and ap2-9 alleles, medial 1st whorl organs are converted into carpels or carpelloid sepals, and 2nd whorl organs are absent (Deyholos and Sieburth, 2000), suggesting that AP2 represses AG. All strong wuschel (wus) alleles lack carpels and most stamens (Laux et al., 1996), suggesting that WUS upregulates AG. Moreover, in AP3:WUS transgenic plants, 2nd whorl organs are carpelloid stamens instead of petals (Lenhard et al., 2001; Lohmann et al., 2001), whereas in AP3:WUS ag plants, 2nd and 3rd whorl organs do not differentiate into carpelloid stamens (Lenhard et al., 2001). There are data that support that AG is repressed by LEUNIG (LUG) and CURLY LEAF (CLF). First whorl sepals are frequently carpelloid, second whorl organs are staminoid petals in lug or clf mutants, and in the lug ag or clf ag double mutants, the ag mutation is epistatic (Liu and Meyerowitz, 1995; Goodrich et al., 1997). Also, the amount of AG RNA is greater in lug than in wild-type plants (Liu and Meyerowitz, 1995), and in clf mutants, AG mRNA is detected in sepals (Goodrich et al., 1997). Because of the constitutive expression of LUG and CLF (Goodrich et al., 1997; Conner and Liu, 2000), we considered these genes to be always active for the wild-type simulation. AG misexpression is increased by the ap1 mutation in a lug or ap2 background, and AG is occasionally expressed in the outer whorls of ap1-1 mutants. Moreover, whorl 1 organs are sometimes carpelloid, and whorl 2 organs are staminoid in ap1 mutants (Liu and Meyerowitz, 1995), suggesting that AP1 also represses AG. The data above suggest that WUS activity is sufficient in Arabidopsis wild-type flowers to overcome the repression of AG by LUG, CLF, AP1, and AP2 because in AP3:WUS lines there is AG activity in the second floral whorl where LUG, CLF, AP1, and AP2 normally repress AG.
Normal stigmas and styles in terminal flowers in a lfy ap1 background are rescued if the tfl1 mutation is added (Shannon and Meeks-Wagner, 1993), supporting that AG is also repressed by TFL1. The LFY protein binds to an enhancer sequence in AG's first intron (Busch et al., 1999). Also in lfy-6 mutants, the onset of AG expression is delayed, and its final domain of expression is reduced (Weigel and Meyerowitz, 1993). In addition, the expression of a fusion of LFY to a strong activation domain produces increased and ectopic AG expression (Parcy et al., 1998). These facts indicate that LFY directly upregulates AG. Finally, we assumed that AG activates itself either directly or indirectly and that this positive feedback loop requires full AG activity (AG = 2) and SEP activity.
WUS
It has been experimentally established that AG represses WUS (Lenhard et al., 2001; Lohmann et al., 2001). We infer that SEP activity is required for WUS downregulation by AG because sep1 sep2 sep3 triple mutant plants bear indeterminate flowers (Pelaz et al., 2000). We also assume that WUS activity depends on WUS state of expression in a previous time step. We assumed this to avoid WUS activity in those system's states where AG is not active (e.g., the petals). SHOOT MERISTEMLESS may mediate WUS self-activation (Mayer et al., 1998).
The Arabidopsis Network Architecture: Novel Predictions on Gene Interactions
In Figure 4, we show the architecture of the floral organ cell fate determination network. Each node corresponds to the concentration of active or functional protein encoded by each gene. The edges represent the regulatory interactions between nodes. Arrows represent positive (activation) interactions, and blunt-end lines represent negative interactions (repression). In this article, when we refer to network architecture we are considering the connections among nodes, the sign of the regulatory interactions (both illustrated in Figure 4), and the relative importance of the input genes in determining the expression state of the target gene. The latter are precisely defined in the tables of logical rules given in Figures 1 to 3. Note that even though the network of gene interactions depicted in Figure 4 only shows either activation (positive) or repression (negative) regulatory interactions, the effect of these on the activation state of the downstream gene depends on the logical rules derived for each gene.
We have incorporated genes that codify for transcription factors (AG, PI, AP3, WUS, etc.; Coen and Meyerowitz, 1991; Mayer et al., 1998), F-box proteins such as UFO (Samach et al., 1999), likely membrane bound signaling molecules such as TFL1 and FT (Bradley et al., 1997; Simpson et al., 1999), positive or negative cofactors probably involved in transcription such as EMF1 (Aubert et al., 2001) and LUG (Conner and Liu, 2000), and CLF (Goodrich et al., 1997), which is a chromatin remodeling protein. To keep the coherence of available experimental data, we incorporated four interactions that have not been documented. These are indicated by dashed lines and constitute novel predictions of the model that should be tested experimentally.
The first assumption concerns the SEP1-3 genes. These are fully expressed after floral induction (Savidge et al., 1995; Mandel and Yanofsky, 1998; Pelaz et al., 2000), but to our knowledge, there is no evidence of positive or negative regulators of these genes that could explain their expression pattern. Therefore, in the model, we have considered that the floral repressor TFL1 negatively regulates these genes. However, the SEP1-3 genes could be inhibited by any other floral repressor or activated by a flower meristem identity gene such as LFY, and results would still hold. The second assumption is that FUL is repressed posttranscriptionally by TFL1 (Ferrándiz et al., 2000; see FUL logical rule). It would be equivalent to assuming that other meristem identity genes regulate FUL.
To recover the observed pattern of expression of PI in the three inner floral whorls, we assumed that PI expression is maintained only when AG or AP3 are active because neither AG or AP3 are expressed in sepals. However, it is most probable that PI is regulated by one or more additional factors that are yet to be discovered.
The final assumption concerns AG and WUS. The repression of WUS by AG has been experimentally established (Lenhard et al., 2001; Lohmann et al., 2001). However, adding this interaction in the model, without assuming a mechanism that maintains the activity of AG, would make AG and WUS activities to cycle on and off because of the negative feedback loop. If we would have modeled this interaction as continuous, WUS and AG levels of activity could either cycle or attain an intermediary steady state in which both AG and WUS would not be fully active. This is unlikely because such intermediary WUS steady state should be lower than the in situ limit of detection because in wild-type flowers WUS mRNA is not detected after AG is turned on (Lenhard et al., 2001) and it should be lower than a threshold activity that would cause floral meristem determinacy. Furthermore, at the same time such intermediary levels of WUS activity would have to be strong enough to maintain AG level of activity and C function. Alternatively, one may predict that AG has a direct or indirect positive feedback loop that maintains its expression even in the absence of WUS. But such a loop has not been documented experimentally and thus constitutes a prediction of this model that should be tested experimentally.
The Steady States of the Network Model Coincide with Experimental Gene Expression Profiles
The network had 139,968 possible initial conditions, and it attained only 10 fixed-point attractors or steady gene expression states (see supplemental data online for complete basins of attraction). These steady gene states (Table 1) predicted by the model coincide with the gene expression profiles that have been documented experimentally in cells of wild-type Arabidopsis inflorescence meristems and floral organ primordia. For example, in the Infl steady states, floral meristem identity genes (LFY, AP1, and AP2) and floral organ identity genes (AP1, AP2, AP3, PI, SEP, and AG) are off, whereas the inflorescence identity genes (EMF1 and TFL1) are on. Interestingly, all four possible combinations of UFO and WUS activity states determine the existence of the four Infl meristem attractors. These correspond to four regions observed in Arabidopsis wild-type meristems with the same combinations of UFO and WUS expression states (Laux et al., 1996; Mayer et al., 1998; Samach et al., 1999; Haecker and Laux, 2001; Brand et al., 2002).
Table 1.
