

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic age-related lung disease with high mortality that is characterized by abnormal scarring of the lung parenchyma. There has been a recent attempt to define the age-associated changes predisposing individuals to develop IPF. Age-related perturbations that are increasingly found in epithelial cells and fibroblasts from IPF lungs compared with age-matched cells from normal lungs include defective autophagy, telomere attrition, altered proteostasis, and cell senescence. These divergent processes seem to converge in mitochondrial dysfunction and metabolic distress, which potentiate maladaptation to stress and susceptibility to age-related diseases such as IPF. Therapeutic approaches that target aging processes may be beneficial for halting the progression of disease and improving quality of life in IPF patients.
Linking aging and chronic lung diseases
One of the most remarkable medical accomplishments in the last century has been the increasing longevity of the human population. A decrease in birth rates, accompanied by a reduction in mortality among the elderly population, has collectively increased the percentage of the aged population, particularly in developed countries. Globally, it is estimated that the proportion of the population over the age of 60 will increase from 11% to 22% by 2050, and 28% by 2100. Currently, 18% of the US population is over 60 years of age (57 million), which is predicted to rise to 27% (107 million) in 2050 and up to 31% (149 million) by 2100 (1). The aging population has propelled an increase in the overall prevalence of chronic conditions. Several lung diseases, including acute lung injury and asthma, and some infectious diseases, such as pneumonia, increase in severity and mortality with age. Additionally, there has been a significant increase in incidence of chronic lung diseases — including chronic obstructive pulmonary disease, most forms of lung cancer, and idiopathic pulmonary fibrosis (IPF) — resulting in aging-related increases in prevalence.
IPF is the most common form of idiopathic interstitial lung diseases. Its overall incidence in the US is 7 to 17 per 100,000 persons with a prevalence between 13.2 and 63 per 100,000 persons (2). A 1996 study initially demonstrated a higher incidence and prevalence of IPF in patients over 65 (3). These observations were confirmed more recently by Raghu et al. (4) and others, using both broad and narrow case-finding criteria for IPF diagnosis. These studies estimated that in the US, the prevalence of IPF increases with age, ranging from 4 per 100,000 for population aged between 18 and 34 years old to 227.2 per 100,000 among those 75 or older (4). This aging-related increase in cumulative incidence and prevalence of IPF has been corroborated by several studies that include populations in Europe and Asia (5–9). Accordingly, the median age at diagnosis of sporadic IPF ranges between 50 and 85 years old, and patients younger than 50 are more commonly associated with familial, rather than sporadic, forms of the disease (2). Mortality rates from IPF are steadily increasing worldwide, and a positive association has also been observed with advanced age (10). Examination of US government health insurance data for people aged 65 and older shows an increasing prevalence of IPF among older age groups (11). These studies confirm the growing concern that aging, and consequently age-related diseases, will drive up health care expenditures. As the elderly population in developed countries is expected to double in the next 25 years, there is an urgent need to understand the pathogenesis of IPF and develop interventions to attenuate or reverse lung fibrosis.
The physiology of aging and the lung
Evolutionary theories of aging include the concept of selection shadow that permits the accumulation of mutations with late deleterious effects, and the theory of antagonistic pleiotropy, which supports the selection of genes that benefit early reproductive life even if they have a deleterious effect at later ages. The disposable soma theory proposes that to maximize reproduction and survival, the organism optimally allocates its metabolic resources. This comes at the cost of limited resources for somatic maintenance and repair, allowing for accumulation of molecular and cellular damage. This concept supports the observation that there is individual plasticity, and the aging process is stochastic in nature (12, 13). Despite the identification of conserved longevity pathways and major environmental factors, the potentially major role of stochastic factors remains poorly defined. Exploring these factors has the potential to increase our knowledge of the variability in the aging process between cells, tissues, and organisms, and even identify new interventions in the aging process (14).
Lung maturity peaks between 20 and 25 years of age. Thereafter, an age-progressive decline in lung function occurs, which is often marked by diminution of forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC). In addition, the residual volume increases and the transport of respiratory gases across the alveolar wall declines, affecting the arterial oxygen tension. A study conducted with a limited number of healthy elderly subjects showed a high frequency of basal subpleural reticular patterns and cysts on CT, as well as bronchial dilation and bronchial wall thickening (15). However, systematic imaging and functional studies in asymptomatic elderly patients have not been conducted to determine the spectrum of normal aging in the lung, which leaves the clinical relevance of these findings unclear.
At a cellular level, only limited knowledge of the factors that define healthy aging of different lung cells is available, and the majority of these studies use animal models. Reporter mouse models that monitor critical factors in the induction and maintenance of senescence, such as p19ARF, demonstrate that accumulation of senescence cells in the lung becomes apparent at 12 months of age associated with decreased lung function characterized by higher compliance or lower capacity to expand at a given pressure and lower elastance, a measure that captures the increase in rigidity and stiffness of the aging lungs (16). In the same set of studies, elimination of p19ARF-expressing cells using a toxin receptor knockout mouse model shows reduction in the loss of elastic fibers in the aging lung tissue, and lower levels of MMP10 and MMP12, two matrix metallopeptidases with elastase activity, and the concomitant improvement in parameters of lung function (16). Altered mitochondrial homeostasis is found in the healthy aging lung (summarized in Table 1). Our group has described a higher frequency of enlarged mitochondria with a bias toward mitochondrial fusion and an increased mitochondrial area in aging type II alveolar epithelial cells (AECIIs) (17). In addition, impaired respiration has been shown in lungs from aged rats (18) and mice (17), with a concomitant increase in mitochondrial ROS (18). Analyses of autophagy activity in aging murine lungs show markers of autophagy flux blockade (17, 19). Most recently, we have shown that dysfunctional autophagy activity also occurs in aging lung fibroblasts from healthy humans (20). Senescence, mitochondrial dysfunction, and insufficient autophagy in the aging lung might have implications in maladaptive responses to stress. For instance, elderly mice are more vulnerable to fibrosis after injury or stress (17, 21, 22) and have a lower capacity for normal repair after damage (23). In this context, our group has shown that the lung’s protective mechanisms against injury, such as mesenchymal stem cell differentiation, are also impaired with age (24).
Table 1. Aging mitochondria in the lung.

Cellular perturbations in the IPF lung
Genomic instability.
Throughout life, DNA is exposed to a wide variety of exogenous and endogenous damaging stimuli. Genetic stability has to be preserved to ensure proper cell replication, and even the smallest imbalance between DNA damage and repair can typically lead to cell death (25). With age, the activity of the cellular DNA repair systems diminishes, allowing accumulation of errors (26) that, when perpetuated as genetic anomalies, can destabilize the genome and dysregulate homeostasis of the cell bearing it.