Gene Expression in Each of the 10 Steady Gene Activation States Attained for All Possible Initial Conditions (139,968) in the Wild-Type Arabidopsis Gene Network
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | Infl4 |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | Sep |
| 1 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | Pe1 |
| 1 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | Pe2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | St2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | Car |
Infl, inflorescence meristematic cells; Sep, sepal primordial cells; Pe, petal primordial cells; St, stamen primordial cells; Car, carpel primordial cells.
The experimentally documented ABC gene expression profiles are reproduced in the steady states corresponding to cells of each organ type (Bowman et al., 1991; Coen and Meyerowitz, 1991; Jofuku et al., 1994). The same is true for all the non-ABC genes included in the network (for example, SEP1-3, Savidge et al., 1995; CLF, Goodrich et al., 1997; UFO, Samach et al., 1999; LUG, Conner and Liu, 2000).
The model predicted two steady states for each petal and stamen cell type: one with and another one without UFO activity. This implies that the activity of this gene is not critical to maintain petal or stamen cell identity. This has been supported by experimental data because in 35S:AP3 35S:PI double transgenic plants, in which PI and AP3 are expressed in a constitutive manner, B gene determined organs are formed even in the first and fourth floral whorls (Krizek and Meyerowitz, 1996) where there is no UFO expression.
The basins of attraction of the attractors that correspond to inflorescence meristem cell identity (Table 2) indicate that to attain this steady state, in the initial conditions the floral meristem identity genes (e.g., AP1 and LFY) should be off and the floral repressors (TFL1 and EMF1) should be on. This also coincides with the experimental results obtained in Arabidopsis and other plant species, where gain-of-function transgenic plants for the floral meristem identity genes or loss-of-function mutants of the floral repressors (Shannon and Meeks-Wagner, 1993; Coupland, 1995; Mandel and Yanofsky, 1995a; Weigel and Nilsson, 1995; Ratcliffe et al., 1999) bear practically normal terminal flowers that consume the shoot apical meristem.
Table 2.
Sets of Initial Conditions That Lead to Infl Steady Activation States in the Arabidopsis Wild Type Network
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | X | 1 | X | X | 0, 1 | X | X | 0 | X | X | X |
| X | 1 | 2 | 0 | X | 1 | X | X | 2 | X | X | 0 | X | X | X |
| 0 | 1 | 1, 2 | 0 | 0, 1 | 0 | X | X | 0 | X | X | X | X | X | X |
| X | 1 | 1, 2 | 0 | 0, 1 | 0 | X | X | 1, 2 | X | X | X | X | X | X |
X represents any possible value.
The size of the basins of attraction may indicate how stable each morphogenetic response is and which genes are critical to attain each cell fate (see supplemental data online). It is noteworthy that the ratio of the number of initial states leading to floral organ versus inflorescence primordial cells is ∼36:1 (Table 3). This could explain why it has been so hard to find a single gene whose mutation determines a nonflowering phenotype (Koornneef et al., 1998) and why once flowering genes are turned on it is so hard to interrupt flower development. However, this result could be biased because many of the genes controlling the transition to flowering, such as CONSTANS (Simon et al., 1996), SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (Samach et al., 2000), and FLOWERING LOCUS C (Michaels and Amasino, 1999) have not been included yet.
Table 3.
Size of the Basins of Attraction for the Arabidopsis Wild Type Gene Network
| Steady Activation States | N | P | M |
|---|---|---|---|
| Infl1-4 | 3,744 | 1.639 | 3 |
| Sep | 4,026 | 3.575 | 5 |
| Pe1 | 4,698 | 3.717 | 5 |
| Pe2 | 672 | 2.748 | 4 |
| St1 | 63,414 | 3.962 | 8 |
| St2 | 3,024 | 2.674 | 4 |
| Car | 60,390 | 3.959 | 8 |
Mean (P) and maximum (M) number of iterations needed to attain each steady state. N, size of the basins of attraction.
Interestingly, the sizes of the basins of attraction of stamens or carpels are much larger than those of sepals or petals, suggesting that the fates of the reproductive organs (stamens and carpels) are much more stable than the fates of the perianth organs (sepals and petals; Table 3; see also supplemental data online).
Simulations of Loss- and Gain-of-Function Mutants Recover Experimentally Observed Gene Expression Profiles
We simulated loss-of-function mutants (Tables 4, 5, and 6) by turning the node corresponding to the mutated gene permanently off. In all cases, the model recovered steady gene activation states that have been observed experimentally (see references in the tables). We also performed simulations of gain-of-function mutants (Tables 7 and 8) and recovered experimentally observed patterns in all cases. We found a minor discrepancy in the simulation of 35S:AP3 lines that lack carpels but have sepals in Arabidopsis, but in our simulation sepal identity was not recovered. This is because of the assumption made to recover PI expression in the inner three whorls (AP3 is on in sepals and this turns PI on hereto because of the B gene positive feedback loop) and hence indicates that this might not be the way PI is regulated in Arabidopsis; instead, an additional network element has to be invoked.
Table 4.
Steady States Predicted by the Model When ap2 Mutants Are Simulated and Relationship with Cell Types Found in Structures of the Mutant Arabidopsis Plants (Liu and Meyerowitz, 1995; Deyholos and Sieburth, 2000)
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | Infl4 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 | 0 | 1 | 1 | 1 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | St2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | Car |
Infl, inflorescence meristematic cells; St, stamen primordial cells; Car, carpel primordial cells.
Table 5.
Steady States Predicted by the Model When lug Mutants Are Simulated and Relationship with Cell Types Found in Structures of the Mutant Arabidopsis Plants (Liu and Meyerowitz, 1995)
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | Infl4 |
| 1 | 0 | 0 | 2 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | CS |
| 1 | 0 | 0 | 2 | 1 | 1 | 2 | 2 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | SP1 |
| 1 | 0 | 0 | 2 | 1 | 1 | 2 | 2 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | SP2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 | 1 | 1 | 0 | 1 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 1 | 1 | 0 | 1 | St2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 1 | 1 | 0 | 1 | Car |
Infl, inflorescence meristematic cells; St, stamen primordial cells; Car, carpel primordial cells; CS, carpelloid sepal primordial cells; SP, staminoid petal primordial cells.
Table 6.
Steady States Predicted by the Model when ap3 Mutants Are Simulated and Relationship with Cell Types Found in Structures of the Mutant Arabidopsis Plants (Bowman et al., 1989)
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | Infl4 |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | Sep |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | Sep |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | Car |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | Car |
Infl, inflorescence meristematic cells; Sep, sepal primordial cells; Car, carpel primordial cells.
Table 7.
Steady States Predicted by the Model When 35S:AG Transgenic Lines Are Simulated and Relationship with Cell Types Found in Structures of the Transgenic Arabidopsis Plants (Mizukami and Ma, 1997)
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 1 | 1 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 1 | 1 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 1 | 0 | 0 | 1 | 1 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | 1 | Infl4 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | St2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | Car |
Infl, inflorescence meristematic cells; St, stamen primordial cells; Car, carpel primordial cells.
Table 8.
Steady States Predicted by the Model When 35S:AP3 Transgenic Lines Are Simulated and Relationship with Cell Types Found in Structures of the Transgenic Arabidopsis Plants (Krizek and Meyerowitz, 1996)
| FT | EMF1 | TFL1 | LFY | FUL | AP1 | AP3 | PI | AG | UFO | WUS | AP2 | SEP | LUG | CLF | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | Infl4 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | St2 |
| 1 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | Pe1 |
| 1 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | Pe2 |
Infl, inflorescence meristematic cells; St, stamen primordial cells; Pe, petal primordial cells.