Several studies have reported genome instability in the IPF patient population (27, 28). Half of sputum samples from IPF patients displayed genetic alterations, either microsatellite instability (MSI) or loss of heterozygosity (LOH), when compared with matched controls (29). A follow-up study including peripheral blood analysis determined that LOH was located in key cell cycle genes such as p16 (30). Moreover, MSI was found in the TGF-β receptor II gene from AECIIs isolated from honeycomb structures in IPF lungs (31). Collectively, these data suggest that DNA damage can actually play a significant role in susceptibility to IPF. Recently, a comparative proteomic analysis of lung tissue found higher expression of DNA damage proteins in IPF lungs (32).
Epigenetic changes.
Epigenetic mechanisms are able to modulate gene activity in the absence of DNA sequence changes, including DNA methylation, histone modification, and expression of noncoding RNAs (microRNAs [miRNAs] and long noncoding RNAs [lncRNAs]). Epigenetic changes that are associated with age (33, 34) seem to be highly conserved in mammals (35, 36), especially the global change of DNA methylation patterns, also known as the “epigenetic clock.” This particular loss of methylation and the subtle differences between changes in healthy aging and in age-related pathological processes (37) could be the key to identifying novel biomarkers in IPF, since it exhibits its own particular methylation profile (38).
Histone posttranslational modifications add another layer of regulation to DNA transcription. Age can modify histone acetylation (39), and it has been proposed that sirtuins, a family of deacetylases whose expression patterns are altered with age, could be one of the culprits (40). For example, SIRT1 suppresses the expression of senescence-associated secretory phenotype (SASP) factors via histone deacetylation in their promoter regions (41). Recently, Sanders et al. (42) reported that pharmacological regulation of histone acetylation ameliorated bleomycin-induced pulmonary fibrosis in mice, suggesting a new therapeutic avenue for IPF.
Noncoding RNAs in small or long forms establish a complex network of transcriptional regulators that can control and modulate different cell programs. Several miRNA target genes are associated with age-related pathways, such as p16 and p21 (43). Studies in IPF lungs have identified significant pathogenic changes in the expression of miRNAs (44). Some of those changes are related to fibrotic response and TGF-β1 pathways, such as decreases in the miRNA let-7 (45), while others target senescence pathways (46). In high contrast, many of the miRNAs dysregulated in IPF recapitulate the fingerprint of developmental cell programming (47).
The notion of lncRNAs (over 200 bp in length) is still young (48), but they are the subject of an emerging body of work to uncover their role in aging and pathological processes (49). It is hypothesized that they can function as competing endogenous miRNAs (ceRNA). Huang et al. elegantly showed that the lncRNA n341773 might function as a ceRNA for miR-199–induced increases in collagen expression in IPF lung fibroblasts (50). Also, the expression of miR-29 and that of MRAK088388 were strongly correlated in lung tissue from murine fibrosis models (51).
Telomere attrition.
Familial IPF and sporadic IPF have also been associated with mutations in the telomerase genes TERC and TERT (52, 53). Around 8%–15% of the familiar pulmonary fibrosis and 1%–3% of sporadic IPF are associated with germline mutations in telomerase genes (54). Absence of telomerase function leads to telomere shortening and induction of senescence. In addition, telomere attrition is a common finding in patients with IPF, even in the absence of telomerase gene mutations (55–63) (Table 2).
Table 2. DNA and chromatin changes associated with pulmonary fibrosis.

Our knowledge is still limited regarding the link between telomerase genetic defects and development of lung fibrosis . It is postulated that telomerase mutations could cause senescence of AECIIs by directly affecting their regenerative capacity, leading to increased susceptibility to lung injury (64). In mice, even just accumulation of telomeric damage through telomere dysfunction in the AECIIs is enough to promote a fibrotic phenotype (65). In sharp contrast, human IPF fibroblasts do not always exhibit short telomeres, even when attrition is detected in matching peripheral blood samples. Furthermore, Liu at al. showed induction of telomerase activity in fibroblasts from bleomycin-treated mice and IPF lungs (66). Taking all these findings into account reminds us that cell type and biological context could also affect how telomerase changes can drive the aging process. Finally, it is worth noting that both TERC (the telomerase RNA subunit) and TERRA (telomeric repeat–containing RNA), important regulators of the telomerase heterochromatin, are transcribed as lncRNAs (67).
Cellular senescence and SASP.
Diminution of the replicative capacity of lung cells is a well-known phenomenon in IPF. Early studies performed in the senescence-accelerated mouse strain SAMP8 demonstrated that mice with a senescence phenotype are more susceptible to lung injury and fibrosis (68). The age-related increase in fibrosis and a defective repair capacity after lung injury have been successively confirmed in aged wild-type mice (21). Senescence can affect the lung’s distinct cell types quite differently (Table 3). For example, senescence can be responsible for exhaustion of stem cells (69), secretion of inflammatory mediators in immune cells and fibroblasts (70), and increased myofibroblast differentiation in lung fibroblasts (71). Stress-induced senescence has been observed in isolated mouse lung fibroblasts from fibrotic lungs, although their proliferative capacity vary between studies. Hecker et al. described that the senescence response was transitory and self-limited in young fibroblasts, in contrast to aging lung fibroblasts that have sustained senescence to resist apoptosis (23). Mechanistic analyses indicate that altered redox balance in aging, controlled by NOX4 and NRF2, promoted myofibroblast senescence. High expression of NOX4 in aging fibroblasts was associated with histone modifications secondary to epigenetic changes with enrichment of H4K16Ac and depletion of H4K20Me3 (72).
Table 3. Cellular senescence and SASP in fibrotic models.

It has long been recognized that senescent cells have alterations in the secretome with an increased production of secretory proteins including PAI-1, and a range of cytokines, chemokines, and proteases (73). In the lung, replicative senescence of mouse lung fibroblasts has been shown to induce expression of several inflammatory mediators and growth factors that have the ability to induce proliferation in adjacent cells (74). SASP provokes ER stress and reinforces senescence through autocrine activity rather than by spreading the senescence phenotype to healthy neighboring cells. Activation of the inflammasome has been shown to be relevant in development of lung fibrosis in aging mice, since deficiency of the inflammasome component NLRP3 diminishes bleomycin-induced lung injury in older animals (75). Another important component in the modulation of SASP is the abundance of caveolae and their components, which increases with senescence (76). As a result, mice deficient in caveolin 1 are protected from bleomycin-induced lung injury with a low profile of SASP (77).
Consistent with studies in mouse models, fibroblasts and lung epithelial cells from IPF lungs express markers of senescence, including positive β-gal staining and p21 expression (23, 71, 78). However, the majority of the current studies with human lung fibroblasts lack appropriate age-matched controls to determine whether the senescent and profibrotic phenotype is related to advanced age.
Mitochondrial dysfunction.