As an additional mutational analysis to validate the model in relation to experimental evidence and to evaluate the role of the network architecture in determining the steady states, we systematically removed each of the interactions (activation or inhibition arrows) postulated in the network model (see Methods) and found the new steady states and corresponding basins of attraction (see Supplemental Table 1 online). This procedure also enabled us to test for possible errors in the logical rules and allows for novel predictions on the effects of canceling the input of single interactions. For example, if the regulation of AG by LFY could be deleted without altering other functions of LFY, flowers with sepals and petals instead of carpels and stamens would be expected. More interestingly, when the activation of AP1 by LFY is removed, the model recovers states of normal flowers, suggesting that this interaction is not necessary for floral meristem identity. However, other aspects of the function of both genes are critical because both lfy and ap1 mutant flowers have flower meristem identity defects.
Results summarized in Supplemental Table 1 online clearly show that the network architecture determines the steady states recovered. To test if these are robust to alterations in the details of the laws governing the activation of each network component keeping the overall architecture intact, we performed in silico point perturbations of the outputs in the tables of logical rules.
The Network Steady States Are Robust to Small Alterations of Gene Function
We tested the robustness of the steady states predicted by the model with single in silico random point alterations of the outputs in the tables of the logical rules. At each run, one alteration was done in one of the outputs of a randomly selected network node. In the simulations, we had all possible states of input genes spelled out and altered the output states for only one of the combinations. The probability of altering any particular node was proportional to the number of different possible combinations of expression states of the genes that affect it. After each point alteration, the steady gene states were obtained for all possible initial conditions and compared with the steady states predicted by the wild-type network. This was repeated until the proportion of novel steady states recovered of all altered networks tested reached a plateau (see Supplemental Figure 1 online). The plateau was reached at 6000 trials and we stopped at 7500.
Only 67 (0.89%) of the 7500 altered networks had at least one initial condition that lead to a new steady state that was not obtained in the wild-type simulation. However, most of these 67 networks predicted very similar cell types to those obtained with the original network. For example, in 25 (0.33%) of these 67 networks, the novel steady activation states predicted after the point alteration were different to the original ones only by a change in a single gene activation state, and it usually affected the level of expression of genes with more than two states. In 16 (0.213%) other networks, no novel steady gene activation states were predicted. Instead, limit cycles in which one of the states was equal to a steady activation state from the original network were obtained (although intermittent, the expected gene expression pattern was maintained). Two (0.0266%) other networks corresponded to cases in which the SEP node, which represented three redundant genes (Pelaz et al., 2000) and hence implied a triple mutant, was altered. Hence, out of 7500 random networks generated by in silico point alterations in the logical rules, only 24 (<0.5%) yielded different steady gene activation states to the ones recovered with the original network.
Evolution of Development: Floral Network Alterations Explain the green petals Mutant Phenotype for Petunia
Deciphering network architectures underlying cell differentiation is a first step toward understanding the mechanisms that rule conservation and variation in morphological traits. Although most flowering species have an overall conserved plan of floral organ determination (Rudall, 1987), mutations have revealed some important variations. In the case of petunia, the overall network of cell fate determination during flower organ development seems to be conserved with respect to Arabidopsis (Ferrario et al., 2004). However, mutant analyses have suggested some differences. For example, whereas in Arabidopsis the AP3 mutation yields a homeotic flower with two whorls of sepals and two whorls of inner carpels, in petunia the mutation in the AP3 ortholog results in a flower with two whorls of sepals, a whorl of unaltered stamens, and a whorl of carpels (Vandenbussche et al., 2004). Vandenbussche and collaborators (2004) have shown that another AP3 duplicate upregulated by an AG ortholog in petunia could be partially substituting for the functions of the AP3 ortholog in stamens. Our simulation results suggest that an architecture like the one proposed here for the Arabidopsis network that includes a duplicated AP3 would yield the gene expression patterns observed for the wild type and a single AP3-like gene mutant and provides a prediction for the double loss-of-function mutations of AP3-like genes in petunia (Tables 9 to 11, Figure 5).
Table 9.
Gene Expression in Each of the 10 Steady Gene Activation States Attained for All Possible Initial Conditions (419,904) in the Wild-Type Petunia Gene Network
| FT | EMF1 | TFL1 | LFY | FUL | FBP26 | PhDEF | PhGL01,2 | pMADS3 | UFO | WUS | PhAP2A | FBP2,5 | LUG | PhCLF1,2 | PhTM6 | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | Infl4 |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | Sep |
| 1 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | Pe1 |
| 1 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | Pe2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | 2 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | 2 | St2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | Car |
Note that the names of the Arabidopsis genes are used in those cases in which no ortholog gene in petunia has been described (see Figure 5 for petunia gene names). Infl, inflorescence meristematic cells; Sep, sepal primordial cells; Pe, petal primordial cells; St, stamen primordial cells; Car, carpel primordial cells.
Table 10.
Steady States Predicted by the Model When phdef Mutants Are Simulated and Relationship with Cell Types Found in Structures of Mutant Petunia Plants (Vandenbussche et al., 2004)
| FT | EMF1 | TFL1 | LFY | FUL | FBP26 | PhDEF | PhGL01,2 | pMADS3 | UFO | WUS | PhAP2A | FBP2,5 | LUG | PhCLF1,2 | PhTM6 | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | Infl4 |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | Sep |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | Sep2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 2 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | 2 | St1 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | 2 | St2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | Car |
Table 11.
Steady States Predicted by the Model When phdef phtm6 Double Mutants Are Simulated and Predicted Relationship with Cell Types in Structures of the Mutant Petunia Plants (Vandenbussche et al., 2004)
| FT | EMF1 | TFL1 | LFY | FUL | FBP26 | PhDEF | PhGL01,2 | pMADS3 | UFO | WUS | PhAP2A | FBP2,5 | LUG | PhCLF1,2 | PhTM6 | Cell Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | Infl1 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | Infl2 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | Infl3 |
| 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | Infl4 |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | Sep |
| 1 | 0 | 0 | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | Sep2 |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | Car |
| 1 | 0 | 0 | 2 | 2 | 0 | 0 | 1 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | Car2 |
Figure 5.

Arabidopsis and Petunia Wild Type (left), ap3 Mutant (right) Flowers, and Corresponding Network Models.
Single petunia mutant for PhDEF is shown in the top part of (B) and a scheme of the predicted double mutant for PhDEF and PhTM6 is shown below (see also prediction in Vandenbussche et al., 2004). Arabidopsis is shown in (A). The networks indicate which nodes were turned off (yellow) to simulate mutants. Network architecture as in Figure 4 for Arabidopsis and as in Supplemental Figure 2 online for petunia. Steady states for wild type and mutant simulations found respectively in Tables 1 and 6 for Arabidopsis and Tables 9, 10 (single mutant for PhDEF gene), and 11 (double mutant for PhDEF and PhTM6 genes) for petunia. Note that Arabidopsis gene names are used if the corresponding gene has not been characterized in petunia (also in Tables 9 to 11 and Supplemental Figure 2 online). The Arabidopsis orthologs of the cloned petunia genes are as follows: FLORAL BINDING PROTEIN26 (FBP26) is an AP1 ortholog (Immink et al., 1999), PhDEF (formerly known as GREEN PETALS) is an AP3 and DEFICIENS (DEF; from A. majus) ortholog, PhGLO1 (FBP1) and PhGLO2 (PETUNIA MADS BOX GENE2; pMADS2) are PI and GLOBOSA (GLO; from A. majus) orthologs (Vandenbussche et al., 2004), pMADS3 is an AG ortholog (Kapoor et al., 2002), PhAP2A is an AP2 ortholog (Maes et al., 2001), FBP2 and FBP5 are SEP orthologs (Ferrario et al., 2003; Vandenbussche et al., 2003), PhCLF1 and PhCLF2 are CLF orthologs (Mayama et al., 2003), and PETUNIA HYBRIDA TM6 (PhTM6) is a paleoAP3 gene (Vandenbussche et al., 2004). Drawings are not to scale. Drawings based on photographs from http://www.weigelworld.org ([A], left), http://www.salk.edu/LABS/pbio-w/gallery.html ([A], right), and Van Tunen et al. (1994) ([B], left and top right).
DISCUSSION
Theoretical studies (e.g., Kauffman, 1969, 1993; Thomas, 1991) and the more recent analyses of molecular networks using genomic data (e.g., Jeong et al., 2000; Wagner, 2001) have been fundamental contributions toward the understanding of molecular regulatory networks and cell differentiation. Initial models considered random networks in which all nodes were connected, on average, to the same number of other network nodes (Kauffman, 1993). But recent studies have shown that unequal connectivity among nodes can greatly influence network dynamics (Aldana and Cluzel, 2003), and biological networks at different levels are certainly not uniformly connected (Jeong et al., 2000).
Obtaining the repertoire of the architectures of relatively smaller networks from more detailed experimental data and studying their structures and dynamical behaviors (e.g., Mjolsness et al., 1991; McAdams and Shapiro, 1995; Mendoza and Alvarez-Buylla, 1998; Albert and Othmer, 2003) will be particularly relevant for understanding developmental mechanisms if small, structurally, and dynamically isolated networks of key regulatory genes determine fundamental and specific aspects of cell differentiation (Wagner, 1996; Hartwell et al., 1999). For such networks, the general conclusions derived from analyses of the statistical behavior of large networks may not hold. In this article, we have shown that a relatively small network, yet derived from thorough experimental data (see references in logical rules section above), is canalized to steady gene activation states that coincide with those observed experimentally in meristematic inflorescence cells and in primordial cells of sepals, petals, stamens, and carpels. This network can be the basis to elaborate explicit spatio-temporal dynamic models of cell specification during floral organ determination.
A network may be considered a developmental module if its steady states are robust to initial conditions and small alterations of parameters defining gene interactions (von Dassow et al., 2000). However, this has been tested for very few gene networks grounded on experimental data (von Dassow et al., 2000). Our results strongly suggest that the network put forward here constitutes a robust developmental module for organ cell-fate determination that may partly underlie the conservation of overall floral plan among angiosperms (Ambrose et al., 2000; Ferrario et al., 2004). On the other hand, the fact that using the same overall network with a duplicated AP3-like gene we are able to recover the green petals phenotype of petunia (Vandenbussche et al., 2004) shows that this type of model can also be useful for exploring variations in developmental mechanisms along species lineages. Particularly, we were able to provide additional support for the prediction made by Vandenbussche and collaborators (2004) for the double mutant of the two AP3-like genes and possible regulatory interaction predictions for the other genes included in the network for petunia. This case is also a documented example for maintenance of overall network functionality and predicted steady states after an event of gene duplication and functional divergence. Furthermore, this event enables mutations in AP3-like genes that do not abolish stamen cell differentiation. Studies of the evolution of development in other eukaryotic systems have also started to explore the role of developmental mechanisms defined in terms of gene networks and how these constrain the patterns of morphological variation that emerge during evolution and thus limit the scope of natural selection (Salazar-Ciudad et al., 2000; Goodwin, 2001).
Additionally, the Arabidopsis network model may be used as a framework to find holes or inconsistencies in gene interactions documented experimentally or to test and/or refine hypotheses in silico that can then be tested experimentally in vivo (Eldar et al., 2002). Our simulations predict that (1) the SEP and FUL genes are positively regulated by a flower meristem identity gene or repressed by an inflorescence meristem identity gene. Note that FUL is off in the Infl steady state (Table 1), whereas its mRNA has been observed in cells of the inflorescence meristem. The regulators (positive or negative) of this gene have not been characterized experimentally. Here, we have considered that this gene is posttranscriptionally repressed by TFL1 (see FUL logical rules). (2) A regulator of PI that restricts its domain of expression to petals, stamens, and carpels needs to be discovered. (3) AG self-activates either directly or indirectly. An alternative less parsimonious possibility would be that WUS activates a yet to be discovered gene that should persist in the absence of WUS (maybe through self-activation) and thus maintain AG expression when WUS is off. The possibility of AG self-regulation has been neglected because in ag-1 plants the AG mRNA pattern of expression is as in wild-type Arabidopsis (Gustafson-Brown et al., 1994). However, this data could still be compatible with an AG positive feedback loop because in the ag-1 background the nonactive AG protein is unable to repress WUS that in turn would permanently upregulate AG. To test the positive feedback loop, we would need to show ectopic GUS staining in an AG:GUS × 35S:AG cross. We are presently pursuing this experiment.
To test the dynamical consequences of the AG positive feedback loop, we ran all analyses presented here for a network that lacks the loop for AG as well as the inhibition of WUS by AG (see supplemental data online). Overall results are the same as those obtained for the network in Figure 4. There are, however, some differences that suggest that the network in Figure 4 is more robust than the one in supplemental data online. Also, although for the network of Figure 4 we did not find that steady states are most sensitive to changes at nodes that have the highest number of outgoing links (Albert et al., 2000), this was the pattern found in the same type of simulations for the network in the supplemental data online. These results suggest that the sensitivity of networks' steady states to alterations of genes with different degrees of connectivity may depend on the number and type of network motifs (Milo et al., 2002). It now becomes relevant to explore the presence and dynamic significance of motifs in networks of different sizes grounded on experimental data.
This network includes most published gene interactions during floral organ cell fate determination and recovers steady states that correspond to observed patterns of gene expression in wild-type and mutant backgrounds. In our previous effort to model cell fate determination during flower morphogenesis, we recovered steady states that did not match any observed gene expression patterns (Mendoza and Alvarez-Buylla, 1998), pointing to missing data. Additionally, in this article, we have derived logical rules grounded on Arabidopsis experimental data to estimate parameters of gene interactions and therefore avoided the circular criteria that we used previously. The tables of logical rules also provide a framework to systematically refine and expand them. Finally, in this article, we have achieved a robustness analysis and simulations that illustrate variations in developmental mechanisms among species.
The fact that wild-type and mutant simulations using the whole network recover states that are observed in wild-type and mutant plants suggests that the key cell-autonomous factors at play in Arabidopsis floral organ fate determination have been included. Even though the architecture of the network importantly relied on mutant analyses of the genes included, the global network behavior under wild-type and mutant assumptions cannot be predicted without formal analyses as the ones performed here, such as the robustness simulations. Although real gene networks are likely to be more complex than the one proposed here, it is outstanding that of the many possible initial conditions and developmental routes, this network is canalized to the few steady state gene expression profiles observed upon floral organ primordia determination. This suggests that specific developmental routes starting on few initial conditions elicited by precise signaling mechanisms are not needed to attain proper cell identity. This indicates that the steady gene activation profiles of floral organ primordia are determined not only by previous gene activation states, but also by the overall network architecture and dynamics. Our analyses suggest also that the network steady states are robust to small alterations of gene function.
It is noteworthy that the basins of attraction of reproductive organ primordial cells (stamens and carpels) are much larger than those of perianth organs primordial cells (sepals and petals). This leads to the prediction that variants with altered or lacking perianth organs should be more common than those with affected reproductive whorls, and this pattern indeed coincides with some surveys (Meyer, 1966). Robustness of reproductive organs is advantageous because alterations of reproductive organs are likely to have greater impacts on fitness than perianth modifications. However, it is not straightforward that natural selection is directly responsible for the origin of this robustness. An alternative explanation could lie in the fact that cell fate determination mechanisms of reproductive organs are older. Molecular mechanisms of reproductive cell fate seem in fact to be conserved among gymnosperms and angiosperms (Winter et al., 1999), whereas the perianth originated later and is privative of angiosperms. Hence, it is reasonable to assume that the basins of attraction of the newly evolved attractors that correspond to perianth organs are relatively smaller if network functionality has to be preserved and the older cell types maintained. In this scenario, natural selection may have been important in preserving network functionality so that it continued to generate reproductive structures, but not in making certain states more robust (with larger basins of attraction) than others from a phase space that was originally equally partitioned among states. Nonetheless, the action of natural selection in directly achieving robustness of reproductive organs cannot be ruled out at this moment.