Aging appears to particularly affect mitochondria, and mitochondrial abnormalities including enlargement, loss of cristae, and destruction of inner membranes (79, 80) are often observed with aging. Moreover, ATP production and respiration in mitochondria from older animals are lower than those in mitochondria from younger counterparts, and often, aging mitochondria produce greater amounts of ROS (81). Consequently, mice with more mitochondrial DNA (mtDNA) mutations display accelerated signs of aging. Despite these findings, genetic interventions that impair the expression and function of the electron transport chain components and mitochondrial function have resulted in an increased lifespan. These disparities are most likely associated with the concept of mitohormesis, where mild mitochondrial impairment of function might increase longevity, but severe mitochondrial damage shortens lifespan.
Dysfunctional mitochondria accumulate with age and are a driving aging mechanism in postmitotic cells including muscle cells, neurons, and cardiomyocytes (82, 83). Less is known about mitochondrial dysfunction as a driving mechanism of age-related disease in mitotic tissues. We discovered that AECIIs from human IPF lungs have accumulation of dysmorphic and dysfunctional mitochondria associated with upregulation of markers of ER stress compared with age-matched controls. These findings were replicated in aging mice (20–24 months old) in response to ER stress stimulation. Dysfunctional mitochondria in AECIIs from IPF and aging lungs were found to be related to a low expression of the regulator of mitochondrial homeostasis PTEN-induced putative kinase 1 (PINK1), a kinase linked to age-related neurodegenerative disease. As a result, PINK1-deficient mice exhibited increased susceptibility to apoptosis and spontaneous TGF-β–driven lung fibrosis, developing similar dysmorphic and dysfunctional mitochondria in the AECIIs (17) and an increased susceptibility to lung injury and subsequent fibrosis. Other research groups have also confirmed the role of mitochondrial dysfunction in lung fibrosis. Hawkins and collaborators recently demonstrated that mutations within a surfactant protein that is linked to familial pulmonary fibrosis cause defective clearance of mitochondria (84). Studies by Patel and collaborators confirmed that PINK1-deficient mice have higher susceptibility to lung fibrosis associated with increased apoptosis (85). In addition, studies focused on lung fibroblasts demonstrated that TGF-β induces amelioration of PINK1 expression and low expression of PINK1 resulted in promotion of myofibroblast transformation (19). Similarly, enhanced myofibroblast differentiation and pro-fibrotic signaling pathways were induced by Parkin deficiency, a well-known regulator of mitophagy (86).
New mitochondrial signaling pathways are being studied in relation to increased vulnerability to lung injury, particularly SIRT3. Mitochondrial SIRT3 is reported to control the transformation of fibroblasts into myofibroblasts via suppression of TGF-β1 signaling (87, 88). In concordance, SIRT3-deficient, aging mice were found to develop tissue fibrosis of multiple organs, including heart, liver, kidney, and lungs (87). In the lung, SIRT3 deficiency augmented pulmonary fibrosis after asbestos and bleomycin exposure (88, 89), and SIRT3 expression is attenuated in skin and lung of systemic sclerosis patients (90). It is also proposed that SIRT3 modulates mtDNA damage by regulating 8-oxoguanine-DNA glycosylase-1 (OGG1) acetylation (91, 92). OGG1 is a DNA repair enzyme that hydrolyzes oxidized guanine residues that are modified as a result of excessive ROS production, and deficiency of OGG1 increases susceptibility to pulmonary fibrosis induced by asbestos (89, 92–94). Accumulation of mutations and the deletion of mtDNA can accelerate aging. There is evidence that the aging lung contains a higher proportion of mutated and damaged mtDNA (95, 96), as well as low expression of SIRT3 (97). It has been suggested that the premature aging phenotype of mtDNA mutator mice accumulates high levels of point mutations due to a proofreading deficiency of the mtDNA polymerase POLG (98). Interestingly, expression of SIRT3 diminished significantly with age in these mice, suggesting that this pathway could also regulate aging-associated mitochondrial dysfunction with vulnerability to lung fibrosis. These findings are summarized in Figure 1.
Figure 1. Mitochondrial dysfunction and lung fibrosis.
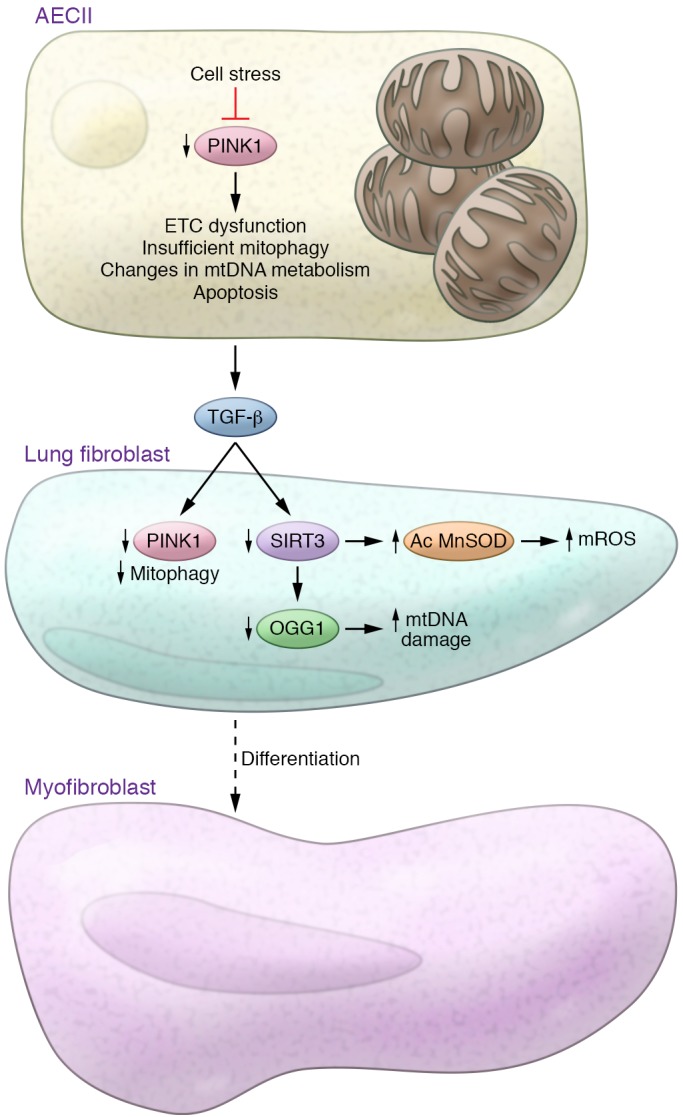
Aging and ER stress cause mitochondrial dysfunction in type II alveolar epithelial cells (AECIIs) by diminishing the expression of the mitochondrial homeostasis regulator PINK1. Deficiency of PINK1 causes mitochondrial dysfunction, which is characterized by low activity of the electron transport chain (ETC) complexes I and IV, alterations in mtDNA metabolism, and insufficient mitophagy, leading to increased susceptibility to apoptosis and induction of TGF-β. TGF-β stimulation of lung fibroblasts has been shown to decrease PINK1 levels and promote insufficient mitophagy and myofibroblast differentiation. Additionally, aging and TGF-β stimulation reduce the expression of SIRT3. SIRT3 deficiency increases levels of acetylated (Ac) MnSOD and consequently increases mitochondrial ROS levels and mtDNA damage, which are also related to low expression of the DNA repair enzyme 8-oxoguanine-DNA glycosylase-1 (OGG1).
Defective proteostasis.
The efficiency of proteostasis declines with age in parallel to defective translation, missense mutations, and oxidative modification. Changes in proteostasis lead to an increase of nonfunctional proteins, accumulation of cytotoxic proteins, and/or aggregation of misfolded proteins. In IPF lungs, evidence of altered proteostasis has been found at different levels, including protein misfolding, markers of ER stress (99–102), defective autophagy, and impaired proteasome activity (103, 104) (Table 4). For instance, familial pulmonary fibrosis is associated with several mutations in surfactant A and C genes (105–108). In addition, recent studies show that genetic alterations outside the coding region of MUC5B can alter MUC5B protein secretion and contribute to cell stress in IPF (109). Alterations in the normal function of the unfolded protein response (UPR), the ubiquitin-proteasome system, or the autophagy-lysosome pathway can increase the pathology caused by impaired proteostasis. For instance, autophagy plays a critical role in antagonizing proteostatic stress by allowing the cellular reorganization and remodeling required for tissue repair. The details of the molecular mechanisms associated with the age-related modifications in proteostasis are not yet clearly understood; however, it is known that younger individuals can enhance proteostasis to compensate for some protein misfolding mutations. Moreover, with advanced age there is an accumulation of cell stress due to environmental exposure (e.g., infections and cigarette smoke) or comorbidities (e.g., metabolic syndrome and sleep deprivation) that might have a negative impact in proteostasis and activate UPR responses in the ER.
Table 4. Dysregulation of proteostasis in IPF.

Markers of ER stress are found in lungs from sporadic IPF (110, 111). Particularly, AECII from areas with dense fibrosis and fibroblast foci have the most notable expression of ER stress markers that colocalize with markers of apoptosis. The cause of ER stress in sporadic IPF is not well understood. Some potential factors involved in induction of ER stress in lung epithelium have been previously associated with IPF, such as herpesvirus infection and exposure to cigarette smoke (112, 113). Our studies using the herpesvirus-induced lung fibrosis murine model have shown that individuals with advanced age respond differently to lung injury, preferentially experiencing lung cell apoptosis and activation of profibrotic pathways (22). We also have shown that ER stress influences mitochondrial function in alveolar epithelial cells by downregulating the critical regulator of mitochondrial homeostasis PINK1 (17). It is possible that additional mechanisms are involved in this crosstalk between ER and mitochondria influencing susceptibility to age-related lung diseases.
Reduced autophagy.
Autophagy, a highly conserved process that participates in maintaining cellular energy resources and quality control by degrading unnecessary proteins and organelles, is an essential anti-aging pathway (25). As an adaptive response, autophagy is able to relieve stressful conditions in the cell, such as starvation, hypoxia, ER stress, oxidative stress, etc. (114). Conversely, IPF lungs have been reported to have a reduced rate of autophagy activity, which may contribute to rapid and sustained myofibroblast differentiation and resistance to apoptosis in lung fibroblasts that perpetuate cell senescence in lung epithelial cells (115–118). Deficient autophagy in IPF lungs has been associated with persistent activation of the serine/threonine protein kinase mammalian target of rapamycin (mTOR), potentially due to low levels of PTEN and consequent hyperactivation of Akt in response to collagen-rich matrices. The transcription factor forkhead box O3a (FOXO3A) is a proposed regulator of autophagy, and it contains conserved Akt phosphorylation sites that create a docking site for 14-3-3, preventing its nuclear translocation (119). Another potential mechanism for the inhibition of autophagy in IPF is related to the high concentration of TGF-β. TGF-β has been found to mediate activation of mTORC1 expression (115). TGF-β stimulation of lung fibroblasts has also been associated with regulation of the expression of LC3 and beclin through a mechanism that includes TGF-β–mediated induction of sphingosine-1 phosphate (120). Controversially, TGF-β also plays a role in the regulation of damaged mitochondria-specific autophagy or mitophagy. In lung fibroblasts, TGF-β has been shown to reduce the expression of PINK1, an important modulator of mitophagy (19), yet it has the opposite effect on PINK1 expression within the lung’s epithelial cells (85).
A more general approach to determine how inhibition of autophagy modulates the fibrotic response has used mice deficient in ATG4B, an essential autophagic factor for autophagosome formation (121). ATG4B expression increases during bleomycin-induced lung fibrosis, but most importantly, its deficiency causes more severe apoptosis, inflammation, and fibrotic responses, confirming that autophagy plays a protective role against lung fibrosis. Finally, we analyzed autophagic activity in young, old, and IPF human lung fibroblasts. Old and IPF lung fibroblasts have dysfunctional autophagy activity with increased LC3 puncta, p62 accumulation, and persistent activation of the mTOR pathway under starvation conditions. However, the activation of mTOR was further enhanced in IPF fibroblasts and associated with greater resistance to apoptosis (20). Altogether, these studies suggest that aberrant activation of mTOR and the resulting defective autophagy in aging IPF fibroblasts play a role in the development of lung fibrosis.
Metabolic dysregulation as a convergent event in the aging cell
One of the most fundamental alterations observed in aging cells is metabolic dysregulation. Mitochondria are the organelles that not only manage the cell’s energy production but can also detect danger and control cell death. Fibrosis results from abnormal tissue repair, and exaggerated remodeling is frequently associated with persistent and/or severe injury and cell apoptosis. Current evidence suggests that mitochondrial dysfunction plays a critical role in the vulnerability to fibrotic conditions (17, 19, 84, 85, 87, 122, 123). Tissue- and cell-specific pathways regulate or respond to mitochondrial function to meet ever-changing physiological needs. During cellular stress, cells adapt to protect mitochondria by activating different signaling pathways to maintain cellular homeostasis. Growing evidence suggests that DNA damage can cause mitochondrial dysfunction by activating nucleus-to-mitochondria signaling (124). Activation of DNA-damage sensors, particularly poly(ADP)ribose polymerase 1 (PARP1), leads to loss of NAD+ and acetyl-CoA, important molecules that regulate the activity of sirtuin and cellular metabolism (125). Loss of sirtuin activity mediates an increase in mitochondrial ROS and inactivation of AMPK, FOXO proteins, and PGC1-α. These alterations dysregulate mitochondrial biogenesis, mitophagy, metabolism, and the expression of mitochondrial antioxidant mechanisms (including superoxide dismutase and uncoupling proteins like UCP2) (126). Additionally, SIRT1 and SIRT6 are required for regulation of DNA repair pathways (127). A vicious cycle of PARP1 activation, NAD+ deficiency, and dysregulation of SIRT1 leads to increases in oxidative stress, and reduced DNA repair capacity might contribute to aging and susceptibility to age-related diseases such as IPF. In concordance, fibroblasts isolated from IPF patients exhibit significantly increased PARP1 expression and activity relative to fibroblasts isolated from control subjects, and PARP1-deficient mice show attenuated pulmonary fibrosis in the bleomycin model (128).