A very similar model to the one proposed here was recently proposed for the pair-rule genes in Drosophila (Albert and Othmer, 2003). Their discrete Boolean network reproduced the results from analyses of an earlier continuous model (von Dassow et al., 2000) and yielded patterns of gene expression coherent with experimental data. Therefore, they concluded that their network is a robust developmental module in which the dynamics of the system depends on the network architecture. Whether most gene networks are canalized and robust is still an unanswered question, but most networks underlying cell differentiation are not autonomous and cell–cell interaction mechanisms, such as protein and hormone diffusion, active transport, and ligand–receptor interactions, likely affect gene network behavior and transitions in cell identity. These mechanisms may be in part responsible for canalization as well. On the other hand, for patterns and/or processes that are more variable or even plastic among individuals of the same species, a discrete approach might not be adequate (e.g., Krakauer et al., 2002; Salazar-Ciudad and Jernvall, 2002). In such instances, specific parameter values of continuous functions may be responsible for alternative behaviors. In addition, cell division is also coupled to cell differentiation during morphogenesis, and the geometry of the spatial domains may constrain the spatial patterns of cell types that emerge (Murray, 1989; Barrio et al., 2004). Therefore, an explicit spatio-temporal framework must be considered to be able to explore the relative role of intercellular signaling mechanisms, patterns of cell division, geometry of the spatial domain, and the network of gene interactions during cell patterning and morphogenesis.
Recent studies have started to propose formal models with explicit spatio-temporal dynamics to explore the mechanisms underlying aerial meristem development in Arabidopsis. Jönsson and collaborators (2003) have considered a small network of genes responsible for stem cell identity and meristematic patterning and have coupled these network dynamics to cell proliferation to recover spatial patterns of gene expression.
Before incorporating spatio-temporal dynamics, we consider that it is important to first derive from experimental data gene network architectures that underlie cell differentiation. This may guarantee that no critical nodes or interactions are left out and avoid ending up with the wrong network and equations with adjusted ad hoc parameters to recover observed gene expression patterns. Once cell-fate determination networks are assembled, hypotheses on the role of cellular dynamics, geometry, growth, and intercellular communication mechanisms in pattern formation and morphogenesis can be tested in a formal framework.
Conclusions
Careful experiments will be fundamental to continue unraveling the mechanistic details in gene transcriptional regulation and translation. Mechanistic details have already proven to be fundamental in being able to precisely understand the molecular nature of biological systems, and they will be critical for future quantitative descriptions and for designing convenient perturbations for clinical or agricultural applications (Hasty et al., 2002). However, the study presented here and other recent modeling approaches for gene control of development (e.g., Mendoza and Alvarez-Buylla, 2000; Albert and Othmer, 2003) show that qualitative data of key elements of a complex biological system may be sufficient to organize genetic interactions into integrative mechanistic explanations that capture critical aspects of the biological systems' global dynamical behavior.
Moreover, only a formal dynamical approach can be used to explicitly address if a developmental network is canalized to particular steady states of gene activation and robust to perturbations of gene functions or not. These are also relevant issues for biotechnological applications of gene network knowledge but also for studies of evolution of development to explore the role of developmental mechanisms in restricting the range of phenotypic variation that may appear during evolution (Salazar-Ciudad et al., 2000) and the role of natural selection in determining network characteristics (Milo et al., 2002) and phenotypic evolution. Finally, once network architecture is validated for a particular study system, it is easier to make explicit predictions on the modifications that may underlie morphological or developmental mechanisms encountered in other systems, as is the case for petunia shown here.
METHODS
The model is discrete. If N is the number of genes involved in the network, then Xn is a vector with expression state for each gene in a space of N dimensions, representing the network state after n iterations as follows:
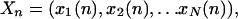 |
(1) |
where xi(n) represents the state of expression of the gene i at the iteration n. We then write:
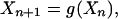 |
(2) |
which indicates that the state at the iteration (n + 1) is determined by the state at the previous iteration.
Each node, except SEP (redundant SEP1, SEP2, and SEP3 genes), stands for the activity of a single gene involved in floral organ fate determination. Most nodes could assume three levels of expression (on, 1 or 2, and off, 0) to enable different activation thresholds when experimental data was available (Thomas, 1991). The system has a finite number of possible initial conditions equal to 139,968, and each one is represented by a vector of dimension 15 in which each column corresponds to the expression state of each network node at initial conditions in the following order: FT EMF1 TFL1 LFY FUL AP1 AP3 PI AG UFO WUS AP2 SEP LUG CLF. The vector of 15 entries that keeps track of the activity level of each node describes the system at each time point. We updated the state of each node synchronously. Starting on each initial condition we iterated the network until it reached an attractor. Thus, we determined the steady gene activation states described in Table 1. All attractors were fixed point attractors (Kauffman, 1993) in which the activity level of all genes remains the same as in the previous iteration. We also kept track of the number of iterations needed for a given initial condition to attain each steady state for future model developments. The set of initial conditions that lead to each of the system's steady states is the basin of attraction of each attractor. The model can be represented by a set of difference equations in which gene interactions are modeled according to logical rules.
Logical rules were grounded on published experimental evidence of gene interactions (see model section above). Expression data can readily be translated into an interaction edge with a particular sign. If a gene's loss-of-function mutation causes ectopic or diminished expression of a second gene, the mutated gene represses or activates the second gene, respectively, and vice versa for a gain-of-function mutation. A similar rationale was used for crosses of promoter:reporter transgenic lines crossed to loss- and gain-of-function mutants. In epistasis analysis, if we have two genes, X and Y, and their corresponding loss of function mutant alleles, x- and y-, when the double mutant (x-y-) has the same phenotype as y-, we say that Y is epistatic over X and that both genes are in the same pathway. If both genes function in the same direction, then we infer a positive regulatory interaction with Y upstream of X. This is the case for emf1 and tfl1 mutants, whose effect on the phenotype is not opposite with the other (Chen et al., 1997; see TFL1 logical rules). However, if the phenotypes of the two gene mutations are opposite, (e.g., ag lug double mutants, in which ag is epistatic; Liu and Meyerowitz, 1995; see AG logical rules), the most parsimonious explanation for epistasis is that the epistatic gene (Y) is repressed by the other one (X) in wild-type plants.
The rationale exemplified above is sufficient to get the sign (inhibition or activation) of regulatory interactions; however, it is not enough to obtain the logical rules. We give here three examples. In the TFL1 logical rule, we concluded from the epistasis data that if the epistatic gene (EMF1) is off, then TFL1 is off, independently of other input states (first row of the logical rule table for TFL1). However, although EMF1 is needed for TFL1 activation, it is not sufficient because in plants that overexpress LFY with a wild-type EMF1 allele, no TFL1 mRNA is detected (Ratcliffe et al., 1999), suggesting that if LFY is fully active (LFY = 2), then TFL1 is inactive even if EMF1 is on. This also occurs in the background of ap1 loss-of-function mutants, and it is also true for overexpression of AP1 itself. Hence, we write the second and third rows of the table of logical rules for TFL1.