It is also recognized that mitochondrial metabolism can influence and modulate nuclear DNA and histone methylation. Existing evidence supports a redox regulation of epigenetics. Consequently, nuclear DNA methylation patterns change with copy number of mtDNA (129), mtDNA variants with different oxidative phosphorylation efficiencies (130), and the content of intracellular ROS (131). In addition, recent studies show the presence of epigenetic modifications in the mtDNA (132–136). Limited information exists about a possible relationship between aging and mitochondrial epigenetic modifications. Preliminary studies show that fibroblasts from old donors exhibit higher levels of mtDNA methylation (137).
Supporting the concept of a bidirectional signaling pathway between the nucleus and mitochondria, telomere attrition has been found to activate p53 with further regulation of mitochondrial function and metabolism by repression of PGC1-α and -β (important regulators of mitochondrial biogenesis). Consequently, generation 4 of mice deficient in TERT exhibit telomere shortening, and reduced number and function of mitochondria (138). Similarly, mitochondrial dysfunction can lead to telomere attrition. Studies using the uncoupling agent FCCP suggest that enhanced production of ROS induces genomic instability and influences telomere length (139). A more recent concept is the interplay between mtDNA and nuclear DNA. Using conplastic mice, mtDNA variants were shown to be sufficient in promoting differences in mitochondrial function and activation of adaptive cellular responses that can lead to alterations in glucose and lipid metabolism, telomere shortening, and even reduced lifespan. This evidence supports the concept that mitochondrial homeostasis is negatively regulated by aging-associated cell perturbations such as genomic instability, deficiency in proteostasis, and impaired autophagy.
Notably, mitochondrial dysfunction can drive aging cell phenotypes like senescence (140). Potential mechanisms of mitochondrial dysfunction–induced senescence include enhanced levels of ROS, sustained activation of AMPK, and alterations of the NAD+/NADH ratio (141–143). Importantly, NAD has been linked to both senescence and aging, and mitochondrial sirtuins such as the NAD-dependent proteins SIRT3 and SIRT5 can suppress senescence and modulate SASP (18, 140). Additional mitochondrial alterations that promote cellular senescence are aberrant mitochondrial dynamics, defective oxidative phosphorylation, and calcium homeostasis (144).
An additional mechanism is the mitochondrial unfolded protein response (UPRmit) that is activated by mitochondrial proteotoxic stress. UPRmit monitors not only the degradation of defective proteins in the mitochondria but also mitochondrial protein import as an indirect indicator of mitochondrial fitness, which is linked to transcription of quality control components (145, 146). As a consequence, synthesis of mitochondrial proteases and mitophagy are adapted accordingly to allow cell survival during UPRmit. In parallel, the dysregulation of mitochondrial proteases has been associated with aging and age-related lung diseases (147).
A possibility exists that mitochondrial dysfunction is a convergent point for aging-related cellular perturbations, eventually becoming a “point of no return” for aging cells and a critical contributor to the pathogenesis of chronic lung diseases such as IPF (Figure 2).
Figure 2. Mechanisms of aging and the pathogenesis of IPF.

Several of the aging cell perturbations associated with IPF lungs converge to produce mitochondrial dysfunction, including DNA damage/genomic instability, altered proteostasis, reduced autophagy, and telomere attrition. Additionally, mitochondrial dysfunction can lead to cellular senescence and stem cell exhaustion. We propose that mitochondrial dysfunction plays a central role in the process of aging and the pathogenesis of IPF.
Perspective on aging regulation in IPF pathology and treatment
Current efforts exist to integrate the normal physiology of aging with the pathobiology of age-related chronic diseases like IPF. A new understanding of how biological systems change with age offers novel pathways that might impact not only the human lifespan but also the extension of human healthspan. Studies in the IPF lung strongly support the concept that aging promotes this disease. As we described above, multiple pathways associated with aging are activated in lung epithelial cells, fibroblasts, and progenitor cells in the IPF lung. Imperative goals in the field are the incorporation not only of aged mice as animal models of lung fibrosis, but of the use of primary human cells and tissues from healthy aging controls in order to clearly illustrate the dynamic connection between aging and disease.
In this new era of gerosciences and with the identification of common pathways in age-related diseases, one of the ultimate goals is to develop preventive and therapeutic approaches to multiple age-related diseases. New data suggest that despite the apparent inevitability of this aging process, interventions such as intermittent fasting, sustained calorie restriction, exercise, and pharmacological treatments (i.e., rapamycin and metformin) will eventually extend the human lifespan (148–154). Additionally, approaches to target specific aging pathways are under development, such as senolytic drugs to preferentially induce apoptosis in senescent cells (155). Our studies support the idea that mitochondria dysfunction can be an important factor in susceptibility to IPF. Mitochondria-targeted therapies have been developed to target (a) specific mitochondrial diseases, or (b) general mitochondrial homeostasis processes that can be applied to a wide spectrum of diseases (156). The second group of strategies includes regulation of mitochondrial biogenesis, activation of mitophagy, inhibition of mitochondrial apoptosis, scavenging of toxic compounds, and bypass of electron transport chain defects. For instance, the peroxisome proliferator–activated receptor (PPAR) and its coactivator PGC1-α play important regulatory role in biogenesis, and this pathway is a potential target for mitochondrial therapy. AMPK agonists that can indirectly activate PGC1-α, or bezafibrate (a PPAR agonist), have been used to stimulate mitochondrial biogenesis and mediate protective effects in myopathy and neurodegenerative disease models associated with mitochondrial dysfunction (157, 158). For instance, a transgenic mouse model of mitochondrial catalase mediates protection against asbestos, and bleomycin induced lung fibrosis associated with prevention of mitochondrial DNA damage (159). Hexafluoro, a novel honokiol derivative, has been shown to induce SIRT3 expression in lung fibroblasts and protect from TGF-β–mediated SIRT3 depletion and ameliorate bleomycin-induced lung fibrosis in mice (90). Our studies on mitochondria homeostasis have shown preferential mitophagy dysregulation, and alterations in fusion/fission dynamics are deregulated in the fibrotic lung. Inhibitors of mitochondrial fission such as the specific DRP1 inhibitor Mdivi-1 have been shown to rescue abnormal mitochondrial dynamics in models of chronic obstructive pulmonary disease (160). Induction of selective mitochondrial turnover of defective mitochondria has been achieved by overexpression of parkin in a model of heteroplasmic cybrid cells (161). Interestingly, studies in humans support the concept that aerobic exercise might increase proteins involved with mitochondrial biogenesis and mitochondrial dynamics (162). Additionally, the fact that mitochondrial dysfunction can be the result of other aging-related cell perturbations, including changes in proteostasis and reduced autophagy, might suggest that therapies targeting these cell dysfunctions can also beneficially affect the mitochondria and promote lung repair. These novel approaches have the potential to be transformative. In the future, innovative drugs may be able to prevent, delay, or reverse multiple or specific aging-related chronic diseases, including IPF, ultimately improving not only an individual’s lifespan but also, and most notably, quality of life.