Another example of how we codified the effect of multiple inputs on the output of the target gene is for AG. LUG, CLF, and AP2 repress AG in the two outer floral whorls (Liu and Meyerowitz, 1995; Goodrich et al., 1997; Deyholos and Sieburth, 2000), and LFY and WUS activate it in the two inner floral whorls (Lenhard et al., 2001; Lohmann et al., 2001). We considered that WUS and LFY activity is sufficient to overcome AG repression by LUG, CLF, and AP2 because in AP3:WUS transgenic plants that express WUS in the second floral whorl where LFY is also expressed (Ferrándiz et al., 2000), there is ectopic AG activity there as well (Lenhard et al., 2001). Hence, we write the third and fourth rows of the table of AG logical rules.
Finally, we give an example for AP3. This gene was modeled as a three-state variable that reaches its highest threshold of activity (AP3 = 2) only if AP3, PI, SEP1, SEP2, or SEP3, and either AP1 (second row of table of AP3 logical rules) or AG (first AP3 logical rule) (Honma and Goto, 2001; Pelaz et al., 2001) are active (i.e., their activity must not be equal to 0). If this does not occur, the first threshold is surpassed (AP3 = 1) only if UFO and LFY are both active (Parcy et al., 1998; third to sixth rows of table of AP3 logical rules); otherwise, AP3 = 0.
In most cases, the logical rules were objectively derived from experimental data. In the few cases that subjective decisions had to be made, we tested if the alternative rules could have significant impacts on the attractors predicted by running the model with the most contrasting rules. The latter are marked with an asterisk in the output value on the rightmost column of the table for the corresponding target node. In all such cases tested, the same attractors and very similar basins of attraction were recovered, suggesting that the different options are equivalent.
We systematically removed each interaction between pairs of network components and obtained the steady states and basins of attraction for each new altered network lacking each of the postulated interactions (see Supplemental Table 1 online). This was achieved by setting to zero the activation state of the input node when the activation level of the target node was being updated. After the updating of the target node, the activation level of the input node was set to its previous level. We simulated loss- and gain-of-function mutants by setting the activation state of the mutated gene to 0 in the first case or 1 or 2 in the second one during the entire simulation.
We performed robustness analyses by testing small changes in the logical rules. We performed random in silico point alterations of the outputs in the tables of logical rules and obtained the ratio of networks that attained steady states that were different to those predicted by the wild-type model and stopped the simulations when this ratio reached a plateau (see Supplemental Figure 1 online). After each alteration, the network was run again for all possible initial conditions to obtain the steady states and basins of attraction, and these were compared with those of the original network. All programs were written in C++ and are available upon request.
Supplementary Material
Acknowledgments
We thank R. Barrio, J. Bowman, G. Cocho, V. Guraieb, P. León, L. Mendoza, P. Miramontes, F. Sánchez-Garduño, F. Vergara-Silva, A. Wagner, six anonymous reviewers, and Grupo de Biomatemáticas (Facultad de Ciencias, Universidad Nacional Autónoma de México) for their useful comments and many suggestions. Many thanks to F. Vergara-Silva for the artwork and to A. Espinosa-Romero for his aid with computers. We thank E. Nuñez and A. Plata for logistical support, the people in Laboratorio de Macroecología, Instituto de Ecología, Universidad Nacional Autónoma de México for the desk space provided to C.E.-S., Departamento de Supercómputo, Dirección General de Servicios de Cómputo Académico, Universidad Nacional Autónoma de México for sharing computational resources, and the Santa Fe Institute for support during final stages of this work. Financial support was from Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica, Universidad Nacional Autónoma de México IN 230002 and IX207104, University of California-MEXUS ECO IE 271, and Consejo Nacional de Ciencia y Tecnología CO1.41848/A-1, CO1.0538/A-1, and CO1.0435.B-1 grants to E.A.B and a PhD scholarship from Consejo Nacional de Ciencia y Tecnología and Universidad Nacional Autónoma de México to C.E.-S.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Elena R. Alvarez-Buylla (ealvarez@miranda.ecologia.unam.mx).
Online version contains Web-only data.
Article, publication date, and citation information can be found at www.plantcell.org/cgi/doi/10.1105/tpc.104.021725.
References
- Albert, R., Jeong, H., and Barabási, A.-L. (2000). Error and attack tolerance of complex networks. Nature 406, 378–382. [DOI] [PubMed] [Google Scholar]
- Albert, R., and Othmer, H.G. (2003). The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J. Theor. Biol. 223, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldana, M., and Cluzel, P. (2003). A natural class of robust networks. Proc. Natl. Acad. Sci. USA 100, 8710–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose, B.A., Lerner, D.R., Ciceri, P., Padilla, C.M., Yanofsky, M.F., and Schmidt, R.J. (2000). Molecular and genetic analyses of the Silky1 gene reveal conservation of floral organ specification between Eudicots and Monocots. Mol. Cell 5, 569–579. [DOI] [PubMed] [Google Scholar]
- Aubert, D., Chen, L., Moon, Y., Martin, D., Castle, L.A., Yang, C., and Sung, Z.R. (2001). EMF1, a novel protein involved in the control of shoot architecture and flowering in Arabidopsis. Plant Cell 13, 1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio, R., Maini, P.K., Padilla, P., Plaza, R., and Sánchez-Garduño, F. (2004). The effect of growth and curvature on pattern formation. J. Dyn. Differ. Equ., in press.
- Bowman, J., Smyth, D., and Meyerowitz, E. (1991). Genetic interactions among floral homeotic genes of Arabidopsis. Development 112, 1–20. [DOI] [PubMed] [Google Scholar]
- Bowman, J.L., Smyth, D.R., and Meyerowitz, E.M. (1989). Genes directing flower development in Arabidopsis. Plant Cell 1, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, D., Ratcliffe, O., Vincent, C., Carpenter, R., and Coen, E.S. (1997). Inflorescence commitment and architecture in Arabidopsis. Science 275, 80–83. [DOI] [PubMed] [Google Scholar]
- Brand, U., Grünewald, M., Hobe, M., and Simon, R. (2002). Regulation of CLV3 expression by two homeobox genes in Arabidopsis. Plant Physiol. 129, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch, M.A., Bomblies, K., and Weigel, D. (1999). Activation of a floral homeotic gene in Arabidopsis. Science 285, 585–587. [DOI] [PubMed] [Google Scholar]
- Chen, L., Cheng, J., Castle, L., and Sung, Z.R. (1997). EMF genes regulate Arabidopsis inflorescence development. Plant Cell 9, 2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen, E.S., and Meyerowitz, E. (1991). The war of the whorls: Genetic interactions controlling flower development. Nature 353, 31–37. [DOI] [PubMed] [Google Scholar]
- Conner, J., and Liu, Z. (2000). LEUNIG, a putative transcriptional corepressor that regulates AGAMOUS expression during flower development. Proc. Natl. Acad. Sci. USA 97, 12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupland, G. (1995). Genetic and environmental control of flowering time in Arabidopsis. Trends Genet. 11, 393–397. [DOI] [PubMed] [Google Scholar]
- Deyholos, M.K., and Sieburth, L.E. (2000). Separable whorl-specific expression and negative regulation by enhancer elements within the AGAMOUS second intron. Plant Cell 12, 1799–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar, A., Dorfman, R., Weiss, D., Ashe, H., Shilo, B., and Barkai, N. (2002). Robustness of the BMP morphogen gradient in Drosophila embryonic patterning. Nature 419, 304–308. [DOI] [PubMed] [Google Scholar]
- Ferrándiz, C., Gu, Q., Martienssen, R., and Yanofsky, M.F. (2000). Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development 127, 725–734. [DOI] [PubMed] [Google Scholar]
- Ferrario, S., Immink, R.G., and Angenent, G.C. (2004). Conservation and diversity in flower land. Curr. Opin. Plant Biol. 7, 84–91. [DOI] [PubMed] [Google Scholar]
- Ferrario, S., Immink, R.G., Shchennikova, A., Busscher-Lange, J., and Angenent, G.C. (2003). The MADS box gene FBP2 is required for SEPALLATA function in Petunia. Plant Cell 14, 914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich, J., Puangsomlee, P., Martin, M., Long, D., Meyerowitz, E.M., and Coupland, G. (1997). A Polycomb-group gene regulates homeotic expression in Arabidopsis. Nature 386, 44–51. [DOI] [PubMed] [Google Scholar]
- Goodwin, B. (2001). How the Leopard Changed Its Spots: The Evolution of Complexity, 2nd ed. (Princeton, NJ: Princeton Science Library).
- Gustafson-Brown, C., Savidge, B., and Yanofsky, M.F. (1994). Regulation of the Arabidopsis floral homeotic gene AP1. Cell 76, 131–143. [DOI] [PubMed] [Google Scholar]
- Haecker, A., and Laux, T. (2001). Cell-cell signaling in the shoot meristem. Curr. Opin. Plant Biol. 4, 441–446. [DOI] [PubMed] [Google Scholar]
- Hartwell, L., Hopfield, J.J., Leibler, S., and Murray, A.W. (1999). From molecular to modular cell biology. Nature 402, C47–C52. [DOI] [PubMed] [Google Scholar]
- Hasty, J., McMillen, D., and Collins, J.J. (2002). Engineered gene circuits. Nature 420, 224–230. [DOI] [PubMed] [Google Scholar]
- Haung, M., and Yang, C. (1998). EMF genes interact with late-flowering genes to regulate Arabidopsis shoot development. Plant Cell Physiol. 39, 382–393. [DOI] [PubMed] [Google Scholar]
- Hill, T.A., Day, C.D., Zondlo, S.C., Thackeray, A.G., and Irish, V.F. (1998). Discrete spatial and temporal cis-acting elements regulate transcription of the Arabidopsis floral homeotic gene APETALA3. Development 125, 1711–1721. [DOI] [PubMed] [Google Scholar]
- Honma, T., and Goto, K. (2000). The Arabidopsis floral homeotic gene PISTILLATA is regulated by discrete cis-elements responsive to induction and maintenance signals. Development 127, 2021–2030. [DOI] [PubMed] [Google Scholar]
- Honma, T., and Goto, K. (2001). Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529. [DOI] [PubMed] [Google Scholar]
- Immink, R.G.H., Hannapel, D.J., Ferrario, S., Busscher, M., Franken, J., Lookeren Campagne, M.M., and Angenent, G.C. (1999). A Petunia MADS box gene involved in the transition from vegetative to reproductive development. Development 126, 5117–5126. [DOI] [PubMed] [Google Scholar]
- Jeong, H., Tombor, B., Albert, R., Oltvai, Z.N., and Barabási, A.-L. (2000). The large scale organization of metabolic networks. Nature 407, 651–654. [DOI] [PubMed] [Google Scholar]
- Jofuku, K.D., den Boer, B.G.W., Van Montagu, M., and Okamuro, J.K. (1994). Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell 6, 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson, H., Shapiro, B.E., Meyerowitz, E.M., and Mjolsness, E. (2003). Signaling in multicellular models of plant development. In On Growth, Form, and Computers, S. Kumar and P.J. Bentley, eds (London: Academic Press).
- Kapoor, M., Tsuda, S., Tanaka, Y., Mayaman, T.M., Okuyama, Y., Tsuchimoto, S., and Takatsuji, H. (2002). Role of Petunia pMADS3 in determination of floral organ and meristem identity, as revealed by its loss of function. Plant J. 32, 115–127. [DOI] [PubMed] [Google Scholar]
- Kardailsky, I., Shukla, V.K., Ahn, J.H., Dagenais, N., Christensen, S.K., Nguyen, J.T., Chory, J., Harrison, M.J., and Weigel, D. (1999). Activation tagging of the floral inducer FT. Science 286, 1962–1965. [DOI] [PubMed] [Google Scholar]
- Kauffman, S.A. (1969). Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 22, 437–467. [DOI] [PubMed] [Google Scholar]
- Kauffman, S.A. (1993). The Origins of Order: Self-Organization and Selection in Evolution. (Oxford: Oxford University Press).
- Kobayashi, Y., Kaya, H., Goto, K., Iwabuchi, M., and Araki, T. (1999). A pair of related genes with antagonistic roles in mediating flowering signals. Science 286, 1960–1962. [DOI] [PubMed] [Google Scholar]
- Koornneef, M., Alonso-Blanco, C., Blankestijn-de Vries, H., Hanhart, C.J., and Peters, A.J.M. (1998). Genetic interactions among late-flowering mutants of Arabidopsis. Genetics 148, 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer, D.C., Page, K.M., and Sealfon, S. (2002). Module dynamics of the GnRH signal transduction network. J. Theor. Biol. 218, 457–470. [PubMed] [Google Scholar]
- Krizek, B.A., and Meyerowitz, E.M. (1996). The Arabidopsis homeotic genes APETALA3 and PISTILLATA are sufficient to provide the B class organ identity function. Development 122, 11–22. [DOI] [PubMed] [Google Scholar]
- Laux, T., Mayer, K.F.X., Berger, J., and Jurgens, G. (1996). The WUSCHEL gene is required for shoot and floral meristem integrity Arabidopsis. Development 122, 87–96. [DOI] [PubMed] [Google Scholar]
- Lenhard, M., Bonhert, A., Jurgens, G., and Laux, T. (2001). Termination of stem cell maintenance in Arabidopsis floral meristems by interactions between WUSCHEL and AGAMOUS. Cell 105, 805–808. [DOI] [PubMed] [Google Scholar]
- Liljegren, S.J., Gustafson-Brown, C., Pinyopich, A., Ditta, G.S., and Yanofsky, M.F. (1999). Interactions among AP1, LFY, and TFL1 specify meristem fate. Plant Cell 11, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z., and Meyerowitz, E.M. (1995). LEUNIG regulates AGAMOUS expression in Arabidopsis flowers. Development 121, 975–991. [DOI] [PubMed] [Google Scholar]
- Lohmann, J.U., Hong, R.L., Hobe, M., Busch, M.A., Parcy, F., Simon, R., and Weigel, D. (2001). A molecular link between stem cell regulation and floral patterning in Arabidopsis. Cell 105, 793–803. [DOI] [PubMed] [Google Scholar]
- Maes, T., Van de Steene, N., Zethof, J., Karimi, M., D'Hauw, M., Mares, G., Van Montagu, M., and Gerats, T. (2001). Petunia AP2-like genes and their role in flower and seed development. Plant Cell 13, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel, A., and Yanofsky, M. (1995. a). A gene triggering flower formation in Arabidopsis. Nature 377, 522–525. [DOI] [PubMed] [Google Scholar]
- Mandel, A., and Yanofsky, M. (1995. b). The Arabidopsis AGL8 MADS-box gene is expressed in inflorescence meristems and negatively regulated by APETALA1. Plant Cell 7, 1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel, A., and Yanofsky, M. (1998). The Arabidopsis AGL9 MADS box gene is expressed in young flower primordia. Sex. Plant Reprod. 11, 22–28. [Google Scholar]
- Mayama, T., Ohtsubo, E., and Tsuchimoto, S. (2003). Isolation and expression analysis of Petunia CURLY LEAF-like genes. Plant Cell Physiol. 44, 811–819. [DOI] [PubMed] [Google Scholar]
- Mayer, K.F.X., Schoof, H., Haecker, A., Lenhard, M., Jürgens, G., and Laux, T. (1998). Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95, 805–815. [DOI] [PubMed] [Google Scholar]
- McAdams, H.H., and Shapiro, L. (1995). Circuit simulation of genetic networks. Science 269, 650–656. [DOI] [PubMed] [Google Scholar]
- Mendoza, L., and Alvarez-Buylla, E.R. (1998). Dynamics of the genetic regulatory network of Arabidopsis thaliana flower morphogenesis. J. Theor. Biol. 193, 307–319. [DOI] [PubMed] [Google Scholar]
- Mendoza, L., and Alvarez-Buylla, E.R. (2000). Genetic regulation of root hair development in Arabidopsis thaliana: A network model. J. Theor. Biol. 204, 311–326. [DOI] [PubMed] [Google Scholar]
- Mendoza, L., Thieffry, D., and Alvarez-Buylla, E.R. (1999). Genetic control of flower morphogenesis in Arabidopsis thaliana: A logical analysis. Bioinformatics 15, 593–606. [DOI] [PubMed] [Google Scholar]
- Meyer, V.G. (1966). Floral abnormalities. Bot. Rev. 32, 165–195. [Google Scholar]
- Michaels, S.D., and Amasino, M. (1999). Flowering Locus C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N., Chklovskii, D., and Alon, U. (2002). Network motifs: Simple building blocks of complex networks. Science 298, 824–827. [DOI] [PubMed] [Google Scholar]
- Mizukami, Y., and Ma, H. (1997). Determination of Arabidopsis floral meristem identity by AGAMOUS. Plant Cell 9, 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjolsness, E., Sharp, D.H., and Reinitz, J. (1991). A connectionist model of development. J. Theor. Biol. 152, 429–453. [DOI] [PubMed] [Google Scholar]
- Monod, J., and Jacob, F. (1961). General conclusions: Telenomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb. Symp. Quant. Biol. 26, 389–401. [DOI] [PubMed] [Google Scholar]
- Murray, J.D. (1989). Mathematical Biology. (Berlin: Springer-Verlag).