Acknowledgments
ALM was funded by NIH R01-HL131789A, R01-HL131789-01, and the Aging Institute, University of Pittsburgh. MR was funded by NIH R01-HL123766-01A1. Special thanks to M.P. Rojas, D. Zank, and K. Fiedler for text editing.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2017;127(2):405–414.
https://doi.org/10.1172/JCI87440.
Contributor Information
Marta Bueno, Email: mbueno@pitt.edu.
Mauricio Rojas, Email: rojasm@upmc.edu.
References
- 1. Bloom DE, Shannon S. The demographic of aging. In: Molecular Aspects of Aging: Understanding Lung Aging. Rojas M, Meiners S, Le Saux CJ, eds. Hoboken, New Jersey, USA: Wiley-Blackwell; 2014:1–12. [Google Scholar]
- 2.Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol. 2013;5(1):483–492. doi: 10.2147/CLEP.S54815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967–972. doi: 10.1164/ajrccm.150.4.7921471. [DOI] [PubMed] [Google Scholar]
- 4.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 5.Fernández Pérez ER, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137(1):129–137. doi: 10.1378/chest.09-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61(11):980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Natsuizaka M, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190(7):773–779. doi: 10.1164/rccm.201403-0566OC. [DOI] [PubMed] [Google Scholar]
- 8.Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011;66(6):462–467. doi: 10.1136/thx.2010.148031. [DOI] [PubMed] [Google Scholar]
- 9.Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48(1):179–186. doi: 10.1183/13993003.01653-2015. [DOI] [PubMed] [Google Scholar]
- 10.Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc. 2014;11(8):1176–1185. doi: 10.1513/AnnalsATS.201404-145OC. [DOI] [PubMed] [Google Scholar]
- 11.Collard HR, et al. Health care utilization and costs of idiopathic pulmonary fibrosis in U.S. Medicare beneficiaries aged 65 years and older. Ann Am Thorac Soc. 2015;12(7):981–987. doi: 10.1513/AnnalsATS.201412-553OC. [DOI] [PubMed] [Google Scholar]
- 12.Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408(6809):233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 13.Kirkwood TB, Rose MR. Evolution of senescence: late survival sacrificed for reproduction. Philos Trans R Soc Lond B Biol Sci. 1991;332(1262):15–24. doi: 10.1098/rstb.1991.0028. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Green FH, Smiley-Jewell SM, Pinkerton KE. Susceptibility of the aging lung to environmental injury. Semin Respir Crit Care Med. 2010;31(5):539–553. doi: 10.1055/s-0030-1265895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copley SJ, et al. Lung morphology in the elderly: comparative CT study of subjects over 75 years old versus those under 55 years old. Radiology. 2009;251(2):566–573. doi: 10.1148/radiol.2512081242. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto M, et al. Elimination of p19(ARF)-expressing cells enhances pulmonary function in mice. JCI Insight. 2016;1(12):e87732. doi: 10.1172/jci.insight.87732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bueno M, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125(2):521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6(4):e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFβ1. Aging Cell. 2015;14(5):774–783. doi: 10.1111/acel.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romero Y, et al. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contribute to apoptosis resistance in IPF fibroblasts. Aging Cell. doi: 10.1111/acel.12514. [published online ahead of print August 26, 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sueblinvong V, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. 2012;344(1):41–51. doi: 10.1097/MAJ.0b013e318234c132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torres-González E, et al. Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am J Respir Cell Mol Biol. 2012;46(6):748–756. doi: 10.1165/rcmb.2011-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecker L, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6(231):231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustos ML, et al. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am J Respir Crit Care Med. 2014;189(7):787–798. doi: 10.1164/rccm.201306-1043OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neri S, et al. Mismatch repair system and aging: microsatellite instability in peripheral blood cells from differently aged participants. J Gerontol A Biol Sci Med Sci. 2005;60(3):285–292. doi: 10.1093/gerona/60.3.285. [DOI] [PubMed] [Google Scholar]
- 27.Fingerlin TE, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathai SK, Schwartz DA, Warg LA. Genetic susceptibility and pulmonary fibrosis. Curr Opin Pulm Med. 2014;20(5):429–435. doi: 10.1097/MCP.0000000000000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vassilakis DA, Sourvinos G, Spandidos DA, Siafakas NM, Bouros D. Frequent genetic alterations at the microsatellite level in cytologic sputum samples of patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2000;162(3 pt 1):1115–1119. doi: 10.1164/ajrccm.162.3.9911119. [DOI] [PubMed] [Google Scholar]
- 30.Demopoulos K, Arvanitis DA, Vassilakis DA, Siafakas NM, Spandidos DA. MYCL1, FHIT, SPARC, p16(INK4) and TP53 genes associated to lung cancer in idiopathic pulmonary fibrosis. J Cell Mol Med. 2002;6(2):215–222. doi: 10.1111/j.1582-4934.2002.tb00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori M, et al. Microsatellite instability in transforming growth factor-β1 type II receptor gene in alveolar lining epithelial cells of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2001;24(4):398–404. doi: 10.1165/ajrcmb.24.4.4206. [DOI] [PubMed] [Google Scholar]
- 32.Korfei M, et al. Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J Proteome Res. 2011;10(5):2185–2205. doi: 10.1021/pr1009355. [DOI] [PubMed] [Google Scholar]
- 33.Bell JT, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8(4):e1002629. doi: 10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsai PC, Spector TD, Bell JT. Using epigenome-wide association scans of DNA methylation in age-related complex human traits. Epigenomics. 2012;4(5):511–526. doi: 10.2217/epi.12.45. [DOI] [PubMed] [Google Scholar]
- 35.Kreiling JA, et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell. 2011;10(2):292–304. doi: 10.1111/j.1474-9726.2010.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiers H, Hannon E, Wells S, Williams B, Fernandes C, Mill J. Age-associated changes in DNA methylation across multiple tissues in an inbred mouse model. Mech Ageing Dev. 2016;154:20–23. doi: 10.1016/j.mad.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christensen BC, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(11):1263–1272. doi: 10.1164/rccm.201408-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalo S. Epigenetic alterations in aging. J Appl Physiol (1985) 2010;109(2):586–597. doi: 10.1152/japplphysiol.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16(1):93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 41.Hayakawa T, et al. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One. 2015;10(1):e0116480. doi: 10.1371/journal.pone.0116480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J. 2014;43(5):1448–1458. doi: 10.1183/09031936.00095113. [DOI] [PubMed] [Google Scholar]
- 43.Abdelmohsen K, Gorospe M. Noncoding RNA control of cellular senescence. Wiley Interdiscip Rev RNA. 2015;6(6):615–629. doi: 10.1002/wrna.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res. 2011;157(4):191–199. doi: 10.1016/j.trsl.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Pandit KV, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182(2):220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Disayabutr S, et al. miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11(6):e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selman M, López-Otín C, Pardo A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J. 2016;48(2):538–552. doi: 10.1183/13993003.00398-2016. [DOI] [PubMed] [Google Scholar]
- 48.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grammatikakis I, Panda AC, Abdelmohsen K, Gorospe M. Long noncoding RNAs(lncRNAs) and the molecular hallmarks of aging. Aging (Albany NY) 2014;6(12):992–1009. doi: 10.18632/aging.100710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang C, Yang Y, Liu L. Interaction of long noncoding RNAs and microRNAs in the pathogenesis of idiopathic pulmonary fibrosis. Physiol Genomics. 2015;47(10):463–469. doi: 10.1152/physiolgenomics.00064.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song X, et al. Analysing the relationship between lncRNA and protein-coding gene and the role of lncRNA as ceRNA in pulmonary fibrosis. J Cell Mol Med. 2014;18(6):991–1003. doi: 10.1111/jcmm.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armanios MY, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 53.Lawson WE, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59(11):977–980. doi: 10.1136/thx.2004.026336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res. 2012;730(1–2):52–58. doi: 10.1016/j.mrfmmm.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alder JK, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(7):729–737. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diaz de Leon A, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5(5):e10680. doi: 10.1371/journal.pone.0010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diaz de Leon A, et al. Subclinical lung disease, macrocytosis, and premature graying in kindreds with telomerase (TERT) mutations. Chest. 2011;140(3):753–763. doi: 10.1378/chest.10-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kannengiesser C, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. 2015;46(2):474–485. doi: 10.1183/09031936.00040115. [DOI] [PubMed] [Google Scholar]
- 60.Silhan LL, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. 2014;44(1):178–187. doi: 10.1183/09031936.00060014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stuart BD, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512–517. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsakiri KD, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsang AR, Wyatt HD, Ting NS, Beattie TL. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell. 2012;11(3):482–490. doi: 10.1111/j.1474-9726.2012.00810.x. [DOI] [PubMed] [Google Scholar]
- 64.Alder JK, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112(16):5099–5104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015;12(2):286–299. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 66.Liu T, Ullenbruch M, Young Choi Y, Yu H, Ding L, et al. Telomerase and telomere length in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49(2):260–268. doi: 10.1165/rcmb.2012-0514OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cusanelli E, Chartrand P. Telomeric repeat-containing RNA TERRA: a noncoding RNA connecting telomere biology to genome integrity. Front Genet. 2015;6:143. doi: 10.3389/fgene.2015.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu J, et al. Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64(7):731–739. doi: 10.1093/gerona/glp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen R, et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J Biol Chem. 2015;290(52):30813–30829. doi: 10.1074/jbc.M115.681619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin C, et al. Protease activated receptor-1 regulates macrophage-mediated cellular senescence: a risk for idiopathic pulmonary fibrosis. Oncotarget. 2015;6(34):35304–35314. doi: 10.18632/oncotarget.6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yanai H, et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 2015;7(9):664–672. doi: 10.18632/aging.100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanders YY, Liu H, Liu G, Thannickal VJ. Epigenetic mechanisms regulate NADPH oxidase-4 expression in cellular senescence. Free Radic Biol Med. 2015;79:197–205. doi: 10.1016/j.freeradbiomed.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 73.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106(40):17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maciel-Barón LA, et al. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age (Dordr) 2016;38(1):26. doi: 10.1007/s11357-016-9886-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stout-Delgado HW, et al. Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am J Respir Cell Mol Biol. 2016;55(2):252–263. doi: 10.1165/rcmb.2015-0222OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Powter EE, et al. Caveolae control the anti-inflammatory phenotype of senescent endothelial cells. Aging Cell. 2015;14(1):102–111. doi: 10.1111/acel.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, Le Saux CJ. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am J Respir Cell Mol Biol. 2012;47(1):28–36. doi: 10.1165/rcmb.2011-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Minagawa S, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L391–L401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herbener GH. A morphometric study of age-dependent changes in mitochondrial population of mouse liver and heart. J Gerontol. 1976;31(1):8–12. doi: 10.1093/geronj/31.1.8. [DOI] [PubMed] [Google Scholar]
- 81.Lambert AJ, et al. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6(5):607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- 82.Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62(3):235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51(5):440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 84.Hawkins A, et al. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L33–L47. doi: 10.1152/ajplung.00217.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Patel AS, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One. 2015;10(3):e0121246. doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kobayashi K, et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol. 2016;197(2):504–516. doi: 10.4049/jimmunol.1600265. [DOI] [PubMed] [Google Scholar]
- 87.Sundaresan NR, et al. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol Cell Biol. 2015;36(5):678–692. doi: 10.1128/MCB.00586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sosulski ML, Gongora R, Feghali-Bostwick C, Lasky JA, Sanchez CG. Sirtuin 3 deregulation promotes pulmonary fibrosis. J Gerontol A Biol Sci Med Sci. doi: 10.1093/gerona/glw151. doi: 10.1093/gerona/glw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci. 2015;16(9):21486–21519. doi: 10.3390/ijms160921486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Akamata K, et al. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. doi: 10.18632/oncotarget.12504. [published online ahead of print October 6, 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng Y, et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013;4:e731. doi: 10.1038/cddis.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim SJ, et al. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem. 2014;289(9):6165–6176. doi: 10.1074/jbc.M113.515130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheresh P, et al. Asbestos-induced pulmonary fibrosis is augmented in 8-oxoguanine DNA glycosylase knockout mice. Am J Respir Cell Mol Biol. 2015;52(1):25–36. doi: 10.1165/rcmb.2014-0038OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Panduri V, et al. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic Biol Med. 2009;47(6):750–759. doi: 10.1016/j.freeradbiomed.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fahn HJ, et al. Age-related 4,977 bp deletion in human lung mitochondrial DNA. Am J Respir Crit Care Med. 1996;154(4 pt 1):1141–1145. doi: 10.1164/ajrccm.154.4.8887618. [DOI] [PubMed] [Google Scholar]
- 96.Lee HC, Lu CY, Fahn HJ, Wei YH. Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. FEBS Lett. 1998;441(2):292–296. doi: 10.1016/S0014-5793(98)01564-6. [DOI] [PubMed] [Google Scholar]
- 97.Kwon Y, Kim J, Lee CY, Kim H. Expression of SIRT1 and SIRT3 varies according to age in mice. Anat Cell Biol. 2015;48(1):54–61. doi: 10.5115/acb.2015.48.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 99.Kropski JA, et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am J Respir Crit Care Med. 2015;191(4):417–426. doi: 10.1164/rccm.201406-1162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lawson WE, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108(26):10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta. 2013;1832(7):940–947. doi: 10.1016/j.bbadis.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yao Y, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24(5):915–925. doi: 10.1038/mt.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baker TA, Bach HH, Gamelli RL, Love RB, Majetschak M. Proteasomes in lungs from organ donors and patients with end-stage pulmonary diseases. Physiol Res. 2014;63(3):311–319. doi: 10.33549/physiolres.932607. [DOI] [PubMed] [Google Scholar]