- Parcy, F., Nilsson, O., Busch, M.A., Lee, I., and Weigel, D. (1998). A genetic framework for floral patterning. Nature 395, 561–566. [DOI] [PubMed] [Google Scholar]
- Pelaz, S., Ditta, G.S., Baumann, E., Wisman, E., and Yanofsky, M.F. (2000). B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405, 200–203. [DOI] [PubMed] [Google Scholar]
- Pelaz, S., Tapia-López, R., Alvarez-Buylla, E.R., and Yanofsky, M.F. (2001). Conversion of leaves into petals in Arabidopsis. Curr. Biol. 11, 182–184. [DOI] [PubMed] [Google Scholar]
- Piñeiro, M., and Coupland, G. (1998). The control of flowering time and floral identity in Arabidopsis. Plant Physiol. 117, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe, O.J., Bradley, D.J., and Coen, E.S. (1999). Separation of shoot and floral identity in Arabidopsis. Development 126, 1109–1120. [DOI] [PubMed] [Google Scholar]
- Rudall, P. (1987). Anatomy of Flowering Plants: An Introduction to Structure and Development. (London: Edward Arnold).
- Ruiz-García, L., Madueño, F., Wilkinson, M., Haughn, G., Salinas, J., and Martínez-Zapater, J.M. (1997). Different roles of flowering-time genes in the activation of floral initiation genes in Arabidopsis. Plant Cell 9, 1921–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Ciudad, I., Garcia-Fernandez, J., and Solé, R.V. (2000). Gene networks capable of pattern formation: From induction to reaction-diffusion. J. Theor. Biol. 205, 587–603. [DOI] [PubMed] [Google Scholar]
- Salazar-Ciudad, I., and Jernvall, J. (2002). A gene network model accounting for development and evolution of mammalian teeth. Proc. Natl. Acad. Sci. USA 99, 8116–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samach, A., Klenz, J.E., Kohalmi, S.E., Risseeuw, E., Haughn, G.W., and Crosby, W.L. (1999). The UNUSUAL FLORAL ORGANS gene of Arabidopsis thaliana is an F-box protein required for normal patterning and growth in the floral meristem. Plant J. 20, 433–445. [DOI] [PubMed] [Google Scholar]
- Samach, A., Onouchi, H., Gold, S.E., Ditta, G.S., Schwarz-Sommer, Z., Yanofsky, M.F., and Coupland, G. (2000). Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 288, 1613–1616. [DOI] [PubMed] [Google Scholar]
- Savidge, B., Rounsley, S.D., and Yanofsky, M.F. (1995). Temporal relationships between the transcription of two Arabidopsis MADS-box genes and the floral organ identity genes. Plant Cell 7, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, E.A., and Haughn, G.W. (1993). Genetic analyses of the floral initiation process (FLIP) in Arabidopsis. Development 119, 745–765. [Google Scholar]
- Shannon, S., and Meeks-Wagner, D.R. (1993). Genetic interactions that regulate inflorescence development in Arabidopsis. Plant Cell 5, 639–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, R., Igeno, M.I., and Coupland, G. (1996). Activation of floral meristem identity genes in Arabidopsis. Nature 384, 59–62. [DOI] [PubMed] [Google Scholar]
- Simpson, G., Gendall, A., and Dean, C. (1999). When to switch to flowering. Annu. Rev. Cell Dev. Biol. 99, 519–550. [DOI] [PubMed] [Google Scholar]
- Thomas, R. (1991). Regulatory networks seen as asynchronous automata: A logical description. J. Theor. Biol. 153, 1–23. [Google Scholar]
- Vandenbussche, M., Zethof, J., Royaert, S., Weterings, K., and Gerats, T. (2004). The duplicated B-class heterodimer model: Whorl-specific effects and complex genetic interactions in Petunia hybrida flower development. Plant Cell 16, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbussche, M., Zethof, J., Souer, E., Koes, R., Tornielli, G.B., Pezzotti, M., Ferrario, S., Angenent, G.C., and Gerats, T. (2003). Toward the analysis of the Petunia MADS box gene family by reverse and forward transposon insertion mutagenesis approaches: B, C, and D floral organ identity functions require SEPALLATA-like MADS box genes in Petunia. Plant Cell 15, 2680–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tunen, A.J., Busscher, M., Colombo, L., Franken, J., and Angenent, G.C. (1994). Molecular control of floral organogenesis and plant reproduction in Petunia hybrida. In Molecular and Cellular Aspects of Plant Reproduction, R.J. Scott and A.D. Stead, eds (Cambridge, UK: Cambridge University Press), pp. 9–16.
- von Dassow, G., Meir, E., Munro, E.M., and Odell, G.M. (2000). The segment polarity network is a robust developmental module. Nature 406, 188–193. [DOI] [PubMed] [Google Scholar]
- Wagner, A. (2001). The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol. Biol. Evol. 18, 1283–1292. [DOI] [PubMed] [Google Scholar]
- Wagner, D., Sablowski, R.W.M., and Meyerowitz, E.M. (1999). Transcriptional activation of APETALA1 by LEAFY. Science 285, 582–584. [DOI] [PubMed] [Google Scholar]
- Wagner, G.P. (1996). Homologues, natural kinds and the evolution of modularity. Am. Zool. 36, 36–43. [Google Scholar]
- Weigel, D., and Meyerowitz, E.M. (1993). Activation of floral homeotic genes in Arabidopsis. Science 261, 1723–1726. [DOI] [PubMed] [Google Scholar]
- Weigel, D., and Nilsson, O. (1995). A developmental switch sufficient for flower initiation in diverse plants. Nature 377, 495–500. [DOI] [PubMed] [Google Scholar]
- Winter, K.U., Becker, A., Münster, T., Kim, J.T., Saedler, H., and Theissen, G. (1999). MADS-box genes reveal that genetophytes are more closely related to conifers than to flowering plants. Proc. Natl. Acad. Sci. USA 96, 7342–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C., Chen, L., and Sung, Z.R. (1995). Genetic regulation of shoot development in Arabidopsis: Role of the EMF genes. Dev. Biol. 169, 421–435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.