- 104.Semren N, et al. Regulation of 26S Proteasome Activity in Pulmonary Fibrosis. Am J Respir Crit Care Med. 2015;192(9):1089–1101. doi: 10.1164/rccm.201412-2270OC. [DOI] [PubMed] [Google Scholar]
- 105.Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem. 2010;285(29):22103–22113. doi: 10.1074/jbc.M110.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 107.Thomas AQ, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 108.Wang Y, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peljto AL, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309(21):2232–2239. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Korfei M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lawson WE, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 112.Jorgensen E, Stinson A, Shan L, Yang J, Gietl D, Albino AP. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer. 2008;8:229. doi: 10.1186/1471-2407-8-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee DY, Sugden B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood. 2008;111(4):2280–2289. doi: 10.1182/blood-2007-07-100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Araya J, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304(1):L56–L69. doi: 10.1152/ajplung.00213.2012. [DOI] [PubMed] [Google Scholar]
- 116.Nho RS, Hergert P. IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS One. 2014;9(4):e94616. doi: 10.1371/journal.pone.0094616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Patel AS, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7(7):e41394. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ricci A, et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J Cell Physiol. 2013;228(7):1516–1524. doi: 10.1002/jcp.24307. [DOI] [PubMed] [Google Scholar]
- 119.Im J, Hergert P, Nho RS. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. Am J Physiol Lung Cell Mol Physiol. 2015;309(6):L552–L561. doi: 10.1152/ajplung.00079.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang LS, et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: role of S1P signalling and autophagy. Thorax. 2015;70(12):1138–1148. doi: 10.1136/thoraxjnl-2014-206684. [DOI] [PubMed] [Google Scholar]
- 121.Cabrera S, et al. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11(4):670–684. doi: 10.1080/15548627.2015.1034409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang SX, Jaurand MC, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health B Crit Rev. 2011;14(1-4):179–245. doi: 10.1080/10937404.2011.556051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kamp DW, Panduri Va, Weitzman SA, Chandel N. Asbestos-induced alveolar epithelial cell apoptosis: role of mitochondrial dysfunction caused by iron-derived free radicals. Mol Cell Biochem. 2002;234-235(1-2):153–160. [PubMed] [Google Scholar]
- 124.Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. 2016;17(5):308–321. doi: 10.1038/nrm.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fang EF, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157(4):882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 127.Toiber D, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. 2013;51(4):454–468. doi: 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hu B, Wu Z, Hergert P, Henke CA, Bitterman PB, Phan SH. Regulation of myofibroblast differentiation by poly(ADP-ribose) polymerase 1. Am J Pathol. 2013;182(1):71–83. doi: 10.1016/j.ajpath.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther. 2008;7(8):1182–1190. doi: 10.4161/cbt.7.8.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bellizzi D, D’Aquila P, Giordano M, Montesanto A, Passarino G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics. 2012;4(1):17–27. doi: 10.2217/epi.11.109. [DOI] [PubMed] [Google Scholar]
- 131.Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med. 2007;43(7):1023–1036. doi: 10.1016/j.freeradbiomed.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009;6(1):65–72. doi: 10.4161/rna.6.1.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rebelo AP, Williams SL, Moraes CT. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 2009;37(20):6701–6715. doi: 10.1093/nar/gkp727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108(9):3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sripada L, Tomar D, Prajapati P, Singh R, Singh AK, Singh R. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PLoS One. 2012;7(9):e44873. doi: 10.1371/journal.pone.0044873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J Biol Chem. 1983;258(15):9078–9085. [PubMed] [Google Scholar]
- 138.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu L, Trimarchi JR, Smith PJ, Keefe DL. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell. 2002;1(1):40–46. doi: 10.1046/j.1474-9728.2002.00004.x. [DOI] [PubMed] [Google Scholar]
- 140.Wiley CD, et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016;23(2):303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013;493(7434):689–693. doi: 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee SM, Dho SH, Ju SK, Maeng JS, Kim JY, Kwon KS. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology. 2012;13(5):525–536. doi: 10.1007/s10522-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 143.Passos JF, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell. 2015;14(1):1–7. doi: 10.1111/acel.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497(7450):451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Quirós PM, Langer T, López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16(6):345–359. doi: 10.1038/nrm3984. [DOI] [PubMed] [Google Scholar]
- 147.Quirós PM, Ramsay AJ, López-Otín C. New roles for OMA1 metalloprotease: From mitochondrial proteostasis to metabolic homeostasis. Adipocyte. 2013;2(1):7–11. doi: 10.4161/adip.21999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Johnson TE. 25 years after age-1: genes, interventions and the revolution in aging research. Exp Gerontol. 2013;48(7):640–643. doi: 10.1016/j.exger.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kenyon CJ. The genetics of ageing. Nature. 2010;464(7288):504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 150.Longo VD, et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell. 2015;14(4):497–510. doi: 10.1111/acel.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19(2):181–192. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Masoro EJ, Shimokawa I, Higami Y, McMahan CA, Yu BP. Temporal pattern of food intake not a factor in the retardation of aging processes by dietary restriction. J Gerontol A Biol Sci Med Sci. 1995;50A(1):B48–B53. doi: 10.1093/gerona/50A.1.B48. [DOI] [PubMed] [Google Scholar]
- 153.Mattson MP, et al. Meal frequency and timing in health and disease. Proc Natl Acad Sci U S A. 2014;111(47):16647–16653. doi: 10.1073/pnas.1413965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Morgan TE, Wong AM, Finch CE. Anti-inflammatory mechanisms of dietary restriction in slowing aging processes. Interdiscip Top Gerontol. 2007;35:83–97. doi: 10.1159/000096557. [DOI] [PubMed] [Google Scholar]
- 155.Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Viscomi C, Bottani E, Zeviani M. Emerging concepts in the therapy of mitochondrial disease. Biochim Biophys Acta. 2015;1847(6–7):544–557. doi: 10.1016/j.bbabio.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 157.Viscomi C, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011;14(1):80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Johri A, et al. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum Mol Genet. 2012;21(5):1124–1137. doi: 10.1093/hmg/ddr541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kim SJ, et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic Biol Med. 2016;101:482–490. doi: 10.1016/j.freeradbiomed.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mizumura K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107(26):11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Konopka AR, Suer MK, Wolff CA, Harber MP. Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J Gerontol A Biol Sci Med Sci. 2014;69(4):371–378. doi: 10.1093/gerona/glt107. [DOI] [PMC free article] [PubMed] [Google Scholar